Advances in Bioscience and Biotechnology
Vol.5 No.4(2014), Article ID:43678,7 pages DOI:10.4236/abb.2014.54046
Effect of the R406H Substitution on the Normal Function of CHEK2
Ahmet Yilmaz1,2, Michael E. Davis3, Weiqiang Zhao4*
1Department of Human Genetics, McGill University, Montreal, Canada
2The Lady Davis Institute, Jewish General Hospital, Montreal, Canada
3Department of Animal Sciences, The Ohio State University, Columbus, USA
4Department of Pathology, The Ohio State University, Columbus, USA
Email: *weiqiang.zhao@osumc.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 18 January 2014; revised 21 February 2014; accepted 7 March 2014
ABSTRACT
CHEK2 (Checkpoint kinase homolog 2) encodes a protein involved in pathways that arrest the cell cycle in response to genomic stress such as DNA damage or replication blocks. Carriers of some of the CHEK2 mutations are at an increased risk of breast cancer. A mutation in the kinase domain of the CHEK2 gene resulting in the R406H substitution has been reported. However, it is currently unknown whether the substitution alters the function of CHEK2 in vitro. We evaluated the effect of the R406H substitution on the normal function of CHEK2 using a yeast complementation assay. The yeast cells lacking Rad53, the yeast homologue of human CHEK2 were transformed with the wild type as well as plasmids carrying mutations resulting in the R406H, 1100delC, and S428F variants. Yeast cells carrying the R406H variant grew at a rate similar to those carrying the wild type CHEK2, whereas the yeast carrying the S428F and 1100delC mutants grew at a slower rate. These results suggest that, unlike the well-known pathogenic alleles such as 1100delC and S428F, the R406H substitution does not abrogate the function of CHEK2. Therefore, this variant is probably not important in development of breast cancer in women.
Keywords:Breast Cancer; CHEK2; Yeast; R406H; French-Canadian Women; Indian Women

1. Introduction
Comprising 31% of all cancers seen in women, breast cancer is one of the most common cancers [1] . Although mostly sporadic, 15% of cases show some type of familial aggregation and 5% are clearly inherited [2] . Germline mutations in BRCA1/2 genes account for approximately 30% of inherited breast cancers [3] . The search for possible genetic causes of the remaining cases of inherited breast cancer without any mutation in BRCA1/2 has resulted in the discovery of rare germline mutations in several genes including CHEK2 (Checkpoint kinase homolog 2) [4] .
CHEK2 encodes a protein that arrests the cell cycle in response to DNA damage. The halt in the cell cycle provides the cell with the time required for cellular repair of DNA, ensuring inheritance of only repaired or intact DNA [5] . The CHEK2 protein is divided into three distinct SQ/TQ, forkhead-associated, and serine/threonine kinase domains. The serine/threonine kinase domain is important in phosphorylating and activating target proteins.
Lost or reduced kinase activity due to deletion of a cytosine at position 1100 of CHEK2 was first reported in Li-Freaumani syndrome patients who did not carry any mutation in TP53 [4] . The 1100delC variant has since been well-studied in patients with breast cancer [6] . The impaired CHEK2 activity is additionally linked to development of other types of cancers including ovarian [7] , prostate [8] , colorectal [9] , and bladder [10] cancer.
Two previous studies identified the R406H variant in Indian [11] and French-Canadian [12] women with breast cancer. Although in silico studies showed the loss of a turn in CHEK2 protein with the R406H substitution [11] , it is currently unknown whether the substitution alters the function of CHEK2 in vitro.
The R406H variant was not associated with a change in breast cancer risk in the French-Canadian women [12] . However, the sensitivity to breast cancer due to mutations in CHEK2 is likely different in varied populations. For example, the well-known 1100delC allele that confers 1.4- to 4.7-fold increase in breast cancer risk in Northern or Eastern European women is not pathogenic in Asian populations [13] [14] . Therefore, the possibility of the R406H allele still being a breast cancer risk in other populations should not be ignored.
Large epidemiologic studies, which are expensive and time consuming, are needed to achieve satisfactory statistical power to investigate such possibility. Instead, a functional assay to measure CHEK2 activity in cells carrying the R406 substitution can be used to evaluate this possibility because both the S428F and 1100delC variants, the two well-known pathogenic alleles, decrease CHEK2 activity in functional assays. A change in function of CHEK2 in cells carrying the R406 variant could then be used to justify the need to screen populations other than the French-Canadian women. The objective of this study was to investigate effects of the R406H substitution on the function of CHEK2 in yeast cells.
