Advances in Bioscience and Biotechnology
Vol.4 No.5(2013), Article ID:32290,10 pages DOI:10.4236/abb.2013.45090
Antibody fragments: Prolonging circulation half-life special issue-antibody research
![]()
1UCL School of Pharmacy, University College London, London, UK
2National Institute for Health Research (NIHR) Biomedical Research Centre at Moorfields Eye Hospital NHS, Foundation Trust and UCL Institute of Ophthalmology, London, UK
Email: steve.brocchini@ucl.ac.uk
Copyright © 2013 Annabelle Patricia Herrington-Symes et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 28 February 2013; revised 20 April 2013; accepted 15 May 2013
Keywords: Antibody Fragment; Half-Life Extension; PEGylation; Fc Fusion
ABSTRACT
Antibodies are currently the fastest growing class of therapeutic proteins. When antibody fragments are included, there are over thirty-five antibody-based medicines approved for human therapy. Many more antibody and antibody-like fragments are being evaluated clinically. Production of antibody fragments can be efficient and their compact size can allows for better tissue extravasation into solid tumors than full antibodies. Unfortunately, a key limitation of antibody fragments for systemic use is their short half-life in circulation. Prolonging their circulation half-life can be accomplished clinically by the covalent conjugation of the antibody fragment to the water-soluble polymer, poly(ethylene glycol) (PEG). Many polymers and strategies are also being pursued to increase antibody fragment half-life.
1. INTRODUCTION
Kohler and Milstein in 1975 described how antibodies could be made by fusing murine B lymphocytes with myeloma cells to generate hybrid cells (hybridomas). These hybridomas propagate indefinitely in culture and produce homogenous antibodies with respect to antibody isotype, primary amino acid sequence, affinity and specificity. This discovery created the ability to produce antibodies for research and clinical development. There are now more than 35 antibodies and antibody fragments that have been approved for clinical use (Table 1).
Antibody production has evolved from murine to chimerisation, humanization, phage display and now transgenic mice. Even with these innovations, supplying large quantities required for therapeutic use can be difficult. Often the doses of the antibody used in therapy can be relatively high compared to other protein-therapeutics. When considering production as a limiting factor, solutions have included the production of antibody fragments such as Fab, Fv and scFv using microbial expression systems. These systems can provide greater amounts of the desired protein compared to earlier eukaryotic systems [1- 3]. Using prokaryotic expression systems can be very efficient to produce antibody fragments due to their small size and lack of glycosylation [3].
Antibody fragments can display improved tissue penetration due to their smaller size compared to full antibodies. The lack of an Fc domain means antibody fragments have reduced half-lives and a lower immunogenicity risk. The Fc domain has two functions in a full IgG, these are: i) neonatal Fc receptor (FcRn) mediated recycling to prolong circulation half-life and ii) recruitment of cytotoxic effector functions through the complement and/or through interactions with γFc receptors [4,5].
There have been many clinically successful monoclonal antibodies including MabThera®, Herceptin®, Erbitux® (Table 1). Antibody fragments have also made their way into the clinic, including Lucentis®, Cimzia®, ReoPro® and Removab® (Table 1). Antibody engineering is expanding to include non-endogenous molecular structures [6]. Many antibody derived scaffolds are in clinical trials, such as over six scFv’s [7]. Additionally the functionality of monoclonal antibodies is being expanded, for example there are over 14 antibody drug conjugates (ADCs) that have been evaluated in the clinic [8]. Antibodies tend to be developed for cancer, immunological and cardiovascular targets [9].
Antibody fragments have some advantages over full IgG molecules. One advantage is that antibody fragments due to their smaller size can penetrate tissue more rapidly than full IgG molecules. This property has been exploited to develop diagnostics, with three in vivo imaging agents currently on the market (CEA-Scan, Myoscint, Verluma) [10,11].
In this review we focus on the recombinant and chemical modifications applied to improve the short-comings of different antibody fragments. We discuss the different modified antibody variants, with particular interest on the techniques used by FDA approved antibody fragments (Table 1). Additionally, we look at alternative methods, which are in their infancy and hold real promise for future therapeutic antibody fragments.
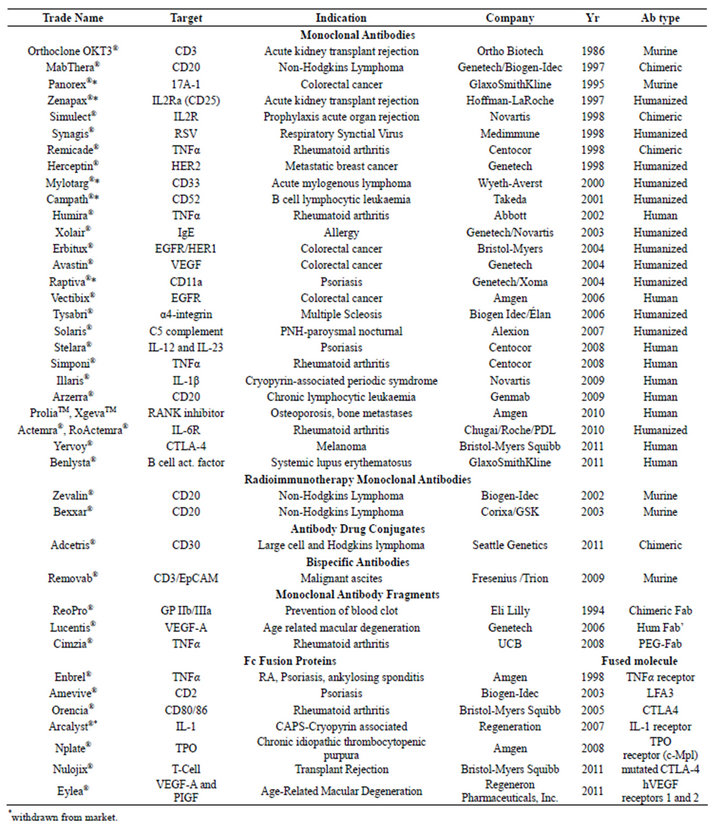
Table 1. Monoclonal antibodies and other antibody derivatives on the market [12].
