Journal of Modern Physics
Vol.2 No.10(2011), Article ID:8207,6 pages DOI:10.4236/jmp.2011.210146
Spin-Orbit Electronic Structure of the ScBr Molecule
Faculty of Science, Beirut Arab University, Beirut, Lebanon
E-mail: *fkorek@yahoo.com
Received April 2, 2011; revised May 3, 2011; accepted June 1, 2011
Keywords: ScBr Molecule, Ab Initio Calculation, Spin-Orbit Calculation, Potential Energy Curves, Spectroscopic Constants
ABSTRACT
A theoretical investigation of the spin-orbit electronic states of the molecule ScBr has been performed via CASSCF and MRCI (single and double excitations with Davidson correction) calculations. Spin-orbit effects have been introduced through semi-empirical spin orbit pseudo-potential for scandium while they have been neglected for bromine. Potential energy curves for 42 electronic states in the representation W(±) have been determined along with the corresponding spectroscopic constants. The comparison of the present results with those available in the literature shows a good agreement. New results have been investigated in present work for 30 electronic states in the representation W(±) for the first time.
1. Introduction
Scandium is the first of the transition metal atoms which has only one d-electron in the ground electronic state. Interest in its compounds thus arises because they can be considered as prototypes for understanding the role that d-orbitals play in chemical bonding and chemiluminescence reactions [1]. The spectra and structure of scandium monochloride ScCl and scandium monofloride ScF have been the subject of many theoretical and experimental studies while scandium monobromide ScB) is the least studied spectroscopically. In literature the experimental and theoretical data in the representation 2s+1L(±) of the molecule ScBr have been previously reported by Fischell et al. [2], Langhoff et al. [3] and Lin et al. [3]. The only investigated electronic states of this molecule with spin orbit effect are those calculated by Bencheikh [4] using the ligand field theory. Recently [5] we studied the electronic structure of 23 electronic states of the molecule ScBr without spin-orbit effect. In the present work, an ab initio investigation of 42 low-lying electronic states with spin-orbit effect of the molecule ScBr has been performed. The potential energy curves (PECs) along with the transition energy with respect to the minimum energy of the ground state Te, the equilibrium internuclear distance Re, the harmonic frequency we, the permanent dipole moment and the rotational constant Be, have been calculated for these spin-orbit electronic states.
2. Computational Approach
The PECs of the lowest-lying electronic states of the molecule ScBr have been investigated via CASSCF method. Multireference CI calculations (single and double excitations with Davidson corrections) were performed to determine the correlation effects. The entire CASSCF configuration space was used as the reference in the MRCI calculations. These calculations have been performed via the computational chemistry program MOLPRO [6] taking advantage of the graphical user interface GABEDIT [7]. Scandium species is treated in all electron scheme, the 21 electrons of the scandium atom are considered using a contracted Gaussian basis set used by Langhoff et al. [3] for s, d, and f functions then we added p function from 6-31G**basis set [8] which has 16 diffused functions contracted set of Gaussian functions. The bromine species is treated as a system with 28 inner electrons taken into account through pseudo potential  [9] together with the corresponding Gaussian basis set from the basis LANL2DZ-ECP for s, p functions and we have added d function from 6-311G basis set [10] which has five d functions contracted to two functions, also we added f function from SDB-aug-cc-pVTZ basis set [11] which has two f diffused functions contracted to one f function given a final [2s 2p 2d 1f ] contracted basis. The energies for W(±) states have been obtained from the diagonalization of the matrix energy corresponding to a total hamiltonian, which is the sum of the electrostatic hamiltonian previously treated at the CASSCF/MRCI level [5] and the SO pseudo-potential
[9] together with the corresponding Gaussian basis set from the basis LANL2DZ-ECP for s, p functions and we have added d function from 6-311G basis set [10] which has five d functions contracted to two functions, also we added f function from SDB-aug-cc-pVTZ basis set [11] which has two f diffused functions contracted to one f function given a final [2s 2p 2d 1f ] contracted basis. The energies for W(±) states have been obtained from the diagonalization of the matrix energy corresponding to a total hamiltonian, which is the sum of the electrostatic hamiltonian previously treated at the CASSCF/MRCI level [5] and the SO pseudo-potential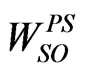 . This matrix was built up on the basis of CASSCF wave functions, while diagonal matrix elements came from CI plus Davidson correction calculations. Among the 28 electrons explicitly considered for ScBr (21 electrons for Sc and 7 for Br) 18 inner electrons were frozen in subsequent calculations so that 10 valance electrons were explicitly treated. The active space contains
. This matrix was built up on the basis of CASSCF wave functions, while diagonal matrix elements came from CI plus Davidson correction calculations. Among the 28 electrons explicitly considered for ScBr (21 electrons for Sc and 7 for Br) 18 inner electrons were frozen in subsequent calculations so that 10 valance electrons were explicitly treated. The active space contains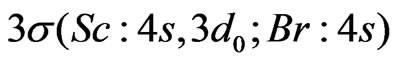 ,
,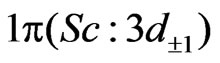 and
and 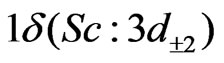 orbitals in the C2v symmetry; this corresponds to 7 active molecular orbitals distributed into irreducible representation a1, b1, b2 and a2 in the following way: 4a1, 1b1, 1b2, 1a2, noted [4,1,1,1], while the doubly occupied orbitals
orbitals in the C2v symmetry; this corresponds to 7 active molecular orbitals distributed into irreducible representation a1, b1, b2 and a2 in the following way: 4a1, 1b1, 1b2, 1a2, noted [4,1,1,1], while the doubly occupied orbitals 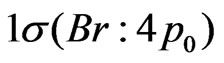 and
and 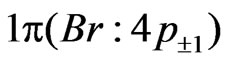 of bromine have been considered as inactive in the CAS-SCF calculations.
of bromine have been considered as inactive in the CAS-SCF calculations.
