Advances in Bioscience and Biotechnology
Vol. 3 No. 8 (2012) , Article ID: 25832 , 12 pages DOI:10.4236/abb.2012.38143
Nitric oxide leads to cytoskeletal reorganization in the retinal pigment epithelium under oxidative stress
![]()
1Department of Biological Sciences, Michigan Technological University, Houghton, USA
2Department of Biomedical Engineering, Michigan Technological University, Houghton, USA
3Retina Proteomics Laboratory, Department of Petroleum Chemistry, American University of Nigeria, Yola, Nigeria
Email: #mcfrost@mtu.edu, #wan.jahng@aun.edu.ng
Received 5 September 2012; revised 17 October 2012; accepted 26 November 2012
Keywords: Oxidative Stress; Nitric Oxide; Retinal Pigment Epithelium; PP2A; Vimentin
ABSTRACT
Light is a risk factor for various eye diseases, including age-related macular degeneration (AMD) and retinitis pigmentosa (RP). We aim to understand how cytoskeletal proteins in the retinal pigment epithetlium (RPE) respond to oxidative stress, including light and how these responses affect apoptotic signaling. Previously, proteomic analysis revealed that the expression levels of vimentin and serine/threonine protein phosphatase 2A (PP2A) are significantly increased when mice are exposed under continuous light for 7 days compared to a condition of 12 hrs light/dark cycling exposure using retina degeneration 1 (rd1) model. When melatonin is administered to animals while they are exposed to continuous light, the levels of vimentin and PP2A return to a normal level. Vimentin is a substrate of PP2A that directly binds to vimentin and dephosphorylates it. The current study shows that upregulation of PP2Ac (catalytic subunit) phosphorylation negatively correlates with vimentin phosphorylation under stress condition. Stabilization of vimentin appears to be achieved by decreased PP2Ac phosphorylation by nitric oxide induction. We tested our hypothesis that site-specific modifications of PP2Ac may drive cytoskeletal reorganization by vimentin dephosphorylation through nitric oxide signaling. We speculate that nitric oxide determines protein nitration under stress conditions. Our results demonstrate that PP2A and vimentin are modulated by nitric oxide as a key element involved in cytoskeletal signaling. The current study suggests that external stress enhances nitric oxide to regulate PP2Ac and vimentin phosphorylation, thereby stabilizing or destabilizing vimentin. Phosphorylation may result in depolymerization of vimentin, leading to nonfilamentous particle formation. We propose that a stabilized vimentin might act as an anti-apoptotic molecule when cells are under oxidative stress.
1. INTRODUCTION
Intense light kills retinal and RPE cells through ireversible apoptotic reactions [1-5]. Paradoxically light may protect the retina and RPE by controlling expressions of anti-apoptotic and anti-oxidative molecules, including erythropoietin, thioredoxin (THR), and heme oxygenase-1 (HO-1) [6]. Excess light may trigger circadian-dependent pathologic signals and oxidative stress by changing expressions of caspases or proteins in the visual cycle that include rhodopsin, cellular retinaldehyde binding protein (CRALBP), and RPE65 [6-8]. The mechanism of oxidative stress-induced apoptosis still remains elusive, but our previous proteomics approach implies that the protein expression profile, including BCL-xL, prohibitin, and vimentin, might be involved in the initiation of apoptosis as a positive or negative signal in the retina [6,8,9]. Our studies suggest that the balance between antiand pro-apoptotic proteins as well as the balance between oxidative stress and anti-oxidative shield may determine the cell fate based on external light/dark cycle environment.
Apoptotic cell death induced by intense or constant light in the retina and RPE may lead to major diseases that cause blindness, including AMD and RP [4,5]. Retinal and RPE cells have chromophores, including rhodopsin, peropsin, and retinal G protein receptor (RGR) that contain 11-cis-retinoid that can be isomerized by photons. Rhodopsin is a mediator of intense light-induced apoptosis and the activation of the transcription factor AP-1 complex (c-Fos/Jun) acts as a central downstream pathway for light-induced apoptosis. However, the mechanism by which light causes morphological alterations that initiate the death signal is elusive. Lack of such knowledge is an important issue, because without it, acquiring the ability to modulate cellular protection is highly unlikely. Thus there is an urgent need for the understanding of the molecular and biochemical mechanisms involved in light-induced cytoskeletal changes. Our previous studies suggest that post-translational nitration or phosphorylation may regulate cytoskeletal reorganization and protein trafficking under oxidative stress [6-10]. Nitric oxide (NO) is a highly reactive free radical gas that is involved in the expression of erythropoietin receptors (EPOR) in neurons from mouse brains [11,12]. Recent studies suggest that NO plays a specific role in the protective pathway; however, it is nearly impossible to establish optimal levels of NO efficacy using current techniques. Clearly defining how much NO is needed and for what duration it is produced to induce up-regulation of protein phosphorylation is critical because excess levels of NO are toxic and will lead to tissue damage and cell death [13,14]. In the current study, NO produced by cells was detected continuously in situ using a chemiluminescence reaction between NO and ozone. We aimed to establish therapeutic levels of NO that induces upregulation of protecting molecules.