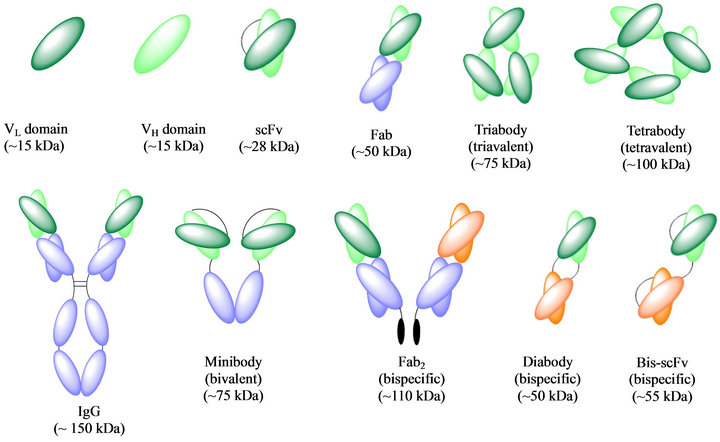
Figure 1. Schematic representation of different antibody formats [1].
2. ANTIBODY FRAGMENT DESIGN, THEIR APPLICATIONS AND LIMITATIONS
2.1. Antibody Fab Fragments
Fab fragments (~50 kDa) (Figure 1) can be produced by two methods: i) proteolysis of intact antibodies yielding monovalent Fab fragments and ii) direct expression from bacterial (E. coli, Bacillus megaterium), mammalian cells (CHO cells), or yeast (Pichia pastoris) [13,14]. Kunert et al., compared the expression of Fab fragments in CHO and Pichia pastoris, where they found that CHO cells were better for expressing glycosylated Fab fragments but that the Pichia system was more efficient during development to make clinical grade products [15]. Purification by Protein A, Protein G and Protein L of Fab fragments of different isotypes are standard methods used to isolate Fabs. Ion exchange and size exclusion chromatography are also used [16,17].
To improve antigen-binding affinity of antibody fragments, two approaches can be taken: i) the creation of large libraries of randomly mutated complementaritydetermining regions (CDRs) and ii) the creation of small libraries focused on mutagenesis that increase the chance to identify higher affinity variants [6].
Fab fragments are often produced to overcome the unwanted Fc effector functions for cytokine inactivation, receptor blockade or viral neutralisation [2]. The smaller size of Fab fragments compared to IgGs is often used to explain the improved tissue penetration of Fabs, which can be observed at tumour sites [2,5,18]. Additionally the smaller size of Fab fragments may allow interactions with cryptic epitopes such as enzyme active sites which mAbs may be unable to access [19]. Fab fragments have full binding specificity due to the antigen-binding site being unaltered; however due to Fab monovalency, it has been observed that Fab fragments have fast off-rates and poor retention times on the target [20].
2.2. Multivalent Antibody Fab Fragments
Multivalent Fab species can be built from univalent Fab fragments using genetic or chemical cross-linking Weir et al. formed a trivalent tri-Fab’-maleimide (TFM) and compared its binding, internalisation and pharmacokinetics (PK) properties to that of native IgG. They found that TFM showed a 3-fold improvement in the binding off-rate and there were increased rates of internalisation compared to IgG. These are promising characteristics when considering a therapeutic for drug delivery to tumour cells. The PK profiles of the monomeric Fab and TFM were found to be comparable [18].
2.3. Single-Chain Variable Region (scFv)
Single-chain, scFv (~28 kDa) (Figure 1) are comprised of both VH and VL domains joined by a flexible polypeptide linker that prevents dissociation. Many scFv formats are being evaluated preclinically. This is attributable to scFv’s having more favourable PKs for tissue penetration due to their even smaller size compared to Fab, while also retaining specific, monovalent, antigen-binding affinity [1,2]. scFv’s can bind a variety of antigens such as haptens, proteins, whole pathogens and can be fused to proteins which can be used in an effort to control infectious disease. Additionally, scFv’s can be used alone or as fusions in enzyme-linked immunosorbent assays (ELISA) [21]. However, scFv’s often exhibit fast off-rates and poor retention times on the target; therefore multivalent scFv’s have been engineered [20].
To produce scFv antibodies, mRNA can be obtained from hybridomas, spleen cells, immunised mice and B lymphocytes and then by reverse transcription spliced into cDNA to serve as a template for antibody gene amplification (PCR) [20]. From here, several types of library can be made: phage (immune, naïve and synthetic) or ribosomal display libraries to create a diverse range of antibody VH and VL genes. To date, scFv antibodies have been successfully expressed in various expression systems such as mammalian cell, yeast, plant and insect cells [22-24].
The most promising system is E.coli as this system has well-studied physiology and genetics, plus the capability to produce scFv rapidly, at high yields, low cost and with robust methodology [21]. As already discussed, CDR optimisation is a standard process in antibody engineering. This process can be used with scFv’s to improve their specific activity, which can allow for lower therapeutic doses to be used to reduce cost and possible toxicities [6].
2.4. Multimeric scFv’s
Mutivalent scFv’s have been obtained by both chemical and genetic cross-linking. The most successful strategy is to genetically reduce the scFv linker length to between zero to five residues to give bivalent dimers (diabodies, ~50 kDa), trivalent trimers (triabodies, 75 kDa) and tetravalent tetrabodies (110 kDa) (Figure 1). These multivalent scFv’s show potential in cancer imaging and radiotherapy applications. They have improved tissue penetration and faster blood clearance compared to the larger native IgG (150 kDa) [1,2]. IgG molecules have been shown to penetrate tumours more slowly than scFv fragments; however due to their monovalent binding they are cleared readily. The ideal tumour targeting reagent would have rapid tumour penetration, high target retention and rapid blood clearance, this has been observed with bivalent diabodies [1]. Another tumour therapy option is minibodies (~75 kDa) (Figure 1) which have shown promise due to their higher tumour intake compared to IgG [1,2].