The calculations have been performed for 100 internuclear distances in the range 2.00 Å ≤ R ≤ 3.00 Å for 42 states in the representation![]() . The PECs for the symmetries W = 0+, 0-, 1, 2, 3, 4 are drawn respectively in the Figures 1-5.
. The PECs for the symmetries W = 0+, 0-, 1, 2, 3, 4 are drawn respectively in the Figures 1-5.
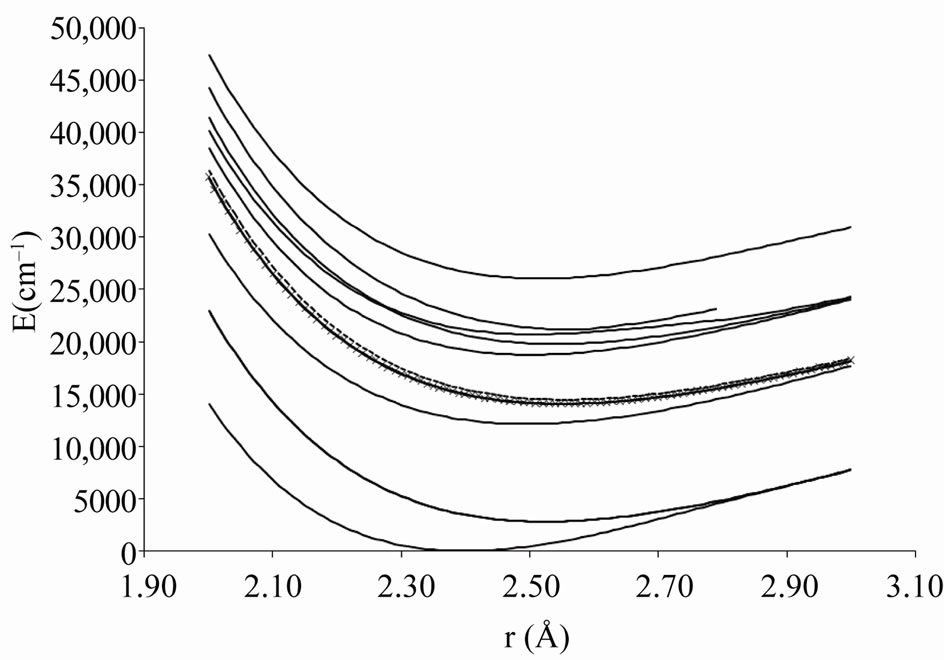
Figure 1. Potential energy curves of the lowest electronic states W = 0+ of the molecule ScBr.
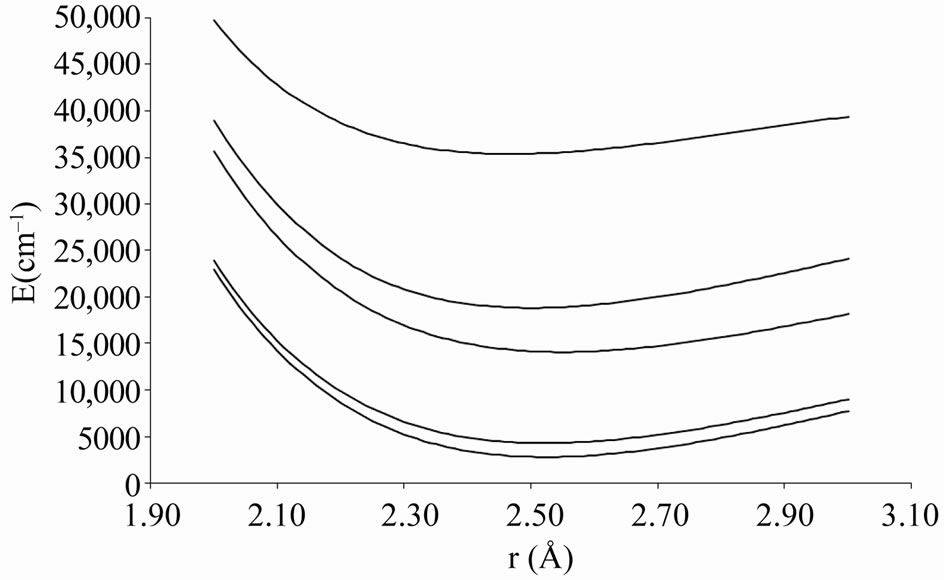
Figure 2. Potential energy curves of the lowest electronic states W = 0– of the molecule ScBr.
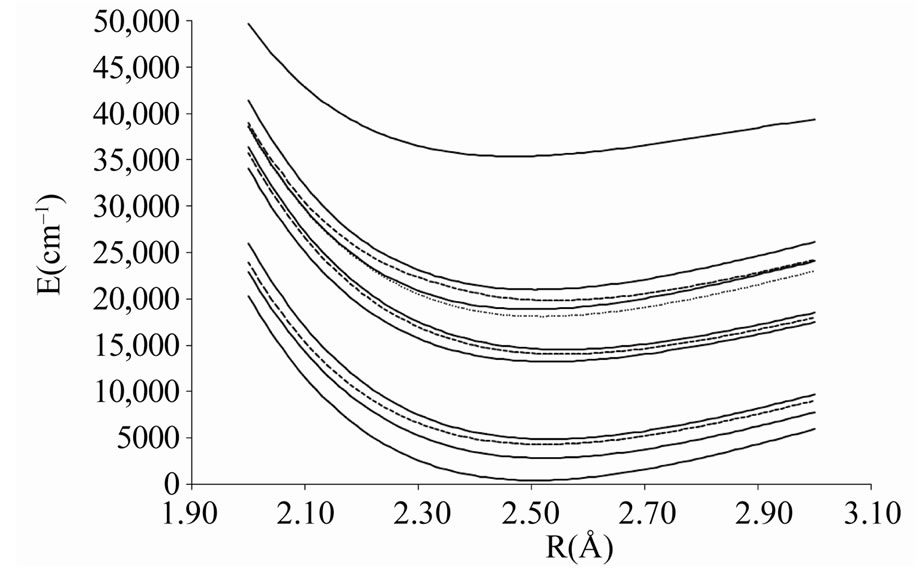
Figure 3. Potential energy curves of the lowest electronic states W = 1 of the molecule ScBr.