The objective of our study is to answer the question whether NO prevents retinal degeneration by inducing or inhibiting phosphorylation signaling of target proteins. To attain the objective of this aim, we tested the working hypothesis that NO may have a dual role as a physiological neuroprotectant and a pathological cytotoxic compound depending on concentration and location. The levels of NO produced by RPE cells were directly measured under different levels of light or oxidative stimulation. We have quantitatively established what level and duration of NO accounts for its protective properties and at what level NO becomes pathological in RPE cell death.
Our results suggest that NO is generated in RPE cells under various oxidative stress conditions in a time-dependent manner. Appropriate levels of NO control phosphorylation of vimentin and PP2A in the RPE that may lead to regulate stress-dependent apoptotic pathways.
2. MATERIALS AND METHODS
2.1. ARPE-19 Cell Culture
Human retinal pigment epithelial cells (ARPE-19) were obtained from American Type Culture Collection (Manassas, VA). Cells were cultured in 25 cm2 flask (Corning, St. Louis, MO) or 56.7 cm2 dishes (Nunc, Naperville, IL) containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO) and 1% penicillin/streptomycin
(Hyclone, Logan, UT) and cells were maintained in humidified 5% CO2 incubator at 37˚C.
2.2. Light, Oxidative Stress, Reoxygenation, and LPS Treatment
Confluent ARPE-19 cells were exposed to bright fluorescent light (7000 lux), tert-butyl hydroperoxide (200 μM), reoxygenation (2 hrs hypoxic condition by N2 then 21% O2) and LPS (5 - 10 μg/mL) in serum free medium for different time intervals in CO2 incubator. Cells grown in 25 cm2 flasks were used to measure NO release in situ. Cells were harvested to extract total proteins using ice cold 1X RIPA lysis buffer [0.5 M Tris-HCl (pH 7.4), 1.5 M NaCl, 2.5% deoxycholic acid, 10% NP-40, and 10 mM EDTA (Millipore, Darmstadt, Germany)] containing protease inhibitor cocktail (Pierce, Rockford, IL). Cell lysates were centrifuged at 10,000 × g for 30 min at 4˚C to pellet cell debris. Proteins were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDSPAGE), followed by Western blotting analysis.
2.3. Direct NO Measurement
NO release from ARPE-19 cells was measured via chemiluminescence detection with Sievers 280i Nitric Oxide Analyzer (GE Instruments, Boulder, CO) using 200 mL/min ambient air with added 5% CO2 sweep gas. NO analyzer was equilibrated with 5% CO2 incubator for 30 min before the experiment. Prior to NO analysis, the instrument was calibrated three times using NO (45 PPM) and air by passing through a Sievers NO zero filters, followed by zero calibration. Confluent cells grown in T25 flask under oxidative environment (light; exposed to 7000 lux of fluorescent light, oxidative stress; treated with 200 μM of t-butyl hydroperoxide, reoxygenation; 21% O2 added at 2 hrs, LPS; treated with 5 μg/mL of LPS) were directly connected to Nitric Oxide Analyzer using custom made adaptor. The amount of released NO is measured in PPB (parts per billion) and is converted to mole per surface area by a calibration factor (nmole·cm–2·min–1) then represented by graphs using MATLAB software.
To determine intracellular NO formation using a fluorescent molecule, cells were incubated in media containing 4,5-diaminofluorescein-2-diacetate (DAF-2DA, 5 μM, Sigma) for 15 min after stress treatment (12,000 lux light, 16 - 24 hrs). After washing three times with PBS, cells were fixed with 2% glutaraldehyde for 3 min. Images were acquired using Zeiss Axiovert 200 M Apo Tome fluorescent microscope with 63× objective lens magnification [14].