3. APPROACHES TO EXTEND THE HALF-LIFE OF ANTIBODY FRAGMENTS
Different strategies have been developed to modify antibody fragments (e.g. Fab, scFv and Fv) [25]. Both recombinant and chemical approaches allow many modifications, especially to prolong circulation time [26]. Recombinant approaches include modification of antibody fragments that could lead to increased binding specificity and affinity (e.g. bispecific antibody Figure 1), to prolong the serum half-life (e.g. fusion with Fc or albumin) of therapeutics by using the endocytic recycling mechanism [27- 29], or by increasing the size of the protein molecule in circulation (e.g. glycoengineering methods).
Another recombinant approach to increase the apparent molecular size is multimerisation of recombinant antibody fragments. This method increases the molecular mass leading to enhancement in the overall size and improvement of binding affinity and valency [30]. However, multimerisation can result in heterogeneous complex formation and crosslinking of the target receptor that can lead to agonist functions that may not be desirable.
The chemical conjugation of poly(ethylene glycol) (PEG) to Fabs is a clinically proven method to extend half-life. Protein PEGylation has been shown to be clinically viable for several classes of protein-based medicine including cytokines, hemapoietics and enzymes. A hydrophilic, flexible polymer such as PEG has a large hydrodynamic volume in solution so that when conjugated to an antibody fragment increases its overall size [31]. The benefits are longer blood half-life, improved bioavailability, protease protection, stability, solubility, reduced immunogenicity and toxicity [32-36]. Several other polymers have also been conjugated to antibody fragments including polysicalic acid (PSA), N-(2-hydroxypropyl)methacrylamide (HPMA) and dextran [35,37].
3.1. Conjugation of PEG and Other Polymers PEGylation
Protein PEGylation is recognised as the leading method to prolong blood residence times of proteins with the potential to also reduce immunogenicity [33,34,37,38]. Although many more non-antibody examples exist (e.g., PEGylated IFN-2a/b, PEGylated erythropoietin), antibody fragments such as scFv, Fab or other domains are also a significant group of proteins that have been PEGylated [32,39].
PEGylation involves the covalent conjugation of a linear or branched PEG molecule to the antibody fragment by either a random or site-specific conjugation reaction [34,40]. The most common random PEG conjugation is via an amine-reactive linker such as N-hydroxy-succinimide (NHS) ester which can conjugate with lysine residues. Such PEG reagents have been used for several antibody fragments including (Fab’)2, Fab and scFv. As expected the PEGylated antibody fragments have shown a prolonged in vivo half-life [3,41-43]. Unfortunately NHS PEG reagents undergo reactions with water as well as different lysine and other nucleophilic groups on the protein. This results in heterogeneous products with PEG often conjugated near or at the binding site [3,32,41,44]. While PEG shielding will decrease “on” rates, direct conjugation of PEG at the binding site may decrease activity too much so that efficacy will be compromised even with the extended half-life. The problem of structural heterogeneity however can lead to a mixture of protein conjugates with varied activity.
An alternative approach is site-specific PEG conjugation, preferably in a position distal from the antigen binding site on an antibody fragment. One example is PEG chain conjugation through a maleimide linker to a free cysteine residue in the hinge region of a Fab’ fragment, or to a genetically engineered antibody fragment with a free cysteine [3,36,45-53]. Maleimide linkers can undergo hydrolysis and also PEG exchange reactions with other proteins, while incorporating a free cysteine into a protein can lead to disulfide scrambling and protein aggregation [54].
Nonetheless, since thiol conjugation is much more efficient compared to amine conjugation, it has been used as a basis to develop a PEG-Fab product. The first market-approved PEGylated Fab fragment is the anti-TNF-α PEGylated Fa-Certolizumab Pegol (Cimzia®) (Table 1). It was approved in April 2008 to treat rheumatoid arthritis [55-57].
Other examples under development are an anti-VEGFR2 PEGylated di-Fab for the solid tumors (Phase II) [58], an anti-CD40L monovalent PEGylated Fab (CDP-7657) to treat patients with systemic lupus erythematosus (Phase I) [59], an anticalin-based binding protein against VEGF, chemically PEGylated with a 40 kDa polymer for solid tumor (Angiocal-PRS-050; Phase I) [60] and a 40 kDa PEGylated adnectin (fibronectin domain-based binding protein being developed by Adnexus/Bristol Myers Squibb, Phase I, CT322) [61].
Another cysteine-directed site-specific coupling approach is PEG conjugation via two sulfur atoms in the disulfide bond of Fab fragments. This has been demonstrated using a thiol-specific PEG mono-sulfone for antiCD4 Fab, ranibizumab (Lucentis®) and Fabs and Fab’ obtained by the proteolytic digestion of IgGs such as bevacizumab (Avastin®) and trastuzumab (Herceptin®). Using this method, the PEGylated Fab showed improved size and retained antigen-binding capacity [62,63].
Another site-specific strategy to PEGylate an antibody fragment involves PEGylating a polyhistidine tag. This is commonly use in recombinant protein expression and purification. It was shown that this approach was able to increase the half-life of an anti-TNF domain antibody 200-fold, using two 20 kDa PEG chains [64].
A protein of interest can be expressed as an N-terminal intein fusion protein. The intein domain induces an N to S acyl shift at this protein-intein junction to form the thioester intermediate. Then C-terminal hydrazide or aminoxy derivatives are then generated through chemical cleavage under aqueous buffered conditions of this thioester intermediate using hydrazine or dioxyamine. The C-terminal hydrazide (or aminoxy) protein is then sitespecifically PEGylated with benzaldehyde-PEG derivatives to form homogenous product [65]. This technology has been applied to extend half-life of single domains [66].