2. Materials and Methods
2.1. Yeast Strains, Plasmids, Cloning, and Site-Directed Mutagenesis
Yeast strain S. Cerevisae lacking Rad53, yeast homolog of CHEK2, was kindly provided by Dr. Rodney Rothstein [15] . In order to suppress lethality due to deletion of Rad53, a second gene, Sml1, an inhibitor of ribonucleotide reductase, was also deleted in this strain. The deletion of Sml1 results in increased dNTP pools available for DNA replication to keep the cells viable and should not affect the results obtained in this study because similar results were obtained when yeast strains carrying Rad53 -/- and/or Sml1 -/- deletion were treated with MMS (methyl methane sulphonate) [16] . The plasmid pMH267 (pBAD101 2 micron LEU2 GAL-CHEK2) carrying the wild type human CHEK2, kindly provided by Dr. Steven Elledge [17] , was used in site-directed mutagenesis to create plasmids carrying mutations resulting in the 1100delC, S428F, and R406H variants.
The site-directed mutagenesis reactions were performed following manufacturer’s instructions (Quik Change XL kit, Stratagene La Jolla CA). A region of 800 bp around the desired mutation of all plasmids was sequenced to verify the identity of the plasmids. The enzyme pfu ultra we used in the site-directed mutagenesis reactions has a negligible rate of introducing an undesired mutation (1.3 × 10−6 per duplication) [18] . Expression of CHEK2 by all plasmids was confirmed using RNA isolated from yeast cells treated with lyticase. The RNA was digested with DNAse I before use in RT-PCR reactions. Absence of contaminating plasmid DNA in RNA preparations was confirmed using at least 60 ng of DNAse I-digested RNA as a template in a 25 µL PCR reaction. No bands were observed when the PCR products were run on an agarose gel.
2.2. Yeast Culture and Transformation
The transformation procedure used in this study has been described previously [19] -[21] . Briefly, the cells grown overnight in 2 X YPDA media were collected by centrifugation and resuspended in 100 µL of water. Salmon sperm DNA was used as the single-stranded carrier DNA. One-hundred µgr of the DNA was added to the cells after being boiled and cooled on ice for five minutes. After vortexing, 500 µL of PLATE solution [PEG 3500 (25% v/v), 1 M LiOAc, 1 M Tris-HCl (pH7.5), and 0.5 M EDTA] was added and the mix was vortexed and incubated overnight at 30˚C. After incubating the samples at 42˚C for 1 hour the next day, the cells were collected by centrifugation, and resuspended in 100 µL of sterile water. One to ten and 1:100 dilutions of the re-suspended yeast cells were made and 100 µL of each dilution, as well as 100 µL of undiluted samples, were plated on His-Ura-Leuplates containing 2% galactose as the carbon source. Plates were incubated at 30˚C for 4 to 6 days until colonies reached approximately 1 mm in size. Three colonies from each plate were picked and inoculated into 2 mL of His-Ura-Leuliquid media in disposable cuvettes (Sigma). The cuvettes were incubated at 30˚C with shaking at 200 or 250 rpm until all cuvettes reached a density of above 0.500 OD600 nm. Yeast cultures were then diluted to 0.500 ± 0.001 OD600 nm and 20 µL of the diluted culture was inoculated into cuvettes containing 2 mL of fresh His-Ura-Leumedia. Twenty µL of the diluted samples were also inoculated into cuvettes containing the same media but with 0.00125% or 0.00250% of MMS (v/v; Sigma) added to induce DNA damage in separate experiments. In addition, we added 20 µL of the diluted samples on His-Ura-Leuliquid media containing glucose, instead of galactose, to suppress CHEK2 expression from the plasmids. All cuvettes were incubated at 30˚C with shaking at 200 or 250 rpm and optical density readings were recorded every 5 hours for approximately 30 hours. Experiments were conducted in duplicates, repeated, and similar results were obtained. As a control for the transformation reactions, the nontransformed yeast was plated on His-Ura-Leuplates or inoculated into His-Ura-Leuliquid media after growing on His-Uraplates in separate experiments. No growth was observed in either case.
3. Results
Cells transformed with the R406H variant grew at a rate slightly lower than those carrying the wild type CHEK2 but higher than both of those carrying S428F or 1100delC variants. Therefore, the CHEK2 protein with the R406H substitution almost fully complemented lost functions due to deletion of Rad53 in the yeast cells (Figure 1).