2.4. SDS-PAGE and Western Blot Analysis
Proteins were quantified using BCA protein assay kit (Pierce) based on the manufacturer’s protocol by plotting BSA standard curve. Samples were dissolved in 4× Laemmli sample buffer (240 mM Tris pH 6.8, 8% SDS, 40% glycerol, 0.4% bromophenol blue, 10% β-mercaptoethanol), heated at 90˚C for 10 min. Proteins were resolved using 10% or 8% - 16% gradient polyacrylamide precast gels (Bio-Rad), followed by electrotransfer on to the methanol activated PVDF membrane (Bio-Rad) via semidry transfer by applying 20 V for 30 min. Non-specific proteins were blocked for 1 hr at 4˚C with 5% nonfat dry milk (Bio-Rad) prepared in TBS with 0.1% (v/v) Tween-20. Membranes were probed with primary antibodies (Santa Cruz Biotechnology Inc, CA), including iNOS (sc-651; 1:100), nitrotyrosine (sc-32731; 1:25), pvimentin (sc-16673; 1:500), p-PP2Ac (sc-12615; 1:500), Bcl-xL (sc-634; 1:500) and β-actin (sc-47778; 1:1000) at 4˚C for overnight and subsequently incubated with corresponding horseradishperoxidase conjugated secondary antibodies for 2 hrs at room temperature. Membranes were developed with West Pico Chemiluminescent Substrate (Pierce) for 2 min and immunoreactive protein bands were visualized using a LAS 4000 mini luminescent image analyzer (GE, Piscataway, NJ). Bands were quantified based on pixel size/intensity/area using Quantity One Software (Bio-Rad) and graphs were plotted based on relative intensity of bands using GraphPad Prism Software.
2.5. Immunocytochemistry
ARPE-19 cells were seeded on sterile glass cover slips placed in 6 well plate (Nunc, 10 cm2 per well) with serum containing DMEM/F12 media supplemented with 1% penicillin-streptomycin (Hyclone) 5% CO2 incubator at 37˚C. Cells were treated with 200 μM of tert-butyl hydroperoxide for 36 or 48 hrs to induce oxidative stress. Cells were fixed with 10% formaldehyde in PBS for 30 min at RT. After fixation, cells were incubated in 0.2% Triton X-100 (Sigma) for 25 min to permeabilize cell membrane and blocked with complete media containing 10% FBS and 0.05% Tween-20 (Sigma) for 1 hr and probed with anti-iNOS, anti-vimentin antibody (1:100; Santa Cruz) for overnight at 4˚C. Subsequently, cells were stained with Alexa Fluor 488-conjugated anti-rabbit IgG (1:700, Molecular Probes) for 2 hrs at RT. Finally, immunostained samples were mounted using Vectashield (Vector Laboratories, Burlingame, CA) mounting medium containing DAPI (4, 6-diamidino-2-phenylindole) to counterstain the nucleus. Images were captured using fluorescent microscope (Zeiss AxioVert 200 M Apo Tome) with 20× magnification.
2.6. Scanning Electron Microscopy (SEM)
ARPE-19 cells were cultured on coverslips in 6 well plate (Nunc, 10 cm2 per well). Cells were treated with 100 μM of H2O2 in serum free media for 2 hrs to induce oxidative stress. Sample preparation protocol was followed as previously reported [16]. After brief wash with PBS, cells were immersed immediately into fixative solution containing 3% paraformaldehyde and 0.1% glutaraldehyde in phosphate buffer (pH 7.1) at RT for 1 hr. Samples were removed from the fixing solution and washed three times with PBS and then post fixed in 1% osmium tetroxide in 0.2 M phosphate buffer. After three additional brief washings in distilled H2O, cells were dehydrate in a graded EtOH-H2O series and then critical-point-dried in a CO2 chamber. Cells were coated with a 10 nm layer of platinum-palladium alloy in a vacuum evaporator to examine in the scanning electron microscope (JEOL JSM-6400) at 4000× magnification.
2.7. Statistical Analysis
Values are presented as the mean in three independent experiments. Two-group comparisons were analyzed by the 2-tailed t test. Multiple comparisons were evaluated by ANOVA and Tukey or Dunnet tests, as appropriate. P < 0.05 is considered statistically significant.
3. RESULTS
3.1. NO Production in the RPE under Oxidative Stress
To address the issue of the protective and pathological roles of NO in neurodegeneration, we directly measured NO produced by RPE cells exposed to different stress. First we asked how much NO is generated and what duration it is produced under various environmental stimuli. We aimed to establish therapeutic or cytotoxic levels of NO that induce upor down-regulation of phosphorylation signaling of vimentin and PP2A. NO produced by RPE cells were determined in situ after exposure to each of following; dark, light, oxidative stress, reoxygenation, and lipopolysaccharide (LPS) as shown in Figure 1. Cells exposed to dark conditions did not show any detectable increase of NO levels. Cells treated with light (7000 lux) showed an increase in NO concentration after 19 hrs of exposure. Cells treated with 200 μM tert-butyl hydroperoxide (t-BuOOH, oxidative stress) showed an increase in NO concentration at 16, 18, and 20 hrs. Based on the observation of the increased levels of NO, oxidative stress and light plays a positive regulator role in the production of NO by ARPE-19 cells.
