Advances in Bioscience and Biotechnology
Vol.4 No.4(2013), Article ID:30917,9 pages DOI:10.4236/abb.2013.44077
Inhibition of Lon protease by bacterial lipopolisaccharide (LPS) though inhibition of ATPase
![]()
1Laboratory of Biodefense & Regulation, Osaka University of Pharmaceutical Sciences, Osaka, Japan
2Division of Molecular Genetics, National Institute of Infectious Diseases, Tokyo, Japan
Email: amano@gly.oups.ac.ip
Copyright © 2013 Nahoko Sugiyama et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 3 December 2012; revised 26 December 2012; accepted 2 January 2013
Keywords: ATPase; Lipid A; Lipopolysaccharide (LPS); Lon; Protease
ABSTRACT
Lon protease, an ATP-dependent protease in Escherichia coli, degrades abnormal proteins and regulates several important cellular functions. Here we show novel inhibitory effects of lipopolysaccharide (LPS) on Lon protease activities. LPS inhibited the peptidase, protease, and ATPase activities of Lon; and a dose-response study showed that LPS at low doses more effectively inhibited the ATPase activity than the peptidase one, suggesting different susceptibility to LPS of these activities associated with Lon. Structure-activity relationship studies revealed that ReLPS, detoxified LPS, and mono-phosphoryl as well as diphosphoryl lipid A, also showed similar inhibition, suggesting that neither O-antigen polysaccharide nor O-acyl chain, but rather phosphate groups in the lipid A domain, seem to have been responsible for the inhibitory effects. Besides, LPS was co-precipitated with Lon by an anti-Lon antibody, showing the direct binding of LPS to Lon. These results suggest that LPS bound to Lon and inhibited the protease activity of Lon by inhibiting its ATPase activity. These results also seem to be another example of a negatively charged phosphate group in membrane components of Escherichia coli being involved in the regulation of protease activity of Lon through binding to Lon and inhibiting its ATPase activity, as in the case of cardiolipin.
1. INTRODUCTION
Proteolysis is an important function in cellular processes, regulating stress responses and quality control of proteins by eliminating defective or abnormal proteins which might otherwise lead to defective cell metabolism and other malfunctions. ATP-dependent proteases have been reported to participate in the catabolism of proteins not only in prokaryotes but also in archaic microorganisms as well as in mitochondria of eukaryotes [1,2]. Among them, Lon proteases were the first ones discovered in E. coli [2]. All of them have a similar structure and are high molecular weight complexes with distinct activity sites, an ATPase and a catalytic domain. They require ATP hydrolysis before degradation of the substrate, which is necessary to unfold the substrate proteins before importing them suitably to the catalytic domain of the protease.
Lon protease of E. coli is involved in the acquisition of tolerance to UV or high pressure, bacteriophage development, regulation of cell division, degradation of ribosomal proteins under amino acid starvation, as well as protein quality control [3-7]. Lon is also known to be involved in the regulation of Salmonella pathogenesis [8]. Besides, Lon binds to DNA with high affinity, which binding stimulates the ATPase and proteolytic activities of Lon [9]. Inorganic polyphosphate (polyP) also activates the catalytic activity of Lon through binding to Lon, which activity participates in enhanced degradation of ribosomal proteins under nutritional starvation [10]. On the contrary, cardiolipin, a major component of E. coli inner membrane phospholipids and having a negative charge, binds to Lon and inhibits both its ATPase and protease activities [11]. These results in the previous studies imply regulatory roles of negatively charged phosphate groups of such molecules on the activity of Lon, although the precise mechanisms underlying these interactions at the molecular basis remain unknown.
In this study, we examined the regulatory effects of lipopolysaccharide (LPS) on both protease (including peptidase) and ATPase activities of Lon. LPS, whose basic structure is 1, 4’-diphosphoryl 2, 2’-N-acyl 3, 3’-o-acylethylbiose, is one of the major components of the outer membrane of the Gram (-) bacteria; and it exerts a variety of biological and pathological effects on mammalian cells and animals by interacting with various cellular components through its phosphate groups, which reside in its active domain called lipid A [12-14]. Besides, LPS forms micelles in aqueous solution, with its phosphate groups being outside, which might mimic the repeated chain of phosphate groups in cardiolipin [15], DNA or polyP. Therefore, we examined whether LPS would have regulatory effects on the activities of Lon.
2. MATERIALS AND METHODS
2.1. Materials
Lipopolysaccharide (LPS; E. coli O55:B5, chromatographically purified), detoxified LPS (alkaline hydrolyzed LPS; E. coli O55:B5, chromatographically purified), ReLPS (LPS; Salmonella minnesota, chromatographically purified), and monoand di-phosphoryl lipid A (Salmonella minnesota Re 595(Re mutant)) were obtained from Sigma (St. Louis, MO, USA). Suc-Phe-Leu-Phe-β-D-methoxynaphtylamide (MNA) was obtained from Bachem Bioscience (Torrance, CA, USA); α-casein, from Sigma; and Alexa Fluor 488-labeled LPS, from Invitrogen (Carlsbad, CA, USA). Other chemicals were of the purest chemical grade available.