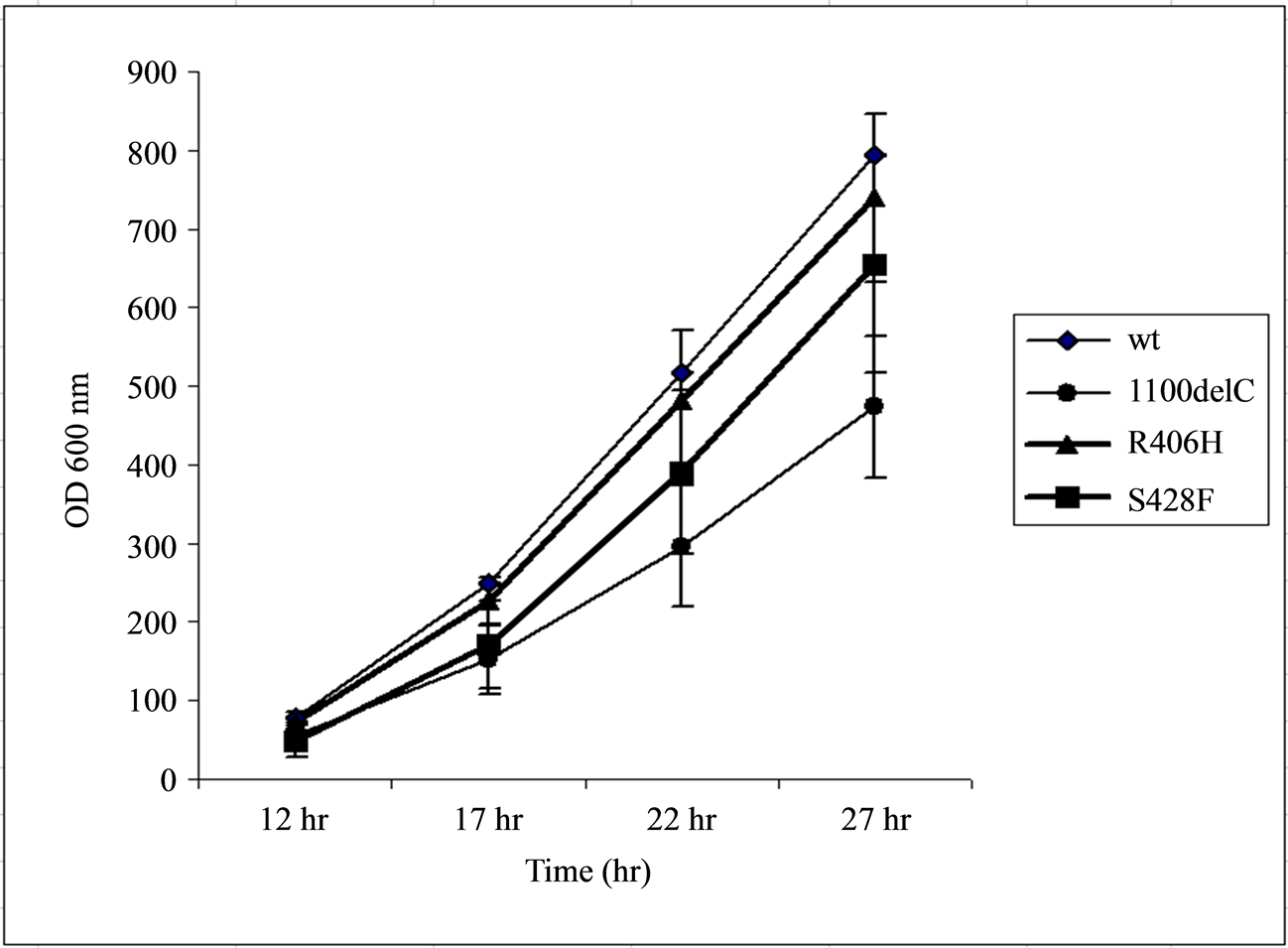
Figure 1. Transformation of the Rad53 -/- Sml1 -/- strain of the yeast S. Cerevisae with wild-type (wt) CHEK2, but not with the 1100delC variant, reverses the reduced growth due to Rad53 deletion. Cells transformed with the R406H variant grew at a rate similar to those transformed with the wild-type CHEK2, suggesting that the R406H substitution did not result in a major change in normal CHEK2 function. The x-axis represents the time of measurement after inoculation of 20 µL of samples diluted to obtain readings of 0.500 ± 0.001 OD600 nm into 2 mL of fresh media.
Cells transformed with the 1100delC variant grew at a rate lower than those transformed with the S428F variant. This result does not agree with the observation by Shaag et al. [22] who reported that the S428F and 1100delC variants proliferated at similar rates. However, the results obtained in the current study are more in line with the observation that the 2% risk conferred by S428F is approximately half of the 5% risk conferred by the 1100delC variant in breast cancer patients with wild type BRCA1/2 [22] .