NO generation positively correlates with our Western blotting analysis of iNOS under light and oxidative stress. Cells treated with reoxygenation after hypoxic conditions (nitrogen stream for 2 hours) before being exposed back to normoxia (21% O2) showed NO up-regulation, immediately after reoxygenation at 2 hrs and remained active
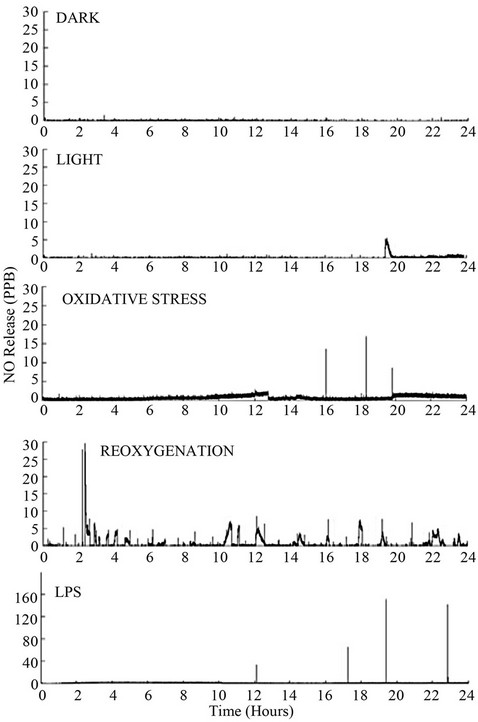
Figure 1. Dark, light, oxidative stress, reoxygenation, and LPS induced NO production in vitro. Confluent ARPE-19 cells cultured in T-25 flasks were incubated under intense light (7000 lux), oxidative stress (200 μM t-BuOOH), reoxygenation (hypoxia by N2 flow for 2 hrs then 21% O2) and lipopolysaccharide (LPS, 5 μg/mL) in humidified 5% CO2 chamber. Released NO from cells was detected each second for total 24 hrs using Nitric Oxide Analyzer 280i. X-axis represents time (hrs) and Y-axis represents concentration in parts per billion (PPB).
for 20 hrs. This data suggests that O2 is the limiting factor for NO synthesis in RPE cells. As a positive control for NO production, cells were treated with LPS (5 μg/mL), which also stimulated NO production at 12, 17, 19.5, and 23 hrs. A quantitative analysis of NO release is shown in Table 1.
Next, light-induced NO in the RPE was detected using 4, 5-diaminofluorescein 2-diacetate (DAF-2DA) as shown in Figure 2. Detection of intracellular NO in living cells using DAF-2DA is a recently developed method in combination with aldehyde fixation. Our fluorescent analysis illustrated that RPE cells exposed to intense light (12,000 lux) generated higher levels of intracellular NO compared to dark control. The control in the dark did not show any detectable fluorescence, provides the direct evidence that RPE cells under intense light generate NO.

Table 1. Quantitative analysis of NO released (PPB) and NO surface flux (nmole·cm–2·min–1) under intense light (7000 lux), oxidative stress (200 mM t-BuOOH), reoxygenation (hypoxia to 21% O2), lipopolysaccharide (5 mg/mL) conditions.

Figure 2. Light induced NO detection by 4, 5-diaminofluorescein 2-diacetate (DAF-2DA). ARPE-19 cells were treated with the dark or intense light (12,000 lux, 16 hrs) conditions. Cells were incubated with DAF-2DA (5 μM, excitation at 470 to 490 nm and emission at 515 nm, green) for 15 min followed by fixation with 2% glutaraldehyde to detect NO levels under light-induced stress.