2.2. Preparation of Lon
Lon was purified by using a maltose-binding protein (MBP) fusion system as described previously [11,16]. In brief, E. coli cells of the JM109 strain carrying the pMAL-c2-Lon vector were cultivated at 37˚C overnight in LB medium containing 50 μg/mL ampicillin and then diluted with fresh LB medium to OD550 = 0.1 and incubated at 37˚C until OD550 = 0.4. The production of the MBP-Lon fusion protein was induced by the addition of 0.3 mM IPTG, followed by cultivation for a further 3 h. The cells were harvested by centrifugation and washed with PBS; and then the resultant precipitates were resuspended in buffer A comprising 0.0025% (w/v) lysozyme, 1 mM EDTA, and 20% (w/v) sucrose in 20 mM TrisHCl, pH 8.0. The cells were disrupted with glass beads (Sigma) in a Cell Beater (BIO 101, Fast PrepTM FP120; Thermo Savant, Holbrook, NY, USA), and the crude extracts were collected by centrifugation. The supernatants were dialyzed against buffer B comprising 200 mM NaCl and 20 mM Tris-HCl, pH 8.0, and then loaded onto a column of amylose resin (New England Biolabs, Beverly, MA, USA) equilibrated with buffer C comprising 200 mM NaCl, 20% (v/v) glycerol, and 20 mM Tris-HCl, pH 8.0. The column was washed with buffer C, and the MBPLon fusion protein was eluted with 10 mM maltose in buffer C, and then concentrated with an Amicon Ultra-4 according to the manufacturer’s instructions (MILLIPOREMA, USA). Protein concentrations were determined by the Bradford method using bovine serum albumin as the standard. For separation of Lon from MBP, the MBPLon fusion protein (400 μg) was incubated with 20 μg of Factor Xa protease (New England Biolabs) at 4˚C for 14 h, and then the Lon protein was further purified with a Sephacryl S - 200 (GE Healthcare, Buckinghamshire, England) column equilibrated with buffer C. The purified Lon protein was stored at −80˚C until used.
2.3. Assays of Peptidase and Protease Activity of Lon
The peptidase activity of Lon protease was determined by using a fluorogenic peptide, Suc-Phe-Leu-Phe-MNA, as a substrate [16]. The reaction, carried out in a 96 - well clustered micro plate at 37˚C for 60 min, contained 0.02 μM and 5 μM of the substrate in the presence or absence of 1 mM ATP in 100 μL of TM buffer (50 mM Tris-HCl, pH 8.0, and 7.5 mM MgCl2). The reaction was terminated by chilling the micro plate on ice, which termination was followed by measurement of the fluorescence of MNA released from the substrate with a spectrofluorometer (Fluoroscan II; Labsystems Oy, Helsinki, Finland) with excitation and emission wavelengths of 335 and 410 nm, respectively.
The proteolytic activity was measured by using casein as a substrate [11,16,17]. Under the standard assay conditions, LPS was pre-incubated with 0.3 μM Lon in 50 mM Tris-HCl, pH 8.0, containing 25 mM MgCl2 on ice for 30 min. Then, the reaction was started by the addition of 5 μM casein in the presence or absence of 4 mM ATP, followed by incubation at 37˚C for 0 - 120 min. The reaction was stopped by mixing 10 μL of the reaction mixture with 5 μL of 3x SDS sample buffer comprising 0.15% (w/v) bromphenol blue, 25% (v/v) glycerol, 6% (v/v) mercaptoethanol, and 3% (w/v) SDS in 150 mM Tris-HCl, pH 7.5. The samples were boiled for 5 min, and then subjected to electrophoresis on 5 - 20% (v/v) polyacrylamide SDS-slab gels (e-PAGEL; ATTO, Tokyo, Japan). Thereafter the gels were stained with 0.025% (w/v) Coomassie Brilliant Blue R - 250 in 10% (v/v) acetic acid and 20% (v/v) methanol solution, and then the densities of the protein bands of casein were determined with an LAS1000 Bio-Image Analyzer (Fuji-film, Tokyo, Japan) running Multi-Gauge software. The results were shown relative to the initial density of the casein band.
2.4. ATPase Activity of Lon Protease in the Presence of LPS
ATPase activity was measured with [γ-32P] ATP (Perkin Elmer, Massachusetts, USA) as a substrate [11,18,19]. Various concentrations of LPS were mixed with 0.3 μM Lon in TM buffer, followed by preincubation on ice for 30 min. Then 4 mM unlabeled ATP with [γ-32P] ATP was added to the reaction mixture, followed by incubation at 37˚C for 0 - 45 min. The reaction was stopped by the addition of ice-cold PBS(-) containing a mixture of 6% (w/v) Norit A (Nacalai Tesque, Kyoto, Japan) and 0.3% (w/v) Dextran T - 500 (GE Healthcare); and the reaction mixture was left on ice for 30 min, and then centrifuged at 11,000 g’ for 15 min. The radioactivity of [32P] orthophosphate in the resultant supernatant was measured by counting it in ACS-II scintillation cocktail (GE Healthcare) and using a Liquid Scintillation Analyzer, Tri-Carb 1600 TR (Perkin Elmer).