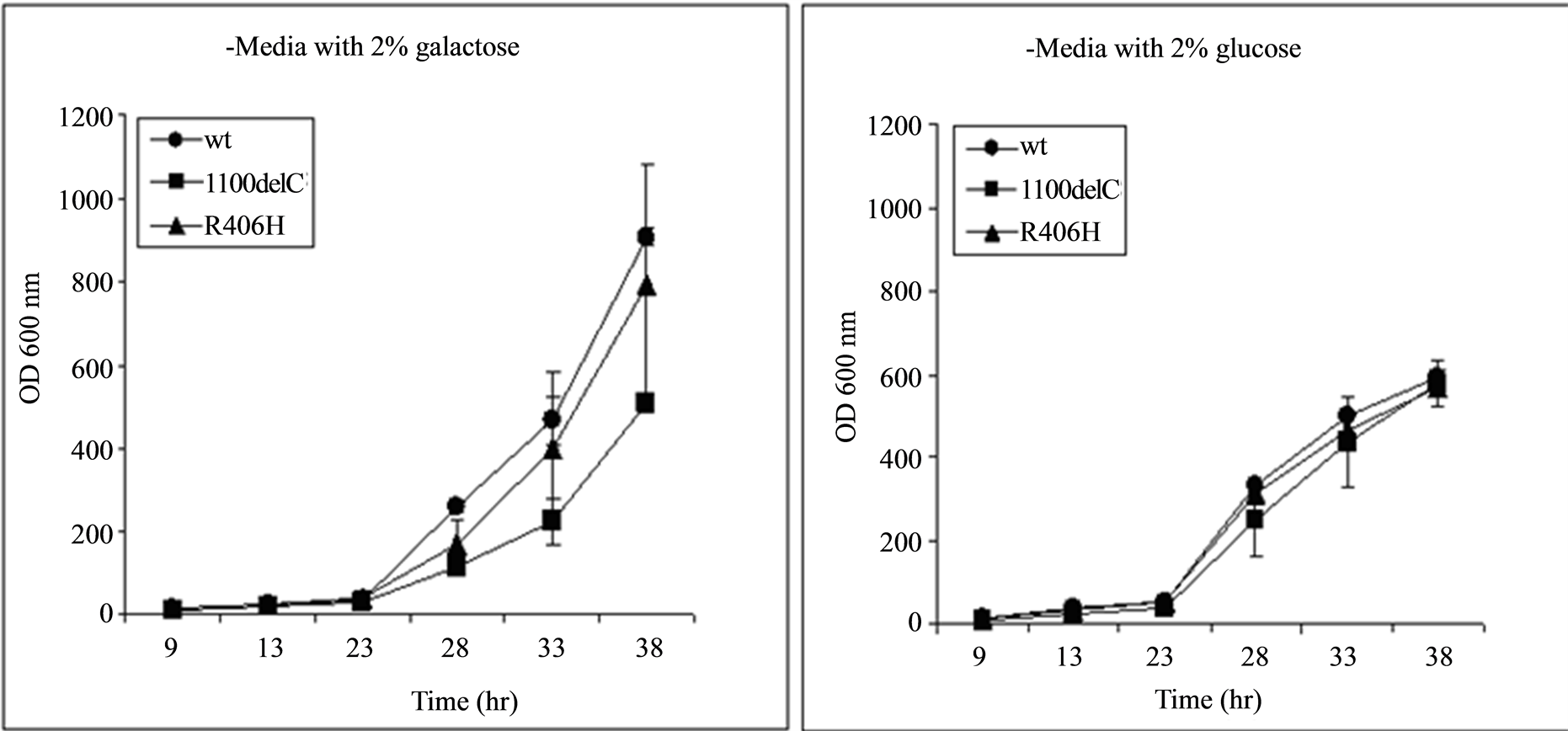
CHEK2 expression by all of the plasmids used in this study was under the control of the GAL promoter, which induces and represses the expression of CHEK2 when galactose and glucose, respectively, are present in the growth media. Growth rate in cells carrying all variants, including those carrying the 1100delC variant and the wild type CHEK2, were similar when the carbon source in the media was switched from galactose to glucose, suggesting that differences observed in growth rates when galactose was present in the media were due to the expression of CHEK2 from the plasmids (Figure 2). Growth seen in cultures containing glucose despite suppression of CHEK2 expression is a result of the shorter doubling time of yeast in glucose than in galactose.
Treatment of cells with genotoxic concentrations of MMS showed that all of the variants, with the exception of 100delC, followed a similar pattern of growth (Figure 3). MMS is an alkylating agent that induces DNA damage by methylating N7-deoxyguanine and N3-deoxyadenine residues [23] . These results are in agreement with previous reports suggesting that cells with reduced Rad53 expression follow a different growth pattern when treated with MMS probably due to stronger stabilization of replication forks [16] .