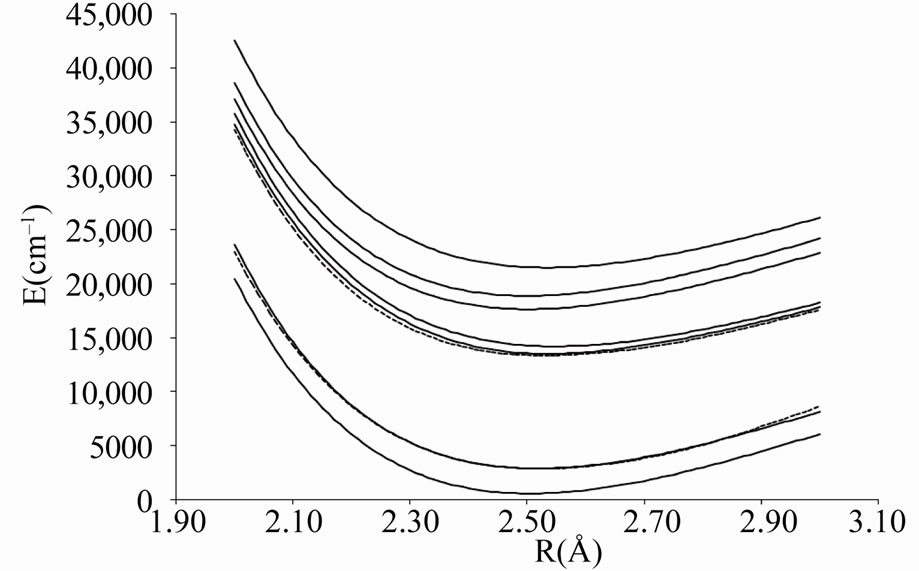
Figure 4. Potential energy curves of the lowest electronic states W = 2 of the molecule ScBr.
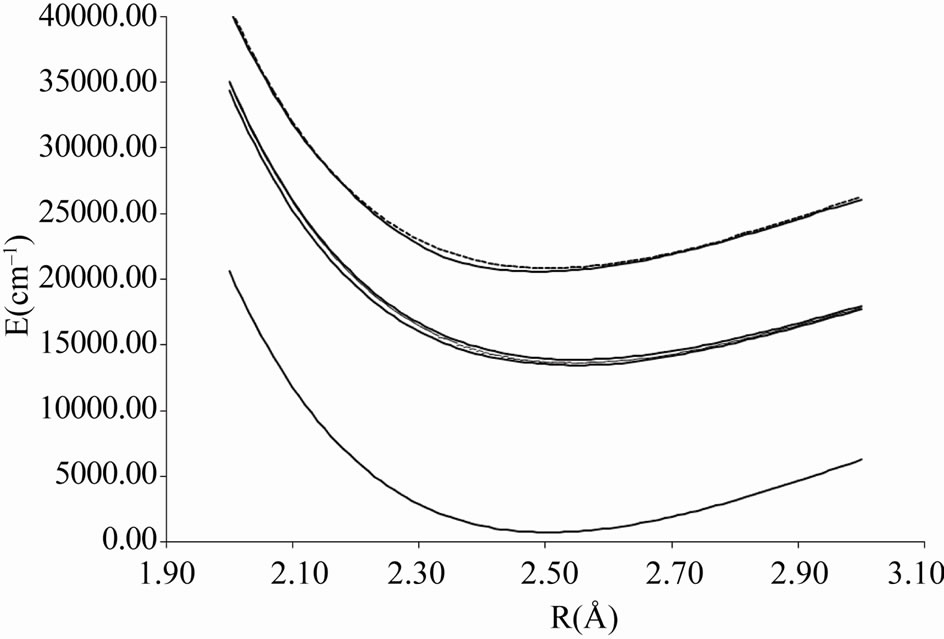
Figure 5. Potential energy curves of the lowest electronic states W = 3, 4 of the molecule ScBr.
In the range of R considered, the avoided crossings that appear between the investigated W-states are displayed in Table 1 together with their positions RAC, the corresponding parent states and the energy difference between two states (n + 1)W/(n)W at these points.
This is illustrated by the avoided crossing at RAC = 2.26 Å for the states (8)0+/(7)0+ in Figure 6 where the parent states are respectively (3)1S+ and (2)3S-.
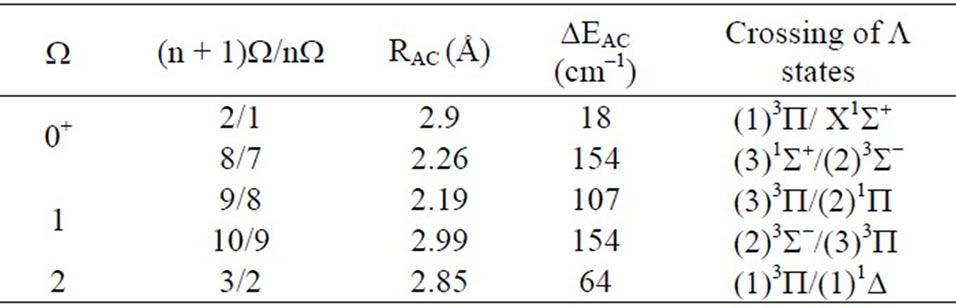
Table 1. Positions of the avoided crossing RAC and the energy difference EAC at these points with the corresponding crossing of L states for W states of ScBr.