2.5. Assay to Measure Binding of Lon to LPS
Alexa Fluor 488 - labeled LPS was incubated at 0.2 μg/μL with or without 0.2 μM Lon, in TM buffer containing 4 mM ATP at 4˚C for 2 hr. An anti-Lon antibody [11] was then added to the reaction mixture, which was then incubated with vortexing at 4˚C for 2 hr. Next, Protein A Sepharose (GE Healthcare) was added, and incubation was continued at 4˚C for 1 hr. Finally, the reaction mixture was centrifuged at 11,000 g’ briefly, and the resultant precipitates were mixed with 200 μL of with buffer D, comprising 0.15 M NaCl, 0.1 mM EDTA, 0.01% Triton X - 100 in 10 mM Tris-HCl, pH 8.0, followed by centrifugation as described above. The precipitates were then suspended in 200 μL of high salt buffer E, containing 1 M NaCl in buffer D, and centrifuged; subsequently the resultant precipitates were suspended in buffer D again. After repeated centrifugation, the precipitates were mixed with 100 μL of buffer C, comprising 1% SDS, 10% glycerol, 2% β-mercaptoethanol in 50 mM Tris-HCl, pH 8.0, and were boiled for 5 min. Finally, the samples were centrifuged at 11,000 g’ for 5 min at 4˚C, and the resultant supernatants were collected and subsequently examined not only for the recovery of Lon by SDS-PAGE/Western blotting, but also for the fluorescence of LPS by using a Fluoroscan II spectrofluorometer with excitation and emission wavelengths of 485 and 538 nm, respectively.
The intensities of the protein bands of Lon were analyzed by using a LAS-1000 Bio-image Analyzer (Fujifilm) and quantified with Multi-Gauge software as described above.
3. RESULTS
3.1. Effects of LPS on Proteolytic Activity of Lon
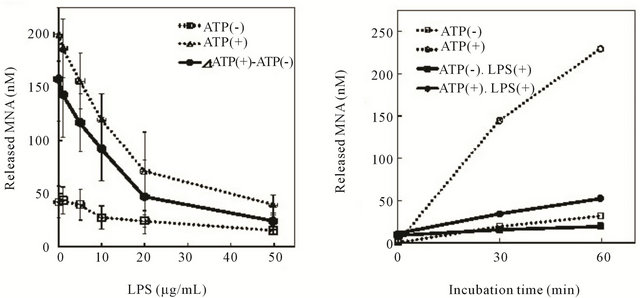
We first investigated the effect of LPS on the peptidase activity of Lon by monitoring the degradation of the fluorescent substrate Suc-Phe-Leu-Phe-MNA. ATP-dependent cleavage of the substrate peptide by Lon was inhibited dose dependently by LPS (Figure 1(a)), and 50 μg/ mL LPS gave almost complete inhibition.
A time-course study of the peptidase activity of Lon revealed that addition of 50 μg/mL LPS significantly and strongly inhibited the ATP-dependent cleavage of the substrate, whereas it showed little inhibition of ATP-independent cleavage (Figure 1(b)). These suggest that LPS selectively inhibited the ATP-dependent peptidase
 (a) (b)
(a) (b)
Figure 1. Effect of LPS on the peptidase activity of Lon. Lon protease (0.02 μM) was incubated with LPS in TM buffer in the presence or absence of 1 mM ATP at 4˚C for 30 min. The reaction was started by the addition of 5 μM Suc-Phe-Leu-Phe-MNA as a substrate, followed by incubation at 37˚C for 0 - 60 min. The fluorescence of the cleaved MNA was measured at excitation 335 nm/emission 410 nm as described in the text. (a) The reaction performed at 37˚C for 60 min after pre-incubation with 0 - 50 μg/mL of LPS as shown on the abscissa. (b) Time-course of the reaction in the presence or absence of 1 mM ATP and/or 50 μg/mL LPS. In these figures, ATP-dependent activity is shown with solid lines.
activity of Lon.
We next examined whether LPS would inhibit the protease activity of Lon by measuring the degradation of casein. Degradation of casein proceeded time dependently in the presence of ATP and was inhibited by LPS (Figures 2(A) and (B)); and this inhibition was dose dependently lessened by it (Figures 2(C) and (D)), with more than 50 μg/mL LPS causing significant inhibition. Comparison of the results shown in Figures 1 and 2 revealed that the extent of LPS-induced inhibition of the proteolytic activity of Lon was greater for the peptidase activity (Figure 1(a)) than for the protease activity (Figure 2(E)).