4. Discussion
This study investigated effects of the R406H substitution on the function of CHEK2. CHEK2 is needed to arrest the cell cycle in response to DNA damage, an important step in ensuring inheritance of only intact DNA.
It is reasonable to assume that the substitution of arginine by histidine in CHEK2 may have an impact on its function because of differences in the chemical characteristics of these amino acids. Histidine has a low pKa of 6, and thus, its charge mostly depends on the cellular pH. Arginine, on the other hand, has a strong pKa of 12 due to a guanidium group at the end of one of its straight chains. Therefore, it is almost always positively charged regardless of the cellular pH. Arginine, but not histidine, can be a target for protein methyltransferases. Arginine methylation is known to be an important regulator of function in proteins carrying this amino acid [24] and the substitution could deprive the protein of such regulations if they did exist. In addition, the geometry of
 (a) (b)
(a) (b)
Figure 2. Cells transformed with all plasmids, including the wild type (wt) CHEK2, grew at similar rates when expression of CHEK2 from the plasmids was repressed by replacing galactose in the media with glucose, suggesting that the differences seen in growth rate of cells transformed with various plasmids in Figure 2(a) are due to expression of CHEK2. The x-axis represents the time of measurement after inoculation of 20 µL of samples diluted to obtain readings of 0.500 ± 0.001 OD600 nm into fresh media.
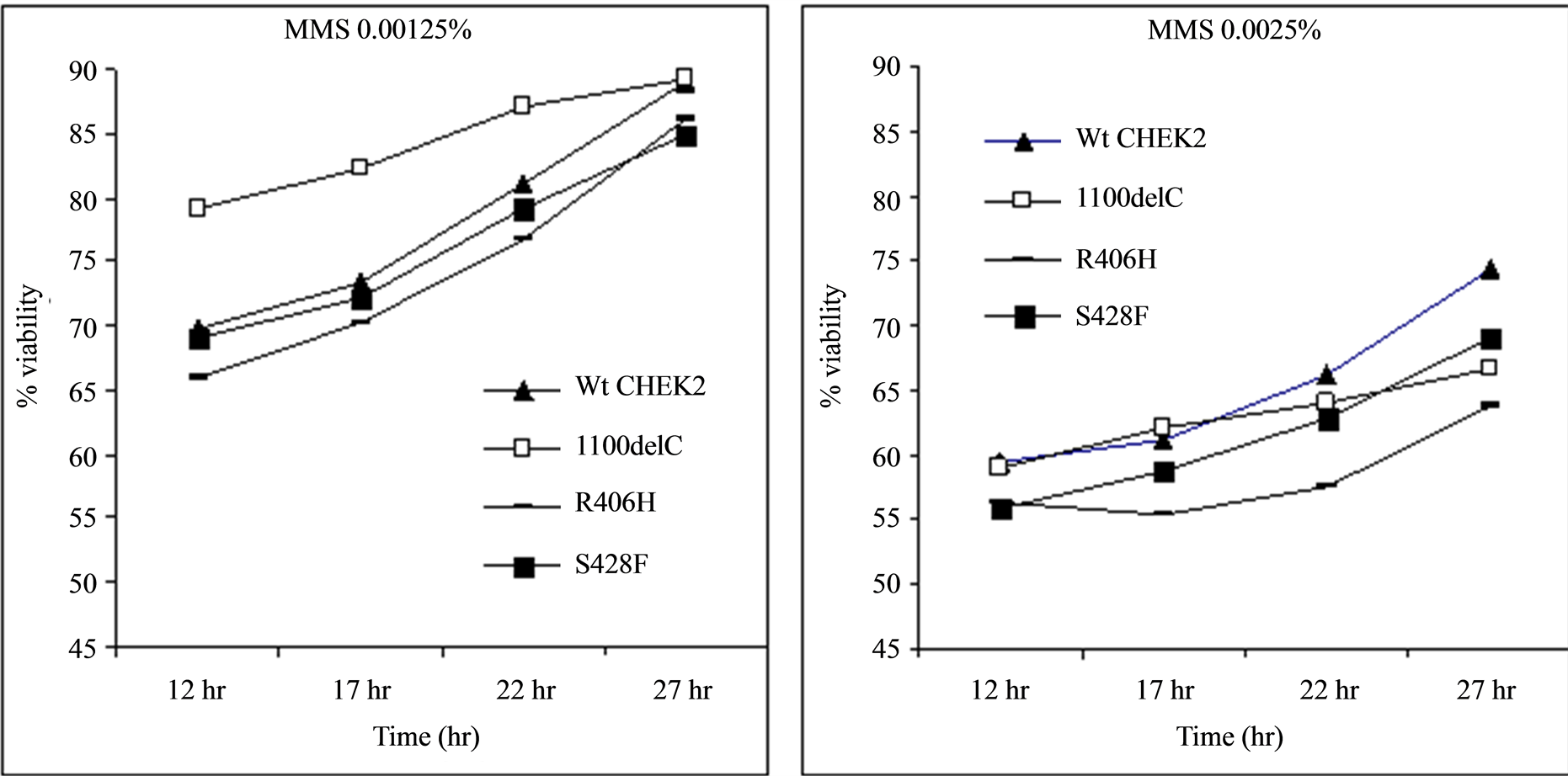
Figure 3. Treatment of transformed Rad53 -/- Sml1 -/- strain of the yeast cells with MMS. The cells transformed with the CHEK2*1100delC variant followed a growth pattern different from the rest of the variants probably due to reduced kinase activity in those cells. The x-axis represents the time of measurement after inoculation of 20 µL of samples diluted to obtain readings of 0.500 ± 0.001 OD600 nm into fresh media.