3.2. Treatment with LPS, Tert-Butyl Hydroperoxide (t-BuOOH) and Light Up-Regulated Inducible Nitric Oxide Synthase (iNOS) in the RPE
Since NO generation is controlled by iNOS, expression changes of iNOS in response to external stimuli were analyzed by Western blotting. As shown in Figure 3, LPS, oxidative stress, and light treatment significantly up-regulated iNOS expression in RPE cells in concentration or time-dependent manner. Levels of iNOS expression are also consistent with NO production in RPE cells. iNOS is responsible for the conversion of L-arginine to NO and citrulline in the cell. ARPE-19 cells were treated with light (7000 lux) for 12 and 18 hrs and a negative control had no light exposure (Figure 4). Light exposed cells had higher levels of iNOS expression compared to time 0 control, with 18 hrs having a higher concentration than 12 hrs and the control showed no visible band of iNOS. Another group of ARPE-19 cells were treated with oxidative stress (200 µM t-BuOOH) or LPS (5 µg/mL or 10 µg/mL). A negative control cells showed no iNOS expression at 130 kDa, while cells treated with oxidative stress and LPS showed iNOS, suggesting an
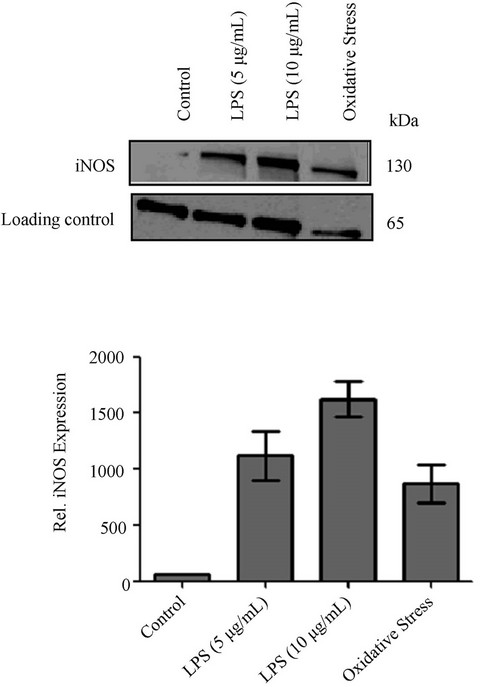
Figure 3. Expression of iNOS under LPS, oxidant, and light treatment. ARPE-19 cells were treated with LPS (5 or 10 μg/mL for 24 hrs), tert-butyl hydroperoxide (200 μM for 24 hrs), and intense light (1000 lux for 24 hrs). Cells were harvested, lysed and proteins were separated by SDS-PAGE. Expression of iNOS (130 kDa) was visualized by Western blotting using rabbit polyclonal anti-iNOS antibody. A low molecular weight non-specific band at 65 kDa was used as a loading control. Oxidative stress induced expression of iNOS in RPE cells.
upregulation of iNOS under these conditions. LPS exposure caused the most significant increase in iNOS upregulation.
3.3. Expression and Localization of iNOS under Oxidative Stress
Then we tested our hypothesis whether oxidative stress controls iNOS expression in RPE cells by using immunocytochemical analysis. Our fluorescent images show that iNOS expression increased under oxidative stress (200 μM, t-BuOOH) compared to untreated control as shown in Figure 5. A negative control without anti-iNOS primary antibody confirmed that our immunocytochemical images are iNOS-specific (Supplement Figure S3). Nucleus staining with DAPI reveals cytoplasmic localization of iNOS.
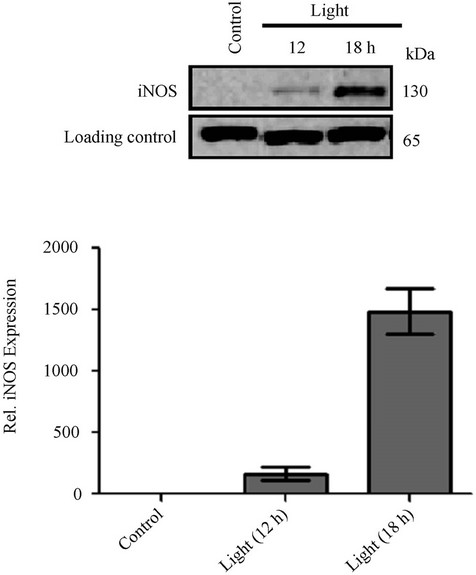
Figure 4. Light induced iNOS expression in RPE cells. Bands were quantified based on pixel number (intensity and area) using Quantity One software (Bio-Rad). Graphs were plotted based on relative iNOS expression using GraphPad Prism software.
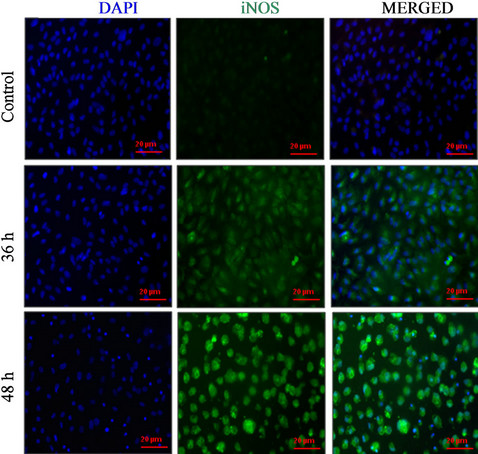
Figure 5. Increased expression of inducible nitric oxide synthase (iNOS) under oxidative stress. ARPE-19 cells treated with tert-butyl hydroperoxide (200 μM for 36 or 48 hrs.) were analyzed by immunocytochemistry. iNOS was visualized by anti-iNOS primary antibody, Alexa Fluor 488-conjugated secondary antibody (green, iNOS) and DAPI (blue, nucleus) staining. Expression of iNOS was observed under oxidative stress in a time-dependent manner. Scale bars represent 20 μm.