3.1.1. Polysialylation
Polysialic acid (PSA) is a polymer derived from N-acetylneuraminic (sialic) acid) and has been used for protein conjugation [67,68]. The hydrophilic nature of PSA is thought to result in similar hydration properties to PEG, giving it a high apparent molecular weight in the blood and therefore increased circulation time. The first PSA antibody fragment conjugate was prepared using aminebased reductive amination by conjugation of PEG-aldehyde to H17E2Fab [69]. Reductive amination is not very efficient as a conjugation procedure and since PSA is charged, in contrast to PEG, the use of ion exchange chromatography can be somewhat tedious during purifications. Another example of PSA conjugation is polysialylation of an antitumor scFv fragment using maleimide chemistry [68]. Currently, no PSA-fragment conjugates are in the clinic.
3.1.2. HPMA
HPMA copolymer is another hydrophilic polymer that has been used extensively [70]. Seymour et al. reported conjugation of HPMA to murine IgG B72.3 and its Fab’ and F(ab’)2 fragments against carcinoembryonic antigen [71]. The half-life of the Fab’ fragment in blood circulation was improved 10-fold (from 35 min to 6 h). Despite the enhanced PK, the random amine-coupled HPMA conjugates showed no tumour-targeting activities. This may be due to steric hindrance of the polymer or disruption of the antigen binding domain by polymer chains. Lu et al. showed that site-specific HPMA conjugation through the maleimide group to the thiol residues created after IgG pepsin digestion results in retained antigen targeting affinity [72]. HPMA conjugation often occurs on pendent chains along the polymer main chain as is the case for PEG where conjugation is at the PEG terminus.
3.1.3. Dextran Modification
Dextran modification has also been applied to improve the PK properties of antibody fragments. Dextran is natural polysaccharide derived from monomers of glucose [73]. In comparison to PEG and PSA, dextran has a more compact structure and possesses multiple primary and secondary hydroxyl groups [35]. Due to its poly-functional character protein conjugation usually yields a heterogeneous mixture of products [73]. One example is dextran conjugation to an anti-CEA Fab fragment. They found that the PK of the conjugate was improved and the immune-reactivity of the Fab molecule was reduced [74].
3.1.4. Albumin Conjugation
Conjugation to albumin can also improve the PK of antibody fragments. Albumin has a naturally extended serum half-life, with a half-life of 19 days in humans [75- 77]. This is due to albumin recycling that is analogous to IgG recycling. A Fab fragment has been site-specifically conjugated to serum albumin using maleimide [78].
3.2. Recombinant Approaches
3.2.1. Recombinantly Modified Glycosylation
Recombiant glycosylation is the incorporation of Nglycosylation sites into the protein sequence, to improve the PK and pharmacodynamics of therapeutic proteins such as EPO, Interferon and blood factors [79,80]. Arensep®, an FDA approved hyper-glycosylated EPO has shown a three-fold increase in circulation time, but a four-fold decrease in receptor binding [79]. Stork et al produced a N-glycosylated bispecific single-chain diabody molecules (scDb) and compared their in vivo halflives to other modification such as PEGylation and fusion to albumin-binding domains (ABD) [81]. They found that the longest circulation time was produced by the fusion of scDb to an ABD (14-fold increase of AUC), followed by a scDb-HSA fusion protein and 40 kDa branched PEGylated scDb (10-fold increase of AUC), while Nglycosylation in HEK293 cells resulted only in a moderate increase in circulation time (2 - 3-fold increase of AUC) [81]. This demonstrates that N-glycosylation can be used as a general method to modulate the PK of antibody fragments.
3.2.2. Recombinant Homo-Amino-Polymers
Inert, hydrophilic polymers represent one of the strategies for improving the PK of proteins of interest; by genetically fusing amino acid polymers to therapeutic proteins to increase the overall hydrodynamic radius of the protein. Schlapsky et al. described the genetic fusion of a glycine rich homo-amino-acid polymer (HAP) to an antiHER2 Fab fragment 4D5. Their in vivo findings found a moderate increase in half-life from 6 h to 2 h with the 200 residue HAPylated Fab fragment compared to the Fab fragment alone [82].
HAPylation has been shown to have easier production with less downstream processing with the polymer being biodegradable, biocompatible and potentially non-immunogenic. Conversely, with HAPylation there are concerns that longer polymers are required for a PK effect. However, shorter polymer chains may have an application in medical imaging [35,82].
3.2.3. Recombinant Polysialylation
Recombinant polysialylation is a novel recombinant glycosylation technique with the aim of improving the halflife of proteins; however this method is still in its infancy. This technique attaches PSA-carrier domains from neural cell adhesion molecules (NCAMs) to antibodies or any other protein of interest. Offering an alternative to chemical conjugation of PSA to proteins, recombinant polysialylation offers easier production without downstream processing. Conjugates are biodegradable, bio-compatible and potentially nonimmunogenic. However, there are concerns over yield and homogeneity [35,83].
3.3. Other Approaches That Can Extend Half-Life and Therapeutic Effect
3.3.1. Bispecific Antibodies (bsMAb)
Currently there are two production approaches: i) the fusion of two hybridoma cell lines into a single “quadroma” cell line, however this is a difficult and time-consuming process; and ii) coupling two Fab fragments together via chemical or genetic conjugation or fusion. Chemical conjugation is still being developed, and there are no products yet in the clinic. However, there has been work described using bifunctional reagents such as 5,5’-dithiobis (2-nitrobenzoic acid) or o-phenylenedimaleimide to generate the bsMAb [84-87].
The key approach for bispecifics to increase half-life is for one binding moiety to bind to circulating albumin as was done with Dabs and diabodies [81]. However bispecific antibodies can either bind to two adjacent targets or can cross-link two different antigens. Thus, they are powerful therapeutic agents, showing great promise in cancer treatment either i) in the recruitment of cytotoxic T cells or ii) to effectively target tumour-associated antigens and deliver a cytotoxic payload to tumour cells [2].