arginine, but not that of histidine, allows for multiple hydrogen bonds and strong binding to negatively charged proteins [25] .
Use of yeast lacking Rad53 to investigate effects of amino acid substitutions on the normal function of CHEK2 has been successful [17] [21] [22] . Introduction of wild type human CHEK2 into Rad53 -/- yeast strain restores decreased growth due to Rad53 deletion. However, cells transformed with the 1100delC variant grow at a significantly lower rate [22] . The deletion of cytosine at position 1100 introduces a premature stop codon, resulting in translation of a shorter CHEK2 protein with impaired kinase activity and potential loss of its functions in controlling the cell cycle.
Yeast can be used as a model system to investigate effects of mutations in human genes important in cell cycle regulation because it possesses most of the essential components of the DNA damage checkpoints found in higher eukaryotes [26] . The G1 checkpoint that prevents replication of damaged DNA and the G2 checkpoint that monitors the chromosome integrity before mitosis are the two main surveillance systems that maintain genomic integrity. Both of these checkpoints have been well studied in yeast [27] . Linkage of ATM to the cell cycle through CHEK2 [17] , as well as most of the CDKs and cyclins, two groups of molecules critical for proper maintenance of the cell cycle, were originally identified in yeast [28] .
The basic mechanisms involved in DNA damage checkpoints in yeast and higher eukaryotes are similar. Briefly, the initial steps in DNA damage checkpoint pathways in yeast are activation of Rad17 or Rad24, and Mec1, in that order. Chek1/Rad53, phosphorylated by Mec1, downregulates CDC5 which may also be ubiquitinylated by APC/C, resulting in cell cycle arrest [29] . Regulation of the cell cycle in higher eukaryotes is more complex but the basic principles to arrest the cell cycle remain the same. CHEK1/2, phosporylated by the ATM/ATR complex, downregulates CDC25B/C which is additionally regulated by APC/C. However, the regulation by APC/C requires activation of additional molecules such as CDK1 [30] .
5. Conclusion
Data presented here indicate that the R406H substitution in CHEK2 identified in recent studies in breast cancer patients does not result in a significant change in the normal function of CHEK2. It is already known that this variant is not a breast cancer risk in French-Canadian women and the results presented in the current study suggest that there is insufficient evidence to warrant studying this variant in other populations as a potential breast cancer risk.
References
- Jemal, A., Siegel, R., Ward, E., Murray, T., Xu, J., Smigal, C. and Thun, M.J. (2006) Cancer Statistics. CA: A Cancer Journal for Clinicians, 56, 106-130. http://dx.doi.org/10.3322/canjclin.56.2.106
- Garcia, M.J. and Benitez, J. (2008) The Fanconi Anaemia/BRCA Pathway and Cancer Susceptibility. Searching for New Therapeutic Targets. Clinical and Translational Oncology, 10, 78-84. http://dx.doi.org/10.1007/s12094-008-0160-6
- Laitman, Y., Kaufman, B., Lahad, E.L., Papa, M.Z. and Friedman, E. (2007) Germline CHEK2 Mutations in Jewish Ashkenazi Women at High Risk for Breast Cancer. Israel Medical Association Journal, 9, 791-796.