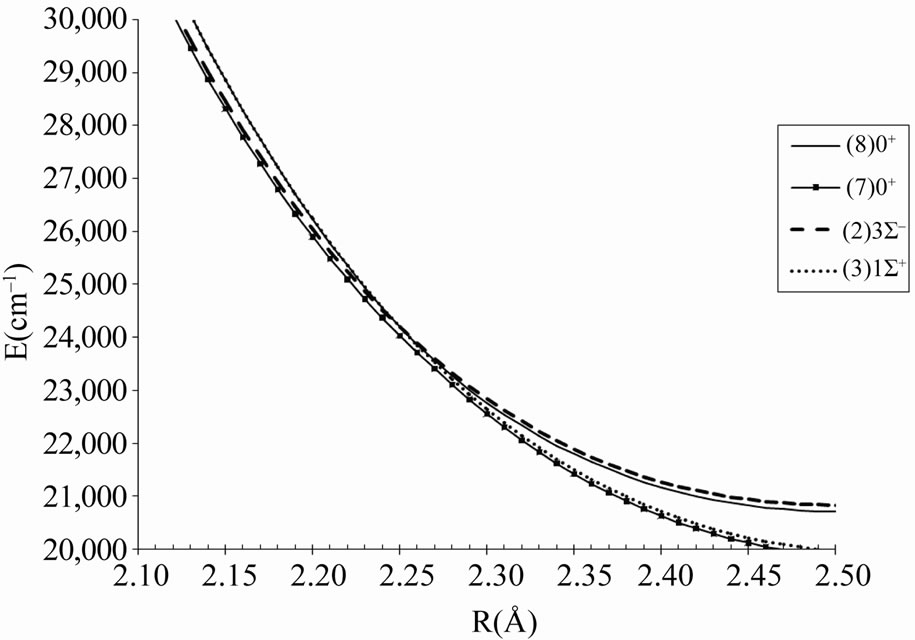
Figure 6. The avoided crossings between the electronic states (7)0+ and (8)0+ with the corresponding crossing between the parent states (3)1S+ and (2)3S- of the molecule ScBr.
The composition in percentage of the W state wave functions in terms of the L states, calculated at R = 2.4Å, is presented in Table 2.
For each state W there is a predominant component L with a contribution larger than 57.54% so that a main parent SL may be identified. Nevertheless, the states where the contribution of the other L is smaller than the dominant also have been obtained. For instance, the wave functions for the states (1)W = 0+ and (2)W = 0+ are mixing of the two crossing states X1S+ and (1)3P, and the states (7)W = 0+ and (8)W = 0+ are mixing of the two states (2)3S- and (3)1S+. The wave functions for the states (8)W = 1 and (9)W = 1 are mixing of the two closest states (2)1P and (3)3P, while those for the states (9)W = 1 and (10)W = 1 are mixing of the two closest states (3)3P and (2)3S-. The wave functions for the states (2)W = 2 and (3)W = 2 are mixing of the two closest states (1)1D and (1)3P. These results are consistent with the avoided crossings and crossings reported in Table 2.
The spectroscopic constants such as the vibrational harmonic constant we, the internuclear distance at equilibrium Re, the rotation constant Be, and the electronic transition energy with respect to the ground state Te have been calculated by fitting the energy values around the equilibrium position to a polynomial in terms of the in-

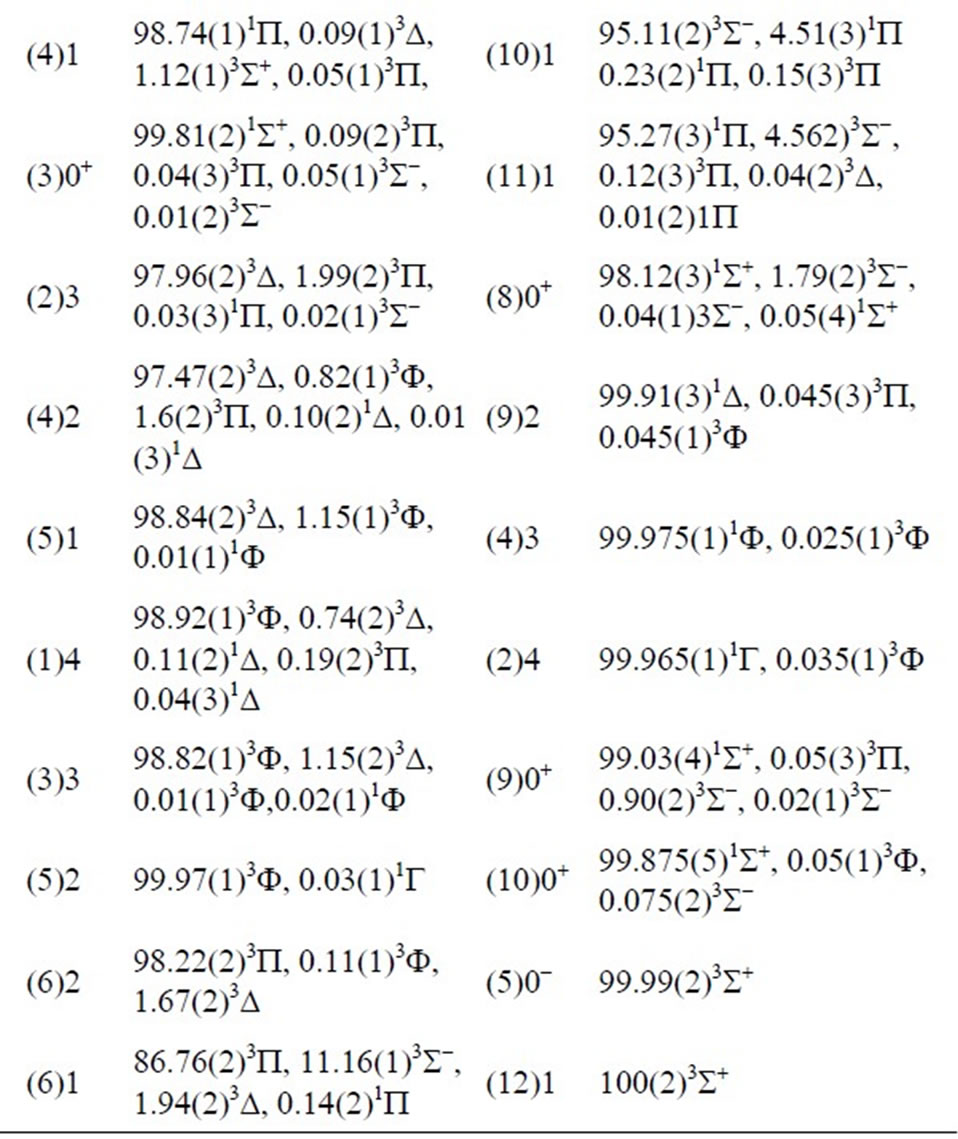
Table 2. Composition of W-states wave functions of the molecule ScBr in terms of L-states (in percentage) at R = 2.4 Å.