A number of bispecific antibodies in clinical trials have one arm targeting the FcγR to enhance tumour killing. Currently the major challenge with bispecifics is production of material in sufficient quantity and purity to meet clinical needs [87]. Nevertheless Removab®, a mouse CD3-human EpCAM bispecific was approved for the treatment of malignant ascites due to epithelial carcinomas in 2009. Removab® kills EpCAM positive tumour cells in the peritoneal cavity which are the known cause of malignant ascites, leaving healthy cells alive since the peritoneum lack EpCAM [88,89]. Other bispecific antibodies in advanced clinical trials (Phase II) include Merrimack’s MM-111, targeting HER2 and HER3 receptors on breast cancer cells, Trion’s Lymphomun/ FBTA0, which targets CD20 on lymphoma cells and CD3 on T cells to treat B cell lymphoma, and Micromet’s blinatumomab which binds on T cells and CD19 which targets lymphoma cells to treat acute lymphoblastic leukaemia (ALL) [90].
3.3.2. Antibody Fusion
Antibody-cytokine fusion proteins have been linked to full length antibodies and antibody fragments such as scFv through the Nor C-termini of the antibody. The hope is to stimulate immune cell types such as monocytes, macrophages, natural killer cells, dendritic cells T and B cells. The Fc fragment of an antibody can be used to extend the half life of other molecules Enbrel® is dimeric fusion protein consisting extracellular domain of p75 TNF receptor linked to the Fc domain of IgG1, with the aim of binding and inactivating cell bound and soluble TNF and lymphotoxin α. Due to the Fc domain of IgG1, the half-life of Enbrel® is 4.8 days and has a high binding affinity making Enbrel® a first line therapy for rheumatoid arthritis [91]. Rheumatoid arthritis patients whose treatment with TNF α is unsuccessful, Orencia® is given. Orencia® is a fusion protein using the Fc domain of IgG1 to extend the half-life of the extracellular domain of CTLA-4 [92].
An alternative strategy is the fusion to albumin or albumin binding domains for example scFV fusion to albumin binding domains [93]. Alternative applications for fusion technology are in tumour therapy, where antitumor efficacy has been shown with Interleukin-12 fused to the N-termini of fibronectin-specific scFv [94]. However antibody-cytokine fusions must be concentrated at the intended site to overcome serious toxicity issues which may arise from cytokines modulating an immune response elsewhere [87].
4. CONCLUSION
This review summarises the strategies used to increase the half-life of therapeutic antibody fragments and the use of antibody fragments to extend the half-life of other molecules. Antibody fragments are an important class of therapeutic medicines. There are Fab fragments in development derived from native IgG as well as a range of single chain fragments and other protein scaffolds. As discussed, if these molecules are to be used as therapeutics there half-lives may need to be modulated; this can be accomplished clinically by the covalent conjugation of poly(ethylene glycol) (PEG) i.e. Cimzia®. Other clinically proven strategies include Fc fusion to increase the half-life of other molecules.
5. ACKNOWLEDGEMENTS
Both AHS and MF are grateful to PolyTherics Ltd for funding their PhD studies. AHS is also grateful to the UCL School of Pharmacy for funding made available for her PhD studies. HK and SB are grateful for funding from NIHR Biomedical Research Centre at Moorfields Hospital and the UCL Institute of Ophthalmology, Moorfields Special Trustees, the Helen Hamlyn Trust (in memory of Paul Hamlyn), Fight for Sight and Freemasons Grand Charity. SB is also grateful for funding from the UK Engineering & Physical Sciences Research Council (EPSRC) for the EPSRC Centre for Innovative Manufacturing in Emergent Macromolecular Therapies. Financial support from the consortium of industrial and governmental users is also acknowledged.
REFERENCES
- Hudson, P.J. and Souriau, C. (2003) Engineered antibodies. Nature Medicine, 9, 129-134. doi:10.1038/nm0103-129
- Chapman, A.P., et al. (1999) Therapeutic antibody fragments with prolonged in vivo half-lives. Nature Biotechnology, 17, 780-783. doi:10.1038/11717
- Bradbury, A., et al. (2003) Antibodies in proteomics I: Generating antibodies. Trends in Biotechnology, 21, 275- 281. doi:10.1016/S0167-7799(03)00112-4
- Holliger, P. and Hudson, P.J. (2005) Engineered antibody fragments and the rise of single domains. Nature Biotechnology, 23, 1126-1136. doi:10.1038/nbt1142
- Nelson, A.L. (2010) Antibody fragments: Hope and hype. Multi-Agent-Based Simulation, 2, 77-83. doi:10.4161/mabs.2.1.10786
- Chowdhury, P.S. and Wu, H. (2005) Tailor-made antibody therapeutics. Methods, 36, 11-24. doi:10.1016/j.ymeth.2005.01.002
- Pucca, M.B., et al. (2011) Therapeutic monoclonal antibodies: scFv patents as a marker of a new class of potential biopharmaceuticals. Brazilian Journal of Pharmaceutical Sciences, 47, 31-38. doi:10.1590/S1984-82502011000100005
- Teicher, B.A. and Chari, R.V. (2011) Antibody conjugate therapeutics: Challenges and potential. Clinical Cancer Research, 17, 6389-6397. doi:10.1158/1078-0432.CCR-11-1417
- Nelson, A.L. and Reichert, J.M. (2009) Development trends for therapeutic antibody fragments. Nature Biotechnology, 27, 331-337. doi:10.1038/nbt0409-331
- Kaur, S., et al. (2012) Recent trends in antibody-based oncologic imaging. Cancer Letters, 315, 97-111. doi:10.1016/j.canlet.2011.10.017
- Leader, B., et al. (2008) Protein therapeutics: A summary and pharmacological classification. Nature Reviews Drug Discovery, 7, 21-39. doi:10.1038/nrd2399
- Wang, W., et al. (2008) Monoclonal antibody pharmacokinetics and pharmacodynamics. Clinical Pharmacology & Therapeutics, 84, 548-558. doi:10.1038/clpt.2008.170
- Steinwand, M., et al. (2010) Production of antibody fragments in the gram-positive bacterium Bacillus megaterium. In: Kontermann, R. and Dübel, S., Eds., Antibody Engineering, Springer Berlin Heidelberg, Berlin, Heidelberg, 293-299.