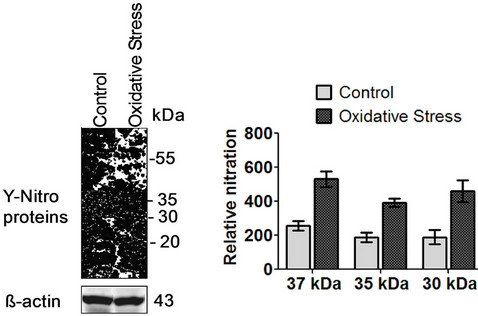
3.4. Oxidative Stress Induced Protein Nitration Shown by 3-Nitrotyrosine in the RPE
Formation of 3-nitrotyrosine is a post-translational modification caused by NO derived peroxynitrite during pathophysiological conditions, which in turn alters protein structure and function. Our Western blotting analysis showed that relative protein nitration increased in 30, 35, 37 kDa proteins and other proteins (55, 32, 20, 18, 10 kDa) under oxidative stress (Figure 6). Reduction (3-nitro tyrosine to 3-amino tyrosine) with disodium dithionite (Na2S2O4) confirmed the increased protein nitration in LPS, light and oxidative stress treated samples (Supplement Figure S1).
3.5. NO Induced Vimentin Dephosphorylation and PP2Ac Phosphorylation in the RPE under Oxidative Stress
Oxidative stress disrupts RPE cell junction and barrier integrity which may cause the pathogenesis of RPE related diseases through the degeneration of the bloodretinal barrier [17]. Increased site-specific phosphorylation of vimentin induces disassembly of vimentin intermediate filaments in vitro. Both cAMP dependent protein kinase and protein kinase C dependent phosphorylation of Ser-38 residue, which is located close to arginine substrate, were reported [18].
 (a) (b)
(a) (b)
Figure 6. Increased protein nitration in RPE under oxidative stress. ARPE-19 cells were treated with tert-butyl hydroperoxide (200 μM for 36 hrs). Cells were harvested, lysed and proteins were separated by SDS-PAGE. (a) Proteins with 3-nitrotyrosine were visualized by Western blotting using anti-3-nitrotyrosine antibody (1:50 dilution). Expression levels of nitrated proteins increased 2 - 3 folds under oxidative stress compared to untreated control. Protein loading was controlled by probing with anti-β actin antibody; (b) Bands represent the expression of nitrated proteins quantified by pixel intensity and area using Quantity One software (Bio-Rad). Quantitative analysis shows 2 - 3 fold increased expressions of nitrated proteins under oxidative stress compared to untreated control. Graphs were plotted using GraphPad Prism software.
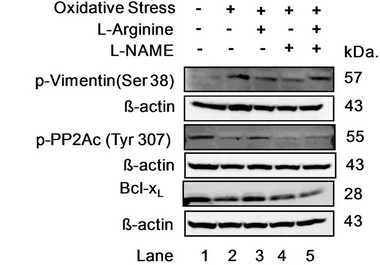
To determine NO-dependent cytoskeletal change that was regulated by PP2A phosphorylation signaling, we examined the interconnection of NO-vimentin-PP2A signaling (Figure 7, Supplement Figure S2). Cells were treated with or without oxidative stress, L-arginine, and a NOS antagonist (N’1-nitro-L-arginine methyl ester, LNAME). Cells treated with oxidative stress showed downregulation of the anti-apoptotic protein p-PP2Ac relative to the control group. Bcl-xL is also an anti-apoptotic protein and is down-regulated in all samples that are exposed to oxidative stress. Bcl-xL expression increased in cells treated with L-arginine (2 mM) under oxidative stress. However, cells treated with oxidative stress, arginine, and L-NAME together at the same time had lowest expression of Bcl-xL. Our data implies that increased levels of NO by L-arginine may have a protective role towards RPE cells under oxidative stress by inducing anti-apoptotic molecules, including Bcl-xL. Vimentin phosphorylation is up-regulated in cells under stress. Cells treated with L-arginine (2 mM) down-regulated vimentin phosphorylation whereas L-NAME along with L-arginine up-regulated vimentin phosphorylation under oxidative stress. This is likely a result of increased cytoskeletal restructuring due to changes of intermediate filaments.
We then asked whether NO can induce PP2Ac catalytic subunit phosphorylation in the RPE under oxidative stress. Stimulation of endothelial cells with LPS and IFNγ increases NO-derived peroxynitrite. Up-regulation of peroxynitrite induces PP2Ac tyrosine nitration and nitrated PP2Ac inhibits PP2Ac phosphorylation. Activation of PP2A by nitration in turn causes endothelial barrier dysfunction [19]. We tested whether phosphorylation of PP2Ac is down-regulated under oxidative stress in RPE cells. Arginine treatment under oxidative stress shows up-regulated PP2Ac phosphorylation compared to oxidative stress only condition. Arginine treatment with or without L-NAME under oxidative stress clearly demonstrated that PP2Ac phosphorylation is down-regulated compared to non-stress control.