3.2. Inhibition of ATPase Activity of Lon by LPS
Because the hydrolysis of ATP precedes the proteolytic activity of Lon [2], we next studied the effect of LPS on


Figure 2. Effect of LPS on the protease activity of Lon. Protease activity of Lon was examined with casein as a substrate. (A), (B), Casein (5 μM) was incubated with 0.3 μM Lon in the presence or absence of 4 mM ATP or with 4 mM ATP and 50 μg/mL LPS at 37˚C for 0 - 120 min. A typical degradation profile of casein is shown, as obtained by quantitative analysis of Coomassie Brilliant Blue (CBB)-stained casein bands (A). The intensity of the casein bands are shown relative to each control at 0 time (B), and the results are the means ± S.E. for 4 independent experiments. (C)-(E), Various concentrations of LPS were added to the reaction mixture, which was then pre-incubated with Lon. Thereafter, the mixture was incubated with casein in the presence or absence of ATP at 37˚C for 60 min, as described in the text. Finally, the reaction mixture was analyzed by SDS-PAGE (C), followed by quantitative analysis using a Bio-image analyzer (D). The results are shown as the amount of casein remaining relative to that for the control without ATP or LPS, or as the degradation of casein (E), calculated from the corresponding results shown in “D.” as relative values. The results are the means ± S.E. of 4 experiments.
 (a) (b)
(a) (b)
Figure 3. Effect of LPS on ATPase activity of Lon. (a) Reaction mixtures containing 0.3 μM Lon and various concentrations of LPS (shown on the abscissa) were pre-incubated in TM buffer at 4˚C for 30 min. Then 4 mM [γ-32P] ATP was added, and incubation was continued at 37˚C for 45 min. The cleaved [γ-32P] phosphate was collected and estimated as described in the text. (b) Time-course of ATP hydrolysis was examined under conditions similar to those in “A” except the LPS concentration was set at 50 μg/mL LPS. The results are shown relative to the control without Lon or LPS, and are the means ± S.E. of 3 independent experiments.
the ATPase activity of Lon by using unlabeled ATP with [γ-32P]ATP as a substrate. LPS inhibited the Lon-mediated hydrolysis of ATP in dose-dependent fashion (Figure 3(a)), and 50 μg/mL LPS inhibited about 60% of it. The results of a time-course study revealed that LPS exhibited little effect during the first 15 min after the addition of ATP at 0 time but that it inhibited the rise in the ATP hydrolysis during the next 15 - 30 min, showing strong inhibition during this time period (Figure 3(b)). These results suggest that LPS acted on Lon at the ATPase sites and inhibited their function not immediately but rather gradually.
3.3. Structure-Activity Relationships of LPS-Induced Inhibition of Lon
In order to know whether the inhibitory effects of LPS on the peptidase, protease, and ATPase activities of Lon were due to phosphate groups associated with the lipid A moiety, we examined the effects of LPS analogs ReLPS, lacking the O-antigen polysaccharide, and detoxified LPS, lacking the O-acyl chains, but having lipid A with diphosphate at its 4, 4’-positions. These analogs showed dose-dependent inhibition of the ATP-dependent peptidase activity of Lon (Figure 4(a)). However, the extents of this inhibition by them were less than those found for LPS, namely, the smooth-type LPS, harboring O-antigen polysaccharide and O-acyl chains [12]. The apparent IC50 values, the concentration achieving 50% inhibition, of LPS and these analogs were about 10 μg/mL for LPS and 50 μg/mL for ReLPS as well as for detoxified LPS.
We next examined the inhibitory effects of LPS and its analogs on the ATPase activity of Lon. As shown in Figure 4(b), none of them were inhibitory at 1 μg/mL; but all of them showed significant inhibition at 50 μg/mL. Contrary to the extents of peptidase inhibition, detoxified LPS showed similar or stronger inhibition than LPS (Figure 4(b)). These results indicate that ReLPS and detoxified LPS inhibited both the peptidase and ATPase activities of Lon similarly as LPS, suggesting that Oantigen polysaccharide or O-acyl chain in LPS did not play any critical role in the inhibition of these activities of Lon.
For further analysis of LPS structure-activity relationships, we examined whether monoor di-phosphoryl lipid A, a core component of LPS, would show similar inhibitory effects on Lon. The results of an assay performed with various concentrations of these lipid A showed dose-dependent inhibition of the ATP-dependent peptidase activity of Lon; and at and more than 0.1 μg/mL di-phosphoryl lipid A and at and more than 0.5 μg/mL mono-phosphoryl lipid A showed significant the inhibition was significant (Figure 5). These results suggest that neither O-antigen polysaccharide nor O-acyl chain, but rather phosphate groups, in the lipid A domain seemed to be needed for the inhibitory effects toward the ATPdependent peptidase activity of Lon.