- Reilly, D. and Yansura, D. (2010) Production of antibodies and antibody fragments in Escherichia coli. In: Kontermann, R. and Dübel, S., Eds., Antibody Engineering, Springer Berlin Heidelberg, Berlin, Heidelberg, 331-344.
- Kunert, R. (2008) Expression of Fab fragments in CHO and Pichia pastoris. BioProcess International. Chapter 5 Supplement, 34-40.
- Zhao, Y., et al. (2009) Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expression and Purification, 67, 182-189. doi:10.1016/j.pep.2009.04.012
- Griep, R. and McDougall, J. (2010) Analysis and purification of antibody fragments using protein A, protein G, and protein L. In: Kontermann, R. and Dübel, S., Eds., Antibody Engineering, Springer Berlin Heidelberg, Berlin, Heidelberg, 301-315.
- Weir, A.N., et al. (2002) Formatting antibody fragments to mediate specific therapeutic functions. Biochemical Society Transactions, 30, 512-516. doi:10.1042/BST0300512
- Singh, G. (2009) The emergence of antibody fragments and derivatives. Innovation in Pharmaceutical Tachnology, BioPharma UK, Biopharma, 46-48.
- Adams, G.P., et al. (2001) High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Research, 61, 4750-4755.
- Ahmad, Z.A., et al. (2012) scFv antibody: Principles and clinical application. Clinical and Developmental Immunology, 2012, Article ID: 980250. doi:10.1155/2012/980250
- Joosten, V., et al. (2003) The production of antibody fragments and antibody fusion proteins by yeasts and filamentous fungi. Microbial Cell Factories, 2, 1. doi:10.1186/1475-2859-2-1
- Hellwig, S. and Melmer, G. (2010) Bioreactor production of scFv fragments in Pichia pastoris. In: Kontermann, R. and Dübel, S., Eds., Antibody Engineering, Springer Berlin Heidelberg, Berlin, Heidelberg, 363-375.
- Conrad, U. and Floss, D. (2001) Expression of antibody fragments in transgenic plants. In: Kontermann, R. and Dübel, S., Eds., Antibody Engineering, Springer Berlin Heidelberg, Berlin, Heidelberg, 377-386.
- Chames, P., et al. (2009) Therapeutic antibodies: Successes, limitations and hopes for the future. British Journal of Pharmacology, 157, 220-233. doi:10.1111/j.1476-5381.2009.00190.x
- Jain, M., et al. (2007) Engineering antibodies for clinical applications. Trends in Biotechnology, 25, 307-316. doi:10.1016/j.tibtech.2007.05.001
- Andersen, J.T. and Sandlie, I. (2009) The versatile MHC class I: Related FcRn protects IgG and albumin from degradation. Drug Metabolism and Pharmacokinetics, 24, 318-332. doi:10.2133/dmpk.24.318
- Mueller, D., et al. (2007) Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. The Journal of Biological Chemistry, 282, 12650-12660. doi:10.1074/jbc.M700820200
- Nguyen, A., et al. (2006) The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Engineering Design & Selection, 19, 291-297. doi:10.1093/protein/gzl011
- Pluckthun, A. and Pack, P. (1997) New protein engineering approaches to multivalent and bispecific antibody fragments. Immunotechnology, 3, 83-105. doi:10.1016/S1380-2933(97)00067-5
- Haraldsson, B. et al. (2008) Properties of the glomerular barrier and mechanisms of proteinuria. Physiological Reviews, 88, 451-487. doi:10.1152/physrev.00055.2006
- Chapman, A.P. (2002) PEGylated antibodies and antibody fragments for improved therapy: A review. Advanced Drug Delivery Reviews, 54, 531-545. doi:10.1016/S0169-409X(02)00026-1
- Jevsevar, S., et al. (2010) PEGylation of therapeutic proteins. Biotechnology Journal, 5, 113-128. doi:10.1002/biot.200900218
- Bailon, P. and Won, C.Y. (2009) PEG-modified biopharmaceuticals. Expert Opinion on Drug Delivery, 6, 1-16. doi:10.1517/17425240802650568
- Chen, C., et al. (2011) Modulating antibody pharmacokinetics using hydrophilic polymers. Expert Opinion on Drug Delivery, 8, 1221-1236. doi:10.1517/17425247.2011.602399
- Palm, T. et al. (2011) The effect of PEGylation on the stability of small therapeutic proteins. Pharmaceutical Development and Technology, 16, 441-448. doi:10.3109/10837450.2010.535830
- Kang, J.S., et al. (2009) Emerging PEGylated drugs. Expert Opinion Emerg Drugs, 14, 363-380. doi:10.1517/14728210902907847
- Payne, R.W., et al. (2011) Product development issues for PEGylated proteins. Pharmaceutical Development Technology, 16, 423-440. doi:10.3109/10837450.2010.513990
- Germershaus, O., et al. (2006) Trastuzumab-polyethylenimine-polyethylene glycol conjugates for targeting Her2-ex-pressing tumors. Bioconjugate Chemistry, 17, 1190-1199. doi:10.1021/bc0601119
- Pasut, G. and Veronese, F.M. (2012) State of the art in PEGylation: The great versatility achieved after forty years of research. Journal of Controlled Release, 161, 461-472. doi:10.1016/j.jconrel.2011.10.037
- Delgado, C., et al. (1996) Enhanced tumour specificity of an anti-carcinoembrionic antigen Fab’ fragment by poly- (ethylene glycol) (PEG) modification. British Journal Cancer, 73, 175-182. doi:10.1038/bjc.1996.32
- Koumenis, I.L., et al. (2000) Modulating pharmacokinetics of an anti-interleukin-8 F(ab’)2 by amine-specific PEGylation with preserved bioactivity. International Journal of Pharmaceutics, 198, 83-95. doi:10.1016/S0378-5173(99)00458-5
- Li, L., et al. (2006) Improved biodistribution and radioimmunoimaging with poly(ethylene glycol)-DOTA-conjugated anti-CEA diabody. Bioconjugate Chemistry, 17, 68-76. doi:10.1021/bc0502614
- Lee, L.S., et al. (1999) Prolonged circulating lives of single-chain fv proteins conjugated with polyethylene glycol: A comparison of conjugation chemistries and compounds. Bioconjugate Chemistry, 10, 973-981.