3.6. NO Induced Anti-Apoptotic Signaling in the RPE under Oxidative Stress
Anti-apoptotic protein Bcl-xL plays a significant role in RPE cell survival under normal and oxidative stress conditions. Reduced Bcl-xL expression levels confer apoptotic cell death in the RPE [20]. Inhibition of Bcl-xL expression alters RPE morphology and diminishes cell survival [21]. We examined whether NO regulates antiapoptotic signaling via Bcl-xL. While arginine addition increased Bcl-xL, NO reduction by L-NAME down-regulates Bcl-xL compared to oxidative stress only. This result suggests that vimentin phosphorylation may regulate
 (a)
(a)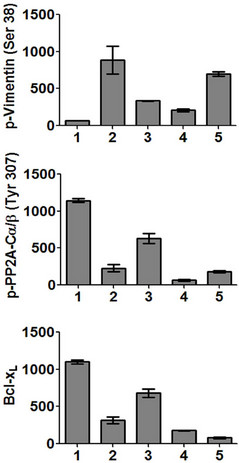 (b)
(b)
Figure 7. (a) NO-dependent changes of p-Vimentin (Ser38), p-PP2Ac (Tyr307) and Bcl-xL under oxidative stress. ARPE-19 cells were incubated with tert-butyl hydroperoxide (200 μM) or L-arginine (2 mM) or L-NAME (2 mM) for 24 hrs in serum free media. Cells were harvested, lysed in RIPA buffer to extract proteins. Proteins were separated by SDS-PAGE and probed with anti-p-vimentin (Ser38), anti-p-PP2Ac (Tyr307) and anti-Bcl-xL antibodies to visualize target proteins by Western blotting; (b) Quantitative analysis of Figure 7(a). Y axis represents relative phosphorylations in arbitrary unit (a.u.). X axis represents each lanes; Lane 1. Control cells without treatments; Lane 2. Cells treated with 200 μM tert-butyl hydroperoxide only (oxidative stress); Lane 3. Cells treated with 200 μM tert-butyl hydroperoxide and 2 mM L-arginine; Lane 4. Cells treated with 200 μM tert-butyl hydroperoxide and 2 mM L-NAME; Lane 5. Cells treated with 200 μM tert-butyl hydroperoxide, 2 mM L-arginine and 2 mM L-NAME.
apoptotic cell death signaling in RPE cells. Moreover, increased NO may play a protective role by enhancing anti-apoptotic signaling in the RPE under oxidative stress.
Our data shows that Bcl-xL expression levels decreased in the RPE under oxidative stress. RPE cells treated with arginine shows increased expression of Bcl-xL under oxidative stress.
3.7. Light Induced Vimentin Filament Disorganization in the RPE
Interactions between microfilaments, intermediate filaments, and microtubules, stabilize structural, cytoskeletal organization of RPE cells. Vimentin is a major intermediate filament, which is responsible for cell adhesion, migration and signaling [22]. To investigate whether light stress may induce morphological changes through vimentin, we performed immunostaining with fluorescently labeled antibodies as shown in Figure 8. Control cells in the dark show that vimentin filaments are extended throughout cytoplasm. Nuclear morphology appears as oval shaped and is intact. Intense light induced dramatic morphological changes as rounded vimentin filaments rather than extended form. Nuclear size decreased; the major radius was reduced by 50% - 60% and the minor radius was diminished by 70% - 80% in length under intense light at 20 hrs compared to control. A patchy green color around the nucleus represents vimentin filament disorganization.
3.8. Apoptosis Mediated Morphological Changes of RPE under Oxidative Stress
Morphological changes of the RPE are associated with various pathological conditions involved in many posterial ocular diseases. Our scanning electron microscopy analysis of RPE cells under oxidative stress demonstrates that the lateral, basal and apical cell surfaces of RPE are changed during apoptosis. RPE cells were treated with 100 μM of H2O2 for 2 hrs. RPE cells under stress show that apoptotic cell surface changes by forming protrusion as apoptotic initiation (Figure 9B); developing bubblelike blebs on their surface (Figure 9C); phagocytic alteration like membrane disruption (Figure 9D); apical, basal and lateral surface changes, including dense projection of microvilli (Figures 9E and F); broken into small,

Figure 8. Vimentin filament changes in the RPE under light induced stress. ARPE-19 cells were exposed to intense light (7000 Lux) for different time points (16 hrs and 20 hrs). Cells were fixed in methanol at –20˚C. Vimentin was probed by anti-vimentin primary antibody and anti-goat Alexa 488 secondary antibody (green, vimentin filaments) and DAPI counterstaining (blue, nucleus). Scale bars represent 20 μm.