ternuclear distance. These data are given in Table 3. The only available data in literature concerning the electronic states of the molecule ScBr in the representation ![]() are the transition energy with respect to the ground state Te of Bencheikh (Bencheikh 1997) calculated by ligand field theory. The comparison of these values to those of the present work showed a good agreement with the relative difference 0.8% ≤ DTe/Te ≤ 11.7 for the states (2, 4)W = 1 and (2, 3)W = 2. This relative difference becomes moderate with 12.8% ≤ DTe/Te ≤ 17.5 for the states (1, 2)W = 0+, (1, 3)W = 1, and (1)W = 3. The agreement deteriorates for the states (1)W = 1, (1)W = 2 where DTe/Te are respectively 33.1% and 24.1%. The calculated values of the splitting of the present work and those of three states investigated by Bencheikh (Bencheikh 1997) are given in Table 4. The comparison between our values and the limited number of data of Bencheikh (Bencheikh 1997) shows an over all acceptable agreement. We are not aiming in the present work to show the advantage of one of the theoretical technique on the other, but the investigation of new valid excited electronic states with spin orbit effect in order to lead the investigation of new experimental works on this molecule.
are the transition energy with respect to the ground state Te of Bencheikh (Bencheikh 1997) calculated by ligand field theory. The comparison of these values to those of the present work showed a good agreement with the relative difference 0.8% ≤ DTe/Te ≤ 11.7 for the states (2, 4)W = 1 and (2, 3)W = 2. This relative difference becomes moderate with 12.8% ≤ DTe/Te ≤ 17.5 for the states (1, 2)W = 0+, (1, 3)W = 1, and (1)W = 3. The agreement deteriorates for the states (1)W = 1, (1)W = 2 where DTe/Te are respectively 33.1% and 24.1%. The calculated values of the splitting of the present work and those of three states investigated by Bencheikh (Bencheikh 1997) are given in Table 4. The comparison between our values and the