- Lu, Y., et al. (2008) Effect of PEGylation on the solution conformation of antibody fragments. Journal of Pharmaceutical Sciences, 97, 2062-2079. doi:10.1002/jps.21170
- Natarajan, A., et al. (2005) Characterization of site-specific ScFv PEGylation for tumor-targeting pharmaceuticals. Bioconjugate Chemistry, 16, 113-121.
- Krinner, E.M., et al. (2006) A highly stable polyethylene glycol-conjugated human single-chain antibody neutralizing granulocyte-macrophage colony stimulating factor at low nanomolar concentration. Protein Engineering Design & Selection, 19, 461-470. doi:10.1093/protein/gzl031
- Xiong, C.Y., et al. (2006) Development of tumor targeting anti-MUC-1 multimer: Effects of di-scFv unpaired cysteine location on PEGylation and tumor binding. Protein Engineering Design & Selection, 19, 359-367. doi:10.1093/protein/gzl020
- Yang, K., et al. (2003) Tailoring structure-function and pharmacokinetic properties of single-chain Fv proteins by site-specific PEGylation. Protein Engineering Design & Selection, 16, 761-770. doi:10.1093/protein/gzg093
- Kubetzko, S., et al. (2006) PEGylation and multimerization of the anti-p185HER-2 single chain Fv fragment 4D5: Effects on tumor targeting. The Journal of Biological Chemistry, 281, 35186-35201. doi:10.1074/jbc.M604127200
- Humphreys, D.P., et al. (2007) Alternative antibody Fab’ fragment PEGylation strategies: Combination of strong reducing agents, disruption of the interchain disulphide bond and disulphide engineering. Protein Engineering Design & Selection, 20, 227-234.
- Albrecht, H., et al. (2003) Production of soluble ScFvs with C-terminal-free thiol for site-specific conjugation or stable dimeric ScFvs on demand. Bioconjugate Chemistry, 15, 16-26.
- Knight, D.M., et al. (2004) Pharmacodynamic enhancement of the anti-platelet antibody fab abciximab by sitespecific pegylation. Platelets, 15, 409-418. doi:10.1080/09537100410001723135
- Roberts, M.J., Bentley, M.D. and Harris, J.M. (2002) Chemistry for peptide and protein PEGylation. Advanced Drug Delivery Reviews, 54, 459-476. doi:10.1016/S0169-409X(02)00022-4
- Choy, E.H.S., et al. (2002) Efficacy of a novel PEGylated humanized anti-TNF fragment (CDP870) in patients with rheumatoid arthritis: A phase II double-blinded, randomized, dose-escalating trial. Rheumatology, 41, 1133-1137.
- Schreiber, S., et al. (2005) A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn’s disease. Gastroenterology, 129, 807-818. doi:10.1053/j.gastro.2005.06.064
- Nesbitt, A., Stephens, S., et al. (2009) Certolizumab pegol: A PEGylated anti-tumour necrosis factor alpha biological agent. In: Veronese, F., Ed., PEGylated Protein Drugs: Basic Science and Clinical Applications, Birkhäuser Basel, Basel, 229-254. doi:10.1007/978-3-7643-8679-5_14
- Ton, N.C., et al. (2007) Phase I evaluation of CDP791, a PEGylated di-Fab’ conjugate that binds vascular endothelial growth factor receptor 2. Clinical Cancer Research, 13, 7113-7188. doi:10.1158/1078-0432.CCR-07-1550
- Dunlap, L. (2013) The role of CD40L in ALS. Drug Discovery News, 9, 5. http://www.drugdiscoverynews.com
- Mross K., F.R., Richly, H., Scharr, D., Buechert, M., Stern, A., Hoth, D., Gille, H., Audoly, L.P. and Scheulen M.E. (2011) First in human phase I study of PRS-050 (Angiocal), a VEGF-A targeting anticalin, in patients with advanced solid tumors: Results of a dose escalation study. American Association for Cancer Research, 10, A212. doi:10.1158/1535-7163.TARG-11-A212
- Tolcher, A.W., et al. (2011) Phase i and pharmacokinetic study of CT-322 (BMS-844203), a targeted adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clinical Cancer Research, 17, 363-371.
- Shaunak, S., et al. (2006) Site-specific PEGylation of native disulfide bonds in therapeutic proteins. Nature Chemical Biology, 2, 312-313. doi:10.1038/nchembio786
- Khalili, H., et al. (2012) Comparative binding of disulfide-bridged PEG-Fabs. Bioconjugate Chemistry, 23, 2262- 2277. doi:10.1021/bc300372r
- Cong, Y., et al. (2012) Site-specific PEGylation at histidine tags. Bioconjugate Chemistry, 23, 248-63. doi:10.1021/bc200530x
- Thom, J., et al. (2011) Recombinant protein hydrazides: Application to site-specific protein PEGylation. Bioconjugate Chemistry, 22, 1017-1020.
- Almac Group Ltd. (2013) Almac protein conjugation technology. https://www.almacgroup.com
- Gregoriadis, G., et al. (2005) Improving the therapeutic efficacy of peptides and proteins: A role for polysialic acids. International Journal of Pharmaceutics, 300, 125- 130.