3.4. Interaction between LPS and Lon
To examine the interaction between LPS and Lon, we investigated the possible binding of Lon to LPS. In this
 (a)
(a) (b)
(b)
Figure 4. Effect of LPS analogs on the peptidase and ATPase activities of Lon protease. (a) and (b) LPS, Re LPS or detoxified LPS was added to Lon protease at the various concentrations shown on the abscissa, and the mixture pre-incubated as described in the text. Then either 5 μM Suc-Phe-Leu-Phe-MNA (a) or 4 mM [γ-32P] ATP (b) was added as a substrate for peptidase or ATPase, respectively, and incubation was carried out at 37˚C for 60 min (a) or 45 min (b). The values are shown relative to the control without ATP or LPS analogs (a) or to that without LPS or its analogs, and are the means ± S.E. of 3 independent experiments.
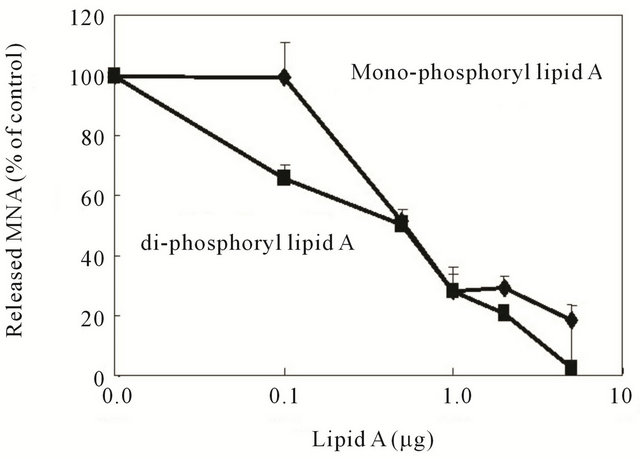
Figure 5. Effect of monoand di-phosphoryl lipid A on the peptidase activity of Lon protease. Various concentrations of monoand di-phosphoryl lipid A were pre-incubated with Lon in the presence or absence of 1 mM ATP in TM buffer at 4˚C for 30 min; and the reaction was started by addition of 5 μM Suc-Phe-Leu-Phe-MNA as a substrate, as described in the legend to Figure 1. After incubation at 37˚C for 60 min. the cleaved MNA was assayed; and the results are shown relative to the control without lipid A. The values are the means ± S.E. of 3 independent experiments.
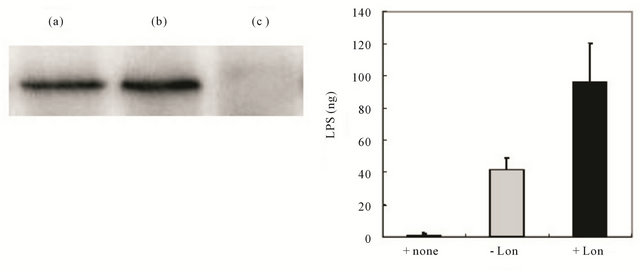
assay, immunoprecipitation with anti-Lon antibody successfully recovered Lon (lane (b) of Figure 6(A)), whereas the control without this antibody showed no precipitated Lon protein (lane (c) of Figure 6(A)), suggesting that Lon had not bound to the Protein A Sepharose beads nonspecifically. Under these conditions, Alexa Fluor 488 - labeled LPS was incubated with Lon in TM buffer containing 4 mM ATP at 4˚C for 2 hr. After immunoprecipitation and subsequent washing, the fluorescent LPS was significantly co-precipitated with Lon at higher levels than the control without Lon (-Lon) or LPS itself (+ none); although non-specific precipitation was observed in the absence of Lon, probably due to affinity of Alexa Fluor 488 - labeled LPS for the Protein A Sepharose beads (Figure 6(B)). These results suggest that LPS bound to Lon and that such binding was resistant to the repeated washing with the buffers containing high salt and 0.01% Triton X - 100.
4. DISCUSSION
Lon has been reported to interact with polymers containing phosphate groups such as DNA and polyP, leading to significant effects on its enzymatic activity [20-24]. Additionally, by using cardiolipin-containing liposomes in in vitro assay systems we showed earlier that Lon interacts specifically with cardiolipin, an important component of E. coli membrane phospholipids [11].