limited number of data of Bencheikh (Bencheikh 1997) shows an over all acceptable agreement. We are not aiming in the present work to show the advantage of one of the theoretical technique on the other, but the investigation of new valid excited electronic states with spin orbit effect in order to lead the investigation of new experimental works on this molecule.
The useful information about the degree of ionic and covalent character in the bonding can be investigated by using an ab initio calculation of the dipole moment operator A best accuracy may be obtained by constructing a multireference configuration interaction (MRCI) wave functions using multi configuration Self-consistent field (MCSCF) active space. By taking the Sc atom as an origin in our calculation, the permanent dipole moments have been investigated for the considered electronic states of the molecule ScBr, the calculated values (in Debye) in terms of the internuclear distance R are plotted in Figures 7 and 8 where these curves tend to zero when R tends to infinity All these calculations have been performed by using the MOLPRO [6] program. One can notice the agreement between the position of the avoided crossing of the potential energy curves (Figure 1 and Table 1) of the (7)0+ and (8)0+ and the crossings of dipole moment curves of these states in Figure 8 which confirm the validity and the accuracy of our results.
3. Conclusions
In the present work, ab initio investigations for 42 low-lying electronic states of ScBr molecule (including spin-orbit effects) have been performed via CAS-SCF/ MRCI method. Multireference CI calculations (single and double excitations + Davidson corrections) in which the entire CAS-SCF configuration space was used as
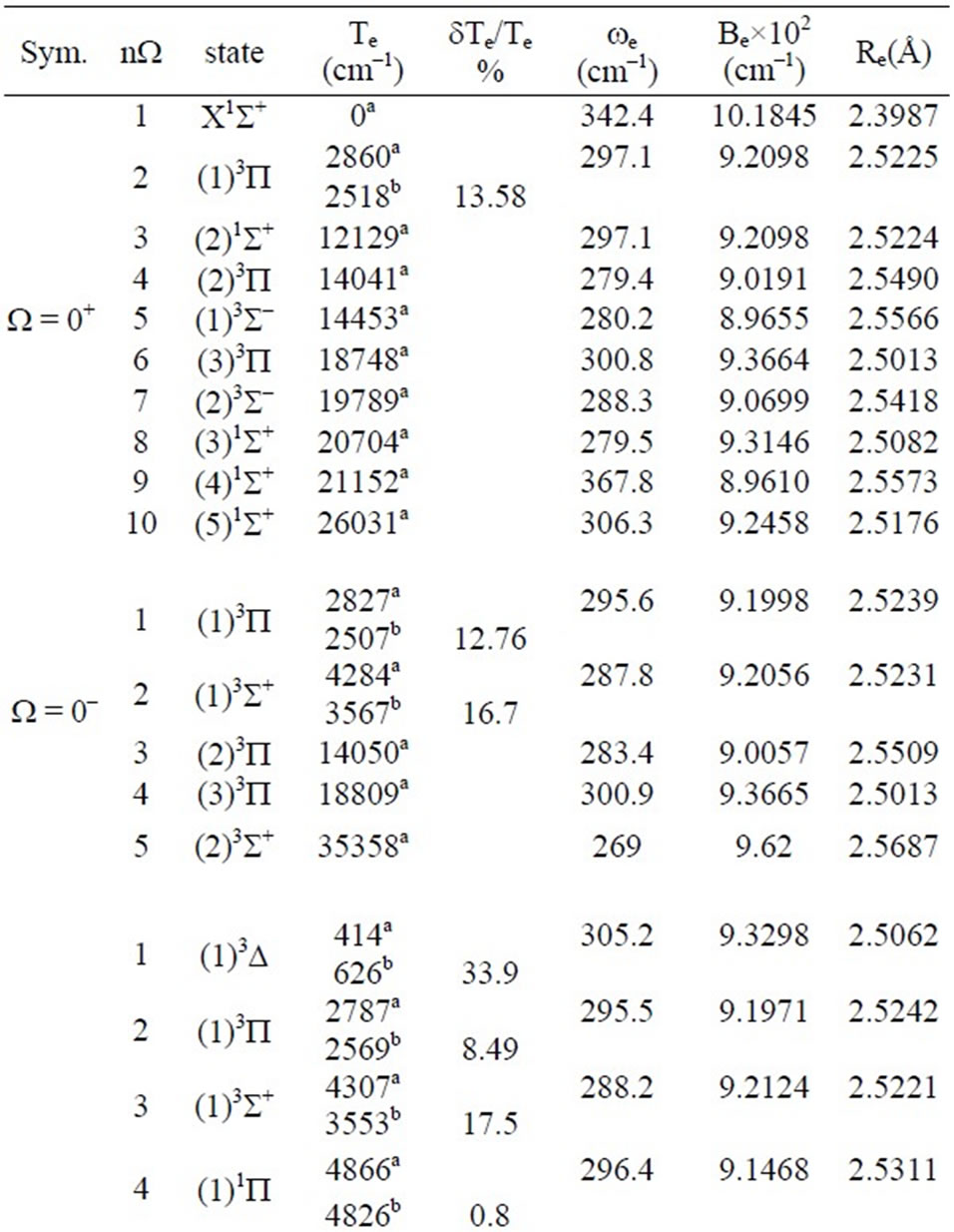
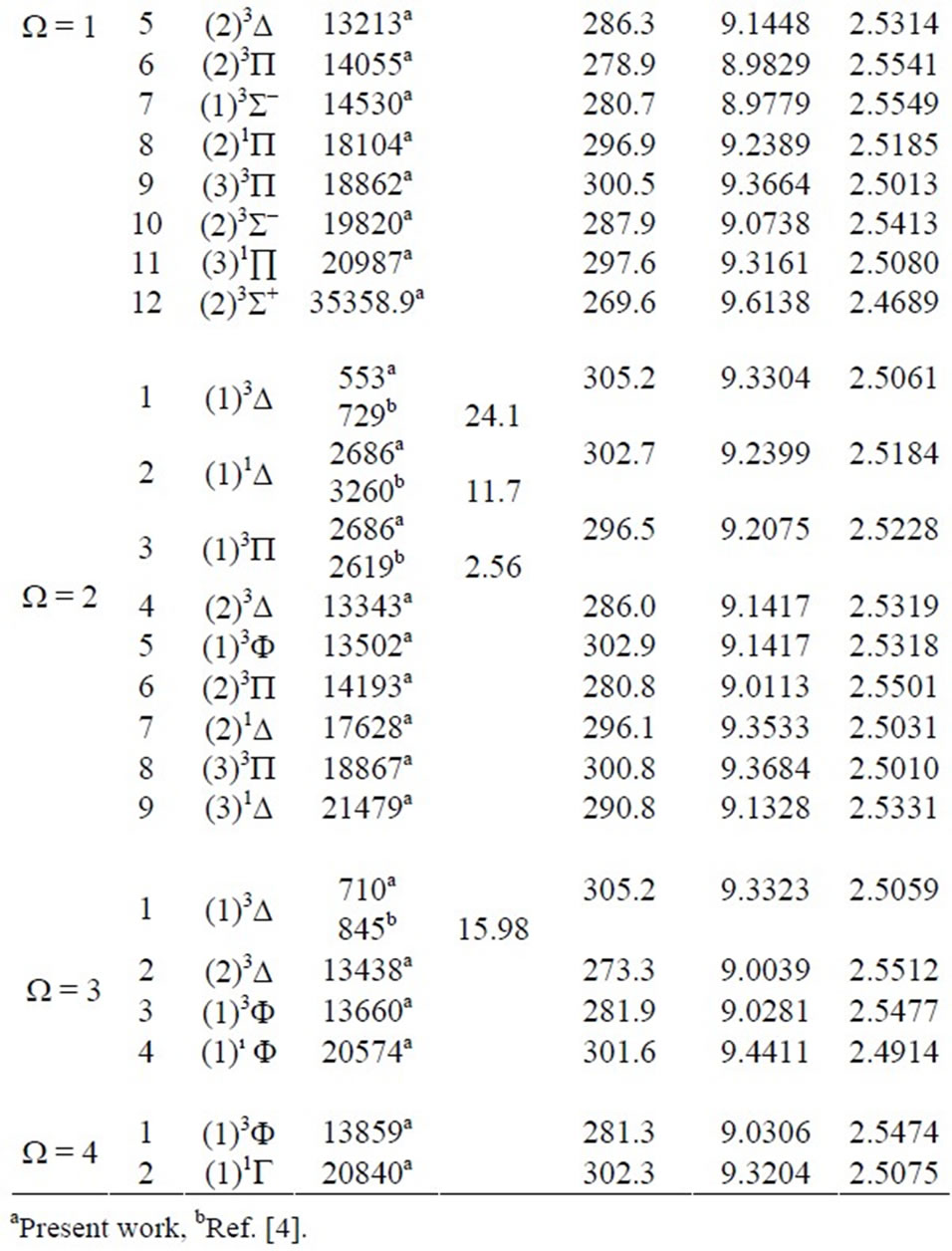
Table 3. The values of transition energy Te, the equilibrium internuclear distance Re, the rotational constant Be and the harmonic frequency we for W-states of the molecule ScBr.