Figure 9. Cytoskeletal changes of RPE cells under oxidative stress identified by scanning electron microscope (SEM). ARPE-19 cells were treated with H2O2 (100 μM for 2 hrs). After fixation with 3% paraformaldehyde and 0.1% glutaraldehyde in phosphate buffer, morphological changes of ARPE- 19 cells were analyzed using JEOL JSM-6400 SEM at 4000× magnification. (A) Control; (B)-(H) Sequential changes of ARPE- 19 cell morphology under oxidative stress. Scale bars represent 8 µm.
membrane-wrapped fragments (Figures 9G and H). These results demonstrate that morphological changes of RPE cells under oxidative stress may lead to cell lysis, cytoskeleton collapse, and ultimately cell death.
4. DISCUSSION
Oxidative stress-induced apoptosis is the final cell death pathway in many irreversible ocular diseases [23,24]. While the end point of apoptosis is well established, the knowledge of early biochemical reactions and molecules has been elusive. We have examined early biosignatures and mechanisms of retinal and RPE cell death under oxidative stress [6-10,25,26]. Our previous studies demonstrated that not only intense light but also constant moderate light and mild oxidative stress may trigger induction of anti-apoptotic Bcl-xL and erythropoietin as well as pro-apoptotic caspases. We noticed that protein modifications, including nitration and phosphorylation, were altered under oxidative stress possibly due to excess NO production.
NO is generated from guanidine oxidation of L-arginine with the stoichiometric formation of L-citrulline by nitric oxide synthase in the presence of oxygen and NADPH. Enzymatic NO formation is involved in physiological processes, including the control of vascular tone, inhibition of platelet aggregation, smooth muscle cell replication, immune response, and wound healing processes [27]. NO has been recognized for its paradoxical role as a promoter of cell growth (neovascularization) and cell death (apoptosis) [28,29].
Exposure to intense light may induce protein nitration via iNOS activation in the retina, which in turn mediates photoreceptor cell death [30]. Elevated tyrosine nitration was observed in the inner segments of photoreceptors and RPE upon intense light exposure due to increased NO production [31]. Inflammatory responses and high oxygen demand in the retina are expected to increase NO generation and oxidative damage to Bruch’s membrane and could lead to pathophysiological protein nitration [32]. NOS is categorized into three isoforms such as calcium-dependent neuronal (nNOS), endothelial (eNOS) isoforms, and the inducible, calcium independent isoform (iNOS), which are related to neuronal communication, vasolidation and inflammation signaling, respectively [33,34]. Optimum expression levels of NOS isoforms are tightly regulated for normal retinal cell homeostasis and RPE phagocytosis. A high concentration of NO could lead to the accumulation of protein aggregates between photoreceptors and RPE cells, which results in photoreceptor degeneration [35]. Light-induced oxygen radicals are mediators of retinal photic injury, based on the observation of the protective effect of free radical scavengers in the light damaged model [36]. However, decreased expressions of eNOS and nNOS were also observed in neuronal and vasoconstrictive changes in AMD choroid. Changes of NOS in AMD suggest that retinal degeneration is directly or indirectly related to local NO concentration in the RPE. NO reacts with superoxide anion ( ) to produce peroxynitrite (ONOO-), a potential cytotoxic and tyrosine nitrating molecule under oxidative stress [37].
) to produce peroxynitrite (ONOO-), a potential cytotoxic and tyrosine nitrating molecule under oxidative stress [37].
Peroxynitrite modulates tyrosine phosphorylation and dephosphorylation during postsynaptic compartments during cell regulation [38]. Exogenous NO exposure also resulted in cytoskeletal reorganization through phosphorylations of VASP (vasodilator-stimulated phosphorprotein) in diabetes model [39]. The current study suggests that NO may regulate phosphorylation of PP2Ac and vimentin as a physiological regulator of cell respiretion to become an agent of cell pathology. In additionthe excessive level of NO or the deficiency of NO may control endothelial dysfunction under oxidative stress. Our results demonstrate that oxidative stress induced disassembly of vimentin intermediate filaments may cause vimentin phosphorylation in the RPE. L-Arginine uptake by RPE cells significantly increases the NO levels in diabetic rat RPE [40]. The current study implies dual roles of NO that 1 - 10 nmole·cm−2·min−1 of NO may protect RPE cell by PP2A and vimentin phosphorylation, whereas higher NO (>100 nmole·cm−2·min−1) generation under oxidative stress may induce apoptosis. Our results show that arginine treatment under oxidative stress significantly decreases vimentin phosphorylation compared to no arginine treatment under oxidative stress. Treatment of NOS antagonist, N’1-nitro-L-arginine methyl ester (L-NAME) along with arginine under oxidative stress, shows that vimentin phosphorylation increased compared to arginine only. Addition of both arginine and L-NAME under oxidative stress reveals that vimentin phosphorylation has positive and negative correlations with