- Bell, D.W., Varley, J.M., Szydlo, T.E., Kang, D.H., Wahrer, D.C.R., Shannon, K.E., Lubratovich, M., Verseli, S.J., Isselbacher, K.J., Fraumeni, J.F., Birch, J.M., Garber, J.E. and Haber, D.A. (1999) Heterozygous Germ Line hCHK2 Mutations in Li-Fraumeni Syndrome. Science, 286, 2528-2531. http://dx.doi.org/10.1126/science.286.5449.2528
- Liang, F. and Wang, Y. (2007) DNA Damage Checkpoints Inhibit Mitotic Exit by Two Different Mechanisms. Molecular and Cellular Biology, 27, 5067-5078. http://dx.doi.org/10.1128/MCB.00095-07
- Weischer, M., Bojesen, S.E., Ellervik, C., Tybjaerg-Hansen, A. and Nordestgaard, B.G. (2008) CHEK2*1100delC Genotyping for Clinical Assessment of Breast Cancer Risk: Meta-Analyses of 26,000 Patient Cases and 27,000 Controls. Journal of Clinical Oncology, 26, 542-548. http://dx.doi.org/10.1200/JCO.2007.12.5922
- Williams, L.H., Choong, D., Johnson, S.A. and Campbell, I.G. (2006) Genetic and Epigenetic Analysis of CHEK2 in Sporadic Breast, Colon, and Ovarian Cancers. Clinical Cancer Research, 12, 6967-6972. http://dx.doi.org/10.1158/1078-0432.CCR-06-1770
- Cybulski, C., Gliniewicz, B., Sikorski, A., Kładny, J., Huzarski, T., Gronwald, J., Byrski, T., Debniak, T., Gorski, B., Jakubowska, A., Wokolorczyk, D., Narod, S.A. and Lubiñski, J. (2007) Epistatic Relationship between the Cancer Susceptibility Genes CHEK2 and p27. Cancer Epidemiology, Biomarkers & Prevention, 16, 572-576. http://dx.doi.org/10.1158/1055-9965.EPI-06-0566
- Wasielewski, M., Vasen, H., Wijnen, J., Hooning, M., Dooijes, D., Tops, C., Klijn, J.G., Meijers-Heijboer, H. and Schutte, M. (2008) CHEK2 1100delC Is a Susceptibility Allele for HNPCC-Related Colorectal Cancer. Clinical Cancer Research, 14, 4989-4994. http://dx.doi.org/10.1158/1078-0432.CCR-08-0389
- Złowocka, E., Cybulski, C., Górski, B., Debniak, T., Słojewski, M., Wokołorczyk, D., Serrano-Fernández, P., Matyjasik, J., van de Wetering, T., Sikorski, A., Scott, R.J. and Lubiński, J. (2008) Germline Mutations in the CHEK2 Kinase Gene Are Associated with an Increased Risk of Bladder Cancer. International Journal of Cancer, 122, 583-586. http://dx.doi.org/10.1002/ijc.23099
- Soumittra, N., Meenakumari, B., Parija, T., Sridevi, V., Nancy, K.N., Swaminathan, R., Rajalekshmy, K.R., Majhi, U. and Rajkumar, T. (2009) Molecular Genetics Analysis of Hereditary Breast and Ovarian Cancer Patients in India. Hereditary Cancer in Clinical Practice, 7, 13. http://dx.doi.org/10.1186/1897-4287-7-13
- Novak, D.J., Chen, L.Q., Ghadirian, P., Hamel, N., Zhang, P., Rossiny, V., Cardinal, G., Robidoux, A., Tonin, P.N., Rousseau, F., Narod, S.A. and Foulkes, W.D. (2008) Identification of a Novel CHEK2 Variant and Assessment of Its Contribution to the Risk of Breast Cancer in French Canadian Women. BMC Cancer, 8, 239. http://dx.doi.org/10.1186/1471-2407-8-239
- Chen, W., Yurong, S. and Liansheng, N. (2008) Breast Cancer Low-Penetrance Allele 1100delC in the CHEK2 Gene: Not Present in the Chinese Familial Breast Cancer Population. Advances in Therapy, 25, 496-501. http://dx.doi.org/10.1007/s12325-008-0057-3
- Zhang, S., Phelan, C.M., Zhang, P., Rousseau, F., Ghadirian, P., Robidoux, A., Foulkes, W., Hamel, N., McCready, D., Trudeau, M., Lynch, H., Horsman, D., De Matsuda, M.L., Aziz, Z., Gomes, M., Costa, M.M., Liede, A., Poll, A., Sun, P. and Narod, S.A. (2008) Frequency of the CHEK2 1100delC Mutation among Women with Breast Cancer: An International Study. Cancer Research, 68, 2154-2157. http://dx.doi.org/10.1158/0008-5472.CAN-07-5187
- Zhao, X. and Rothstein, R. (2002) The Dun1 Checkpoint Kinase Phosphorylates and Regulates the Ribonucleotide Reductase Inhibitor Sml1. Proceedings of the National Academy of Sciences of the USA, 99, 3746-3751. http://dx.doi.org/10.1073/pnas.062502299
- Cordon-Preciado, V., Ufano, S. and Bueno, A. (2006) Limiting Amounts of Budding Yeast Rad53 S-Phase Checkpoint Activity Results in Increased Resistance to DNA Alkylating Damage. Nucleic Acid Research, 34, 5852-5862. http://dx.doi.org/10.1093/nar/gkl741
- Matsuoka, S., Huang, M. and Elledge, S.J. (1998) Linkage of ATM to Cell Cycle Regulation by the Chk2 Protein Kinase. Science, 282, 1893-1897. http://dx.doi.org/10.1126/science.282.5395.1893
- Cline, J., Braman, J.C. and Hogrefe, H.H. (1996) PCR Fidelity of pfu DNA Polymerase and Other Thermostable DNA Polymerases. Nucleic Acids Research, 24, 3546-3551. http://dx.doi.org/10.1093/nar/24.18.3546
- Elble, R. (1992) A Simple and Efficient Procedure for Transformation of Yeasts. Biotechniques, 13, 18-20.