In this present study, we showed that lipopolysaccharides, a major component of the outer membrane of E. coli, inhibited the enzymatic activity of Lon. LPS inhibited both the peptidase and protease activities of Lon in an LPS dose-dependent manner (Figures 1(a) and 2(E), respectively), as well as inhibited the ATPase activity of Lon (Figure 3). Although the inhibition of the peptidase activity by LPS was selective for the ATP-dependent activity of Lon protease (Figure 1), the extents of inhibition of the peptidase activity seemed to be greater in the peptidase assay (Figure 1(a)) than those in the ATPase assay (Figure 3(a)) at each concentration of LPS tested. Especially at concentrations of less than 20 μg/mL, the
(A) (B)
Figure 6. Binding of LPS to Lon protease. (A) The amount of LPS bound to Lon protease was estimated by immunoprecipitation of the reaction mixture containing Lon protease in the presence or absence of Alexa Fluor 485-labeled LPS with anti-Lon antibody, as described in the text. After repeated washing of the precipitate including Protein A Sepharose, the recovered precipitates were boiled, and then subjected to either SDS-PAGE/Western blotting using anti-Lon antibody (A) or quantified for Alexa Fluor fluorescence at Ex 485 nm/Em 538 nm (B), under the conditions described in the text. The recovered LPS bound to Lon protease was determined from a standard curve with known amounts of Alexa Fluor 485 - labeled LPS. In “A,” lane (a) indicates Lon protease; and lanes (b) and (c) indicate a sample after immunoprecipitation with (b) or without (c) anti-Lon antibody. In “B,” the values of LPS amounts are the means ± S.E. of 3 independent experiments.
ATP-dependent peptidase activity seemed to be linearly inhibited to the LPS doses at about −3.5% per μg LPS (Figure 1(a)), whereas ATPase activity was almost linearly inhibited by LPS at about −2.8% per μg LPS (Figure 3(a)). In addition, the extent of inhibition by 50 μg/mL LPS was about 81% and 60% for the peptidase and ATPase activities, respectively.
Contrary to the inhibition of the peptidase activity of Lon, that of the protease activity with casein as a substrate required a much greater amount of LPS; i.e., the IC50 value of LPS for the ATP-dependent degradation of casein was around 70 μg/mL (Figure 2(E)), whereas that for the degradation of Suc-Phe-Leu-Phe-MNA was about 12 μg/mL (Figure 1(a)). Based on the results shown in Figure 3(a), IC50 value of LPS for ATPase was about 30 μg/mL. These results suggest that LPS inhibited ATPdependent peptidase and protease activities as well as the ATPase activity of Lon but that the efficiency of the inhibition depended on the assay systems, including interaction between the substrates and Lon, and that between LPS and Lon, both of which might be influenced by molecular sizes or structures. For instance, Suc-PheLeu-Phe-MNA is a small peptide with MW 681, and is easily accessible to the catalytic domain of Lon; whereas casein is a macromolecule with MW 22.8 kDa, and needs to be unfolded before proteolytic cleavage can occur at the catalytic site of Lon. Therefore it seems feasible that the peptidase activity toward Suc-Phe-Leu-Phe-MNA would be more sensitive to LPS-induced inhibition of Lon than the protease activity toward casein.
Although we found a novel effect of LPS on the activity of Lon, the mechanisms underlying LPS-induced inhibition of enzyme activities remain largely unknown. To elucidate these mechanisms, we examined the structure-activity relationships of LPS analogs and lipid A to determine whether phosphate groups in LPS at the lipid A moiety [12] were involved in the inhibition. The results shown in Figure 4 suggested that O-antigen polysaccharide and O-acyl chains in LPS were not essential for the inhibitory activity of LPS, because ReLPS, lacking O-antigen polysaccharide, as well as detoxified LPS, lacking O-acyl chains, showed inhibition of the peptidase (Figure 4(a)) and ATPase (Figure 4(b)) activities. Besides, monoand di-phosphoryl lipid A’s showed dosedependent inhibition of the peptidase activity of Lon (Figure 5), although stronger inhibition was observed with di-phosphoryl lipid A. These results suggest that phosphate groups in LPS, especially in the lipid A moiety, were involved in the inhibition of the enzymatic activities of Lon. However, it still remains unknown how these phosphate groups in LPS interact with Lon, leading to the inhibition of these activities.
To ascertain the interaction between LPS and Lon, we tested whether LPS bound to Lon under the same conditions used for enzymatic inhibition by performing an immunoprecipitation assay using Lon and an antibody specific for Lon [11]. As was shown in Figure 6(A), Lon was recovered well under these conditions, and Alexa Fluor 488 - labeled LPS was co-precipitated with Lon (Figure 6(b)). These results thus suggest the specific binding of LPS to Lon under the incubation conditions used for the peptidase as well as protease assays.
Taken together, the results in this study suggest that LPS inhibited the peptidase, protease, and ATPase activities of Lon through binding to Lon and that the site(s) of the inhibitory action of LPS seems to include the steps of ATP-binding and probably those of conformational changes in Lon [2,11,20-24].