- Constantinou, A., et al. (2009) Site-specific polysialylation of an antitumor single-chain Fv fragment. Bioconjugate Chemistry, 20, 924-931. doi:10.1021/bc8005122
- Constantinou, A., et al. (2008) Modulation of antibody pharmacokinetics by chemical polysialylation. Bioconjugate Chemistry, 19, 643-650. doi:10.1021/bc700319r
- Kopeček, J. and Kopečková, P. (2010) HPMA copolymers: Origins, early developments, present, and future. Advanced Drug Delivery Reviews, 62, 122-149. doi:10.1016/j.addr.2009.10.004
- Seymour, L.W., et al. (1991) Synthetic polymers conjugated to monoclonal antibodies: Vehicles for tumourtargeted drug delivery. Selective Cancer Therapeutics, 7, 59-73. doi:10.1089/sct.1991.7.59
- Lu, Z.R., et al. (1999) Polymerizable Fab’ antibody fragments for targeting of anticancer drugs. Nature Biotechnol, 17, 1101-1104. doi:10.1038/15085
- Mehvar, R. (2000) Dextrans for targeted and sustained delivery of therapeutic and imaging agents. Journal of Controlled Release, 69, 1-25. doi:10.1016/S0168-3659(00)00302-3
- Fagnani, R., et al. (1995) Altered pharmacokinetic and tumour localization properties of Fab’ fragments of a murine monoclonal anti-CEA antibody by covalent modification with low molecular weight dextran. Nuclear Medicine Communications, 16, 362-369. doi:10.1097/00006231-199505000-00008
- Kratz, F. (2008) Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. Journal Controlled Release, 132, 171-183. doi:10.1016/j.jconrel.2008.05.010
- Dennis, M.S., et al. (2002) Albumin binding as a general strategy for improving the pharmacokinetics of proteins. The Journal of Biological Chemistry, 277, 35035-35043. doi:10.1074/jbc.M205854200
- Dennis, M.S., et al. (2007) Imaging tumors with an albumin-binding Fab: A novel tumor-targeting agent. Cancer Research, 67, 254-261. doi:10.1158/0008-5472.CAN-06-2531
- Smith, B.J., et al. (2001) Prolonged in vivo residence times of antibody fragments associated with albumin. Bioconjugate Chemistry, 12, 750-756. doi:10.1021/bc010003g
- Walsh, G. and Jefferis, R. (2006) Post-translational modifications in the context of therapeutic proteins. Nature Biotechnology, 24, 1241-1252. doi:10.1038/nbt1252
- Elliott, S., et al. (2003) Enhancement of therapeutic protein in vivo activities through glycoengineering. Nature Biotechnology, 21, 414-421. doi:10.1038/nbt799
- Stork, R., et al. (2008) N-glycosylation as novel strategy to improve pharmacokinetic properties of bispecific single-chain diabodies. The Journal of Biological Chemistry, 283, 7804-7812. doi:10.1074/jbc.M709179200
- Schlapschy, M., et al. (2007) Fusion of a recombinant antibody fragment with a homo-amino-acid polymer: Effects on biophysical properties and prolonged plasma half-life. Protein Engineering Design & Selection, 20, 273-284. doi:10.1093/protein/gzm020
- Constantinou, A. (2005) Production and study of polysialylated antibodies for improved cancer therapy. PhD, Imperial College, London.
- Brenner, M.B., et al. (1985) Cross-linking of human T cell receptor proteins: Association between the T cell idiotype β subunit and the T3 glycoprotein heavy subunit. Cell, 40, 183-190. doi:10.1016/0092-8674(85)90321-6
- Glennie, M.J., et al. (1987) Preparation and performance of bispecific F(ab’ gamma)2 antibody containing thioether-linked Fab’ gamma fragments. The Journal of Immunology, 139, 2367-2375.
- Shalaby, M.R., et al. (1992) Development of humanized bispecific antibodies reactive with cytotoxic lymphocytes and tumor cells overexpressing the HER2 protooncogene. The Journal of Experimental Medicine, 175, 217- 225. doi:10.1084/jem.175.1.217
- Presta, L. (2003) Antibody engineering for therapeutics. Current Opinion in Structural Biology, 13, 519-525. doi:10.1016/S0959-440X(03)00103-9
- Chames, P. and Baty, D. (2009) Bispecific antibodies for cancer therapy: The light at the end of the tunnel? Multi-Agent-Based Simulation, 1, 539-547. doi:10.4161/mabs.1.6.10015
- Linke, R., et al. (2010) Catumaxomab: Clinical development and future directions. Multi-Agent-Based Simulation, 2, 129-136. doi:10.4161/mabs.2.2.11221
- Kontermann, R. (2011) Bispecific antibodies: Developments and current perspectives. In: Kontermann, R.E., Ed., Bispecific Antibodies. Springer Berlin Heidelberg, Berlin, Heidelberg, 1-28. doi:10.1007/978-3-642-20910-9_1
- Spencer-Green, G. (2000) Etanercept (enbrel): Update on therapeutic use. Annals Rheumatic Diseases, 59, i46-i49. doi:10.1136/ard.59.suppl_1.i46
- Korhonen, R. and Moilanen, E. (2009) Abatacept, a novel CD80/86-CD28 T cell co-stimulation modulator, in the treatment of rheumatoid arthritis. Basic & Clinical Pharmacology & Toxicology, 104, 276-284. doi:10.1111/j.1742-7843.2009.00375.x
- Hopp, J., et al. (2010) The effects of affinity and valency of an albumin-binding domain (ABD) on the half-life of a single-chain diabody-ABD fusion protein. Protein Engineering Design & Selection, 23, 827-834. doi:10.1093/protein/gzq058
- Peng, L.S., et al. (2001) Mechanism of antitumor activity of a single-chain interleukin-12 IgG3 antibody fusion protein (mscIL-12.her2.IgG3). Journal of Interferon & Cytokine Research, 21, 709-720. doi:10.1089/107999001753124444