- Gietz, R.D. and Woods, R.A. (2006) Yeast Transformation by the LiAc/SS Carrier DNA/PEG Method. Methods in Molecular Biology, 313, 107-120.
- Tischkowitz, M.D., Yilmaz, A., Chen, L.Q., Karyadi, D.M., Novak, D., Kirchhoff, T., Hamel, N., Tavtigian, S.V., Kolb, S., Bismar, T.A., Aloyz, R., Nelson, P.S., Hood, L., Narod, S.A., White, K.A., Ostrander, E.A., Isaacs, W.B., Offit, K., Cooney, K.A., Stanford, J.L. and Foulkes, W.D. (2008) Identification and Characterization of Novel SNPs in CHEK2 in Ashkenazi-Jewish Men with Prostate Cancer. Cancer Letters, 270, 173-180. http://dx.doi.org/10.1016/j.canlet.2008.05.006
- Shaag, A., Walsh, T., Renbaum, P., Kirchhoff, T., Nafa, K., Shiovitz, S., Mandell, J.B., Welcsh, P., Lee, M.K., Ellis, N., Offit, K., Levy-Lahad, E. and King, M.C. (2005) Functional and Genomic Approaches Reveal an Ancient CHEK2 Allele Associated with Breast Cancer in the Ashkenazi Jewish Population. Human Molecular Genetics, 14, 555-563. http://dx.doi.org/10.1093/hmg/ddi052
- Pegg, A.E. (1984) Methylation of the O6 Position of Guanine in DNA Is the Most Likely Initiating Event in Carcinogenesis by Methylating Agents. Cancer Investigation, 2, 223-231. http://dx.doi.org/10.3109/07357908409104376
- Jansson, M., Durant, S.T., Cho, E.C., Sheahan, S., Edelmann, M., Kessler, B. and La Thangue, N.B. (2008) Arginine Methylation Regulates the p53 Response. Nature Cell Biology, 10, 1431-1439. http://dx.doi.org/10.1038/ncb1802
- Betts, M.J. and Russell, R.B. (2003) Amino Acid Properties and Consequences of Subsitutions. In: Gray, I.C., Ed., Bioinformatics for Geneticists Barnes MR, Wiley. http://dx.doi.org/10.1002/0470867302.ch14
- Nurse, P. (2000) A Long Twentieth Century of the Cell Cycle and Beyond. Cell, 100, 71-78. http://dx.doi.org/10.1016/S0092-8674(00)81684-0
- Sanchez, Y., Desany, B.A., Jones, W.J., Liu, Q., Wang, B. and Elledge, S.J. (1996) Regulation of Rad53 by the ATM-Like Kinases MEC1 and TEL1 in Yeast Cell Cycle Checkpoint Pathways. Science, 271, 357-360. http://dx.doi.org/10.1126/science.271.5247.357
- Simon, I., Barnett, J., Hannett, N., Harbison, C.T., Rinaldi, N.J., Volkert, T.L., Wyrick, J.J., Zeitlinger, J., Gifford, D.K., Jaakkola, T.S. and Young, R.A. (2001) Serial Regulation of Transcriptional Regulators in the Yeast Cell Cycle. Cell, 106, 697-708. http://dx.doi.org/10.1016/S0092-8674(01)00494-9
- Visintin, C., Tomson, B.N., Rahal, R., Paulson, J., Cohen, M., Taunton, J., Amon, A. and Visintin, R. (2008) APC/CCdh1-Mediated Degradation of the Polo Kinase Cdc5 Promotes the Return of Cdc14 into the Nucleolus. Genes & Development, 22, 79-90. http://dx.doi.org/10.1101/gad.1601308
NOTES

*Corresponding author.