In relation to the inhibitory effects of cardiolipin on Lon [11], there are some similarities and differences. While cardiolipin is one of the important inner membrane phospholipids, containing 2 phosphate groups per molecule, LPS is located in the outer membrane, comprising a hydrophobic lipid A portion with 2 phosphate groups, a core saccharide portion, and O-saccharide chains that reside outside of the cell membrane [12,14]. The molecular structures of lipid A and cardiolipin resemble each other not only by possessing the 2 phosphate groups in their hydrophilic portion, but also by constructing a three-dimensional architecture with 4 (or 6 in the case of lipid A) acyl chains, which interact with other membrane lipids [12,13,15]. In addition, studies on the biosynthetic pathway of LPS have revealed both the structures of and the enzymes involved in the synthesis of the intermediates in the pathway, including lipid A and related compounds, such as 2’,3’-diacylglucosamine-(β,1’-6)-2, 3,-diacylglucosamine-1-phosphate (DSMP), which has a structure similar to that of mono-phosphoryl lipid A [25]. It is of interest that monoand diphosphoryl lipid A showed inhibitory effects on the peptidase activity of Lon (Figure 5), because these compounds and other biosynthetic intermediates of LPS, together with synthetic enzymes such as LpxB seem to be associated with the membrane periphery [25,26]. Lon would seem to have the chance to interact with these compounds related to lipid A.
Besides, our preliminary observations showed that a pgsApgsB mutant strain of E. coli [27] became transformed into filamentous long cells under cultivation conditions where phosphatidylglycerol as well as cardiolipin synthesis were decreased, resulting in the accumulation of precursors of lipid A, lipid X, and lipid Y, in the cells. The morphology of the filamentous cells resembled that of lon-mutants, which lack synthesis of Lon and thus accumulate a substrate protein of Lon, SulA, leading to cell division arrest after UV irradiation [28]. It seems feasible to think that accumulation of lipid A precursors, which have phosphate moiety as diacylglucosamine 1 - phosphate, would interfere with protease activity of Lon, leading to inhibition of SulA breakdown and thus the formation of filamentous cells. We are currently testing whether lipid A and its precursors bind to Lon and thus inhibit proteolytic activity toward SulA protein in vivo.
This study has generated important and interesting evidence supporting the idea that the proteolytic activity of Lon is regulated by components or biological intermediates of membrane lipids, not only cardiolipin [11] but also lipid A and LPS precursors. Further analysis of and studies on such regulation of Lon might help not only in the elucidation of mechanisms underlying cell division control but also in the characterization of pathogenic bacteria.
5. ACKNOWLEDGEMENTS
This work was supported by a grant-in-aid for High Technology Research from the Ministry of Education, Science, Sports, and Culture of Japan. We thank Drs. Ko Fujimori and Atsushi Koike for their useful discussion and comments.
REFERENCES
- Gottesman, S. (1989) Genetics of proteolysis in Escherichia coli. Annual Review of Genetics, 23, 163-198. doi:10.1146/annurev.ge.23.120189.001115
- Goldberg, A.L. (1992) The mechanism and functions of ATP-dependent proteases in bacterial and animal cells. European Journal of Biochemistry, 203, 9-23.
- Waxman, L. and Goldberg, A.L. (1982) Protease La from Escherichia coli hydrolyzes ATP and proteins in linked fashion. Proceedings of National Academy of Sciences, 79, 4883-4887. doi:10.1073/pnas.79.16.4883
- Gottesman, S. and Zipser, D. (1978) Deg phenotype of Escherichia coli Lon mutants. Journal of Bacteriology, 133, 844-851.
- Wight, R., Stephens, C., Zweiger, G, Shapiro, L. and Alley, M.R. (1996) Caulobacter Lon protease has a critical rple in cell cycle control of DNA methylation. Genes and Development, 10, 1532-1542. doi:10.1101/gad.10.12.1532
- Brian, D., Gur, E., Gollan, L. and Ron, E.Z. (2000) Control of methionine biosynthesis in Escherichia coli by proteolysis. Molecular Microbiology, 37, 1436-1443. doi:10.1046/U365-2958.2000.02097.x
- Kuroda, A., Nomura, K., Ohtomo, R., Kato, J., Ikeda, T., Takiguchi, N., Ohtake, H. and Kornberg, A. (2001) Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science, 293, 705-708.
- Takaya, A., Suzuki, M., Matsui, H., Tomoyasu, T., Sashinami, N.A. and Yamamoto, T. (2003) Lon, a stress-induced ATP-dependent protease, is critically important for systemic Salmonella enterica serovar Typhimurium infection of mice. Infection and Immunity, 71, 690-696. doi:10.1128/IAI.71.2.690-696.2003
- Christensen, S.K., Maenhaut-Michel, G., Mine, N., Gottesman, S., Gerdes, K. and Van Melderen, L. (2004) Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefMyoeB toxin-antitoxin system. Molecular Microbiology, 51, 1705-1717. http://dx.doi.org/10.1046/i.1365-2958.2003.03941.x
- David, T., Stephen, A., Theresa, W., Smith, T. and Goldberg, A.L. (1988) Sequence of the lon gene in Escherichia coli. Journal of Biological Chemistry, 263, 11718- 11728.
- Minami, N., Yasuda, T., Ishii, Y., Fujimori, K. and Amano, F. (2011) Regulatory role of cardiolipin in the activity of an ATP-dependent protease, Lon, from Escherichia coli. Journal of Biochemistry, 149, 519-527.
- Rietschel, E.T., Kirikae, T., Schade, F.U., Mamat, U., Schmidt, G., Loppnow, H., Ulmer, A.J., Zahringer, U., Seydel, U. and Di Padova, F. (1994) Bacterial endotoxin: Molecular relationships of structure to activity and function. FASEB Journal, 8, 217-225.
- Hampton, R.Y. and Raetz, C.R.H. (1991) Macrophage catabolism of lipid A is regulated by endotoxin stimulation. Journal of Biological Chemistry, 266, 19499-19509.
- Galanos, C., Freudenberg, M.A., Luderitz, O., Rietschel, E.T. and Westphal, O. (1979) Chemical, physicochemical and biological properties of bacterial lipopolysaccharide. In: Cohen, E. and Bang, F.B., Eds., Biomedical Application of the Horseshoe Crab (Limulidae), Alan R. Liss, Inc., New York, 321-332.
- Bogard Jr., W.C., Siegel, S.A., Leone, A.O., Damiano, E., Sheal, D.J., Ely, T.M., Frederick, B., Mascelli, M.A., Siegel, R.C., Machiels, N.D., Kapian, P.M. and Daddona, P.E. (1993) Human monoclonal antibody HA-1A binds to endotoxin via an epitope in the lipid A domain of lipopolysaccharide. Journal of Immunology, 150, 4438-4449.
- Sonezaki, S., Kondo, A., Oba, T., Ishii, Y., Kato, Y. and Nakayama, H. (1994) Overproduction and purification of Lon protease from Escherichia coli using a maltose-binding protein fusion system. Applied Microbiology and Biotechnology, 42, 313-318. doi:10.1007/BF00902735
- Sonezaki, S., Okita, K., Oba, T., Ishii, Y., Kond, O.A. and Kato, Y. (1995) Protein substrates and heat shock reduce the DNA-binding ability of Escherichia coli Lon protease. Applied Microbiology and Biotechnology, 44, 484-488. doi:10.1007/BF00169948
- Ishii, Y., Sonezaki, S., Iwasaki, Y., Miyata, Y., Akita, K., Kato, Y. and Amano, F. (2000) Regulatory roles of Cterminal residues of SulA in its degradation by Lon protease in Escherichia coli. Journal of Biochemistry, 127, 837-844. doi:10.1093/oxfordiournals.ibchem.a022677
- Ishii, Y. and Amano, F. (2001) Regulation of SulA cleavage by Lon protease by the C-terminal amino acid of SulA, histidine. Biochemical Journal, 358, 473-480. doi:10.1042/0264-6021:3580473
- Chung, C.H. and Goldberg, A.L. (1982) DNA stimulates ATP-dependent proteolysis and protein-dependent ATPase activity of protease La from Escherichia coli. Proceedings of National Academy of Sciences, 79, 795-799. doi:10.1073/pnas.79.3.795
- Charette, M.F., Henderson, G.W., Doane, L.L. and Markovitz, A. (1984) DNA-stimulated ATPase activity on the lon (CapR) protein. Journal of Bacteriology, 158, 195- 201.
- Nomura, K., Kato, J., Takiguchi, N., Ohtake, H. and Kuroda, A. (2004) Effects of inorganic polyphosphate on the proteolytic and DNA-binding activities of Lon in Escherichia coli. Journal of Biological Chemistry, 279, 34406- 34410. doi:10.1074/ibc.M404725200
- Kuroda, A. (2006) A polyphosphate-lon protease complex in the adaptation of Escherichia coli to amino acid starvation. Bioscience Biotechnology and Biochemistry, 70, 325-331. doi:10.1271/bbb.70.325
- Kuroda, A., Nomura, K., Ohtomo, R., Kato, J., Ikeda, T., Takiguchi, N., Ohtake, H. and Kornberg, A. (2001) Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science, 293, 705-708.
- Louis, E., Metzger, I.V. and Raetz, C.R.H. (2009) Purification and characterization of the lipid A disaccharide synthase (LpxB) from Escherichia coli: A peripheral membrane protein. Biochemistry, 48, 11559-11571. doi:10.1021/bi901750f
- Raetz, C.R. and Dowhan, W. (1990) Biosynthesis and function of phospholipids in Escherichia coli. Journal of Biological Chemistry, 265, 1235-1238.
- Nishiiima, M., Bulawa, C.E. and Raetz, C.R. (1981) Two interacting mutations causing temperature-sensitive phosphatidylglycerol synthesis in Escherichia coli membranes. Journal of Bacteriology, 145,113-121.
- Schoemaker, J.M., Gayda, R.C. and Markovitz, A. (1984) Regulation of cell division in Escherichia coli: SOS induction and cellular location of the sulA protein, a key to lon-associated filamentation and death. Journal of Bacteriology, 158, 551-561.