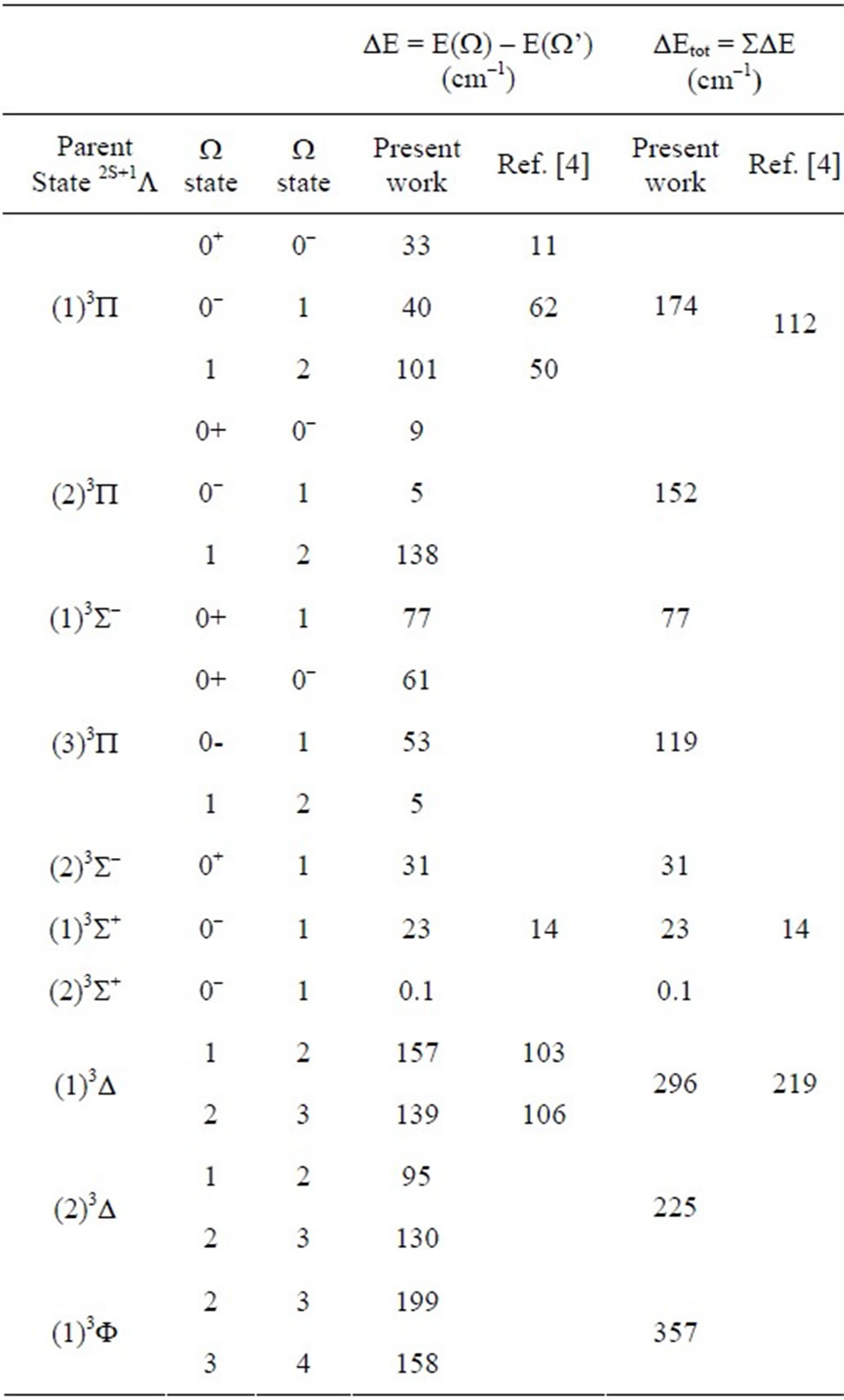
Table 4. Calculation of the splitting of the Ω-states of the molecule ScBr.
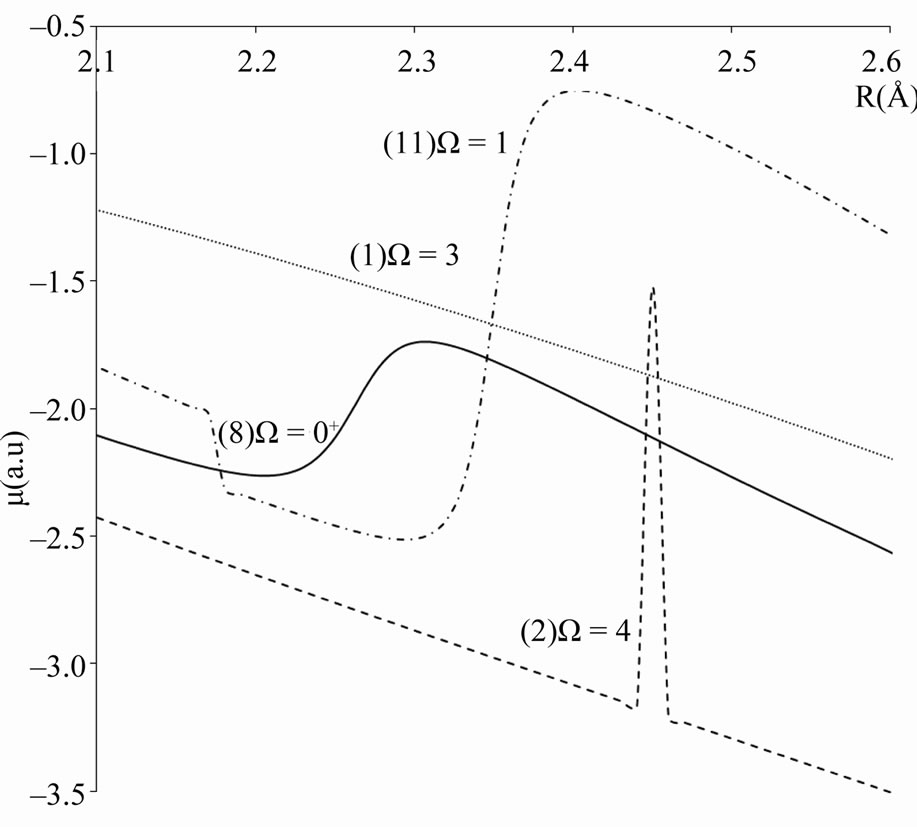
Figure 7. Calculated dipole moment curves of the W-states of the molecule ScBr.
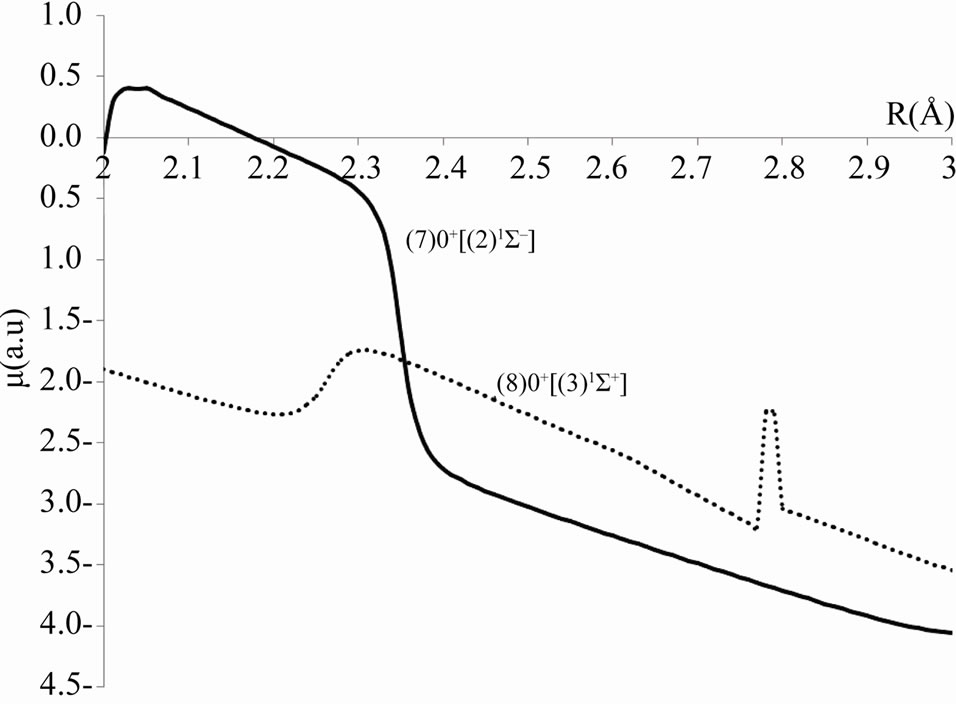
Figure 8. Calculated dipole moment curves for the (7)0+ and (8)0+ states showing the crossing at the position of the avoid crossing of the corresponding potential energy curves of the molecule ScBr.
reference were performed in order to obtain the correlation effects. The potential energy curves and the permanent dipole moment along with the spectroscopic constants Te, Re, we and the rotational constant Be, have been investigated for the lowest-lying electronic states of ScBr including the spin orbit effect. Thirty states have been investigated here for the first time. The comparison of the present results with the few available data in literature for the molecule ScBr has demonstrated a good accuracy. To the best of our knowledge no ab initio study of this molecule including spin orbit effect in literature.
REFERENCES
- W. Lin, C. J. Evans and G. M. C. L. Gerry, “The Pure Rotational Spectrum of ScBr,” Physical Chemistry Chemical Physics, Vol. 2, No. 1, 2000, pp. 43-46. doi:10.1039/a907769c
- D. R. Fischell, H. C. Brayman and T. A. Cool, “Spectroscopic Studies of the Products of Reactions of Yttrium and Scandium Atoms with Halogen Molecules. II. Laser Induced Fluorescence from Yttrium and Scandium Monohalides,” Journal of Chemical Physics, Vol. 73, No. 9, 1980, pp. 4260-4273. doi:10.1063/1.440708
- S. R. Langhoff, C. W. Bauschlicher and H. J. Partridge. “Theoretical Study of the Scandium and Yttrium Halides,” Journal of Chemical Physics, Vol. 89, No. 1, 1988, pp. 396-408. doi:10.1063/1.455481
- M. Bencheikh, “Semiempirical Calculations on Scandium Monohalides,” Journal of Physics B: Atomic, Molecular and Optical physics, Vol. 30, No. 5, 1997, pp. L137-140. doi:10.1088/0953-4075/30/5/002
- M. Korek and A. Hamdan, “Theoretical Electronic Structure of the Molecule ScBr,” International Journal of Quantum Chemistry, Vol. 108, No. 3, 2008, pp. 456-461. doi:10.1002/qua.21524
- H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schütz, P. Celani, T. Korona, G. Rauhut, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, A. W. Lloyd, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklaß, P. Palmieri, R. Pitzer, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni, and T. Thorsteinsson, “MOLPRO,” A Package of Ab Initio Programs.
- A. R. Allouche, “Gabedit—A Graphical User Interface for Computational Chemistry Softwares,” Journal of Computational Chemistry, Vol. 32, No. 1, 2011, pp. 174- 182. doi:10.1002/jcc.21600
- W. J. Hehre, R. Ditchfield and J. A. Pople, “Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules,” Journal of Chemical Physics, Vol. 56, No. 5, 1972, pp. 2257-2262. doi:10.1063/1.1677527
- T. H. Dunning Jr. and P. J. Hay, In: H. F. Schaefer III, Ed., Methods of Electronic Structure Theory, Vol. 2, Plenum Press, New York, 1977.
- R. Krishnan, J. S Binkley, R. Seeger and J. A. Pople, “Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions,” Journal of Chemical Physics, Vo. 72, No. 1, 1980, pp. 650-655. doi:10.1063/1.438955
- J. M. L Martin and A. Sundermann, “Correlation Consistent Valence Basis Sets for Use with the Stuttgart-Dresden-Bonn Relativistic Effective Core Potentials: The Atoms Ga-Kr and In-Xe,” Journal of Chemical Physics, Vol. 114, No. 8, 2001, pp. 3408-3421. doi:10.1063/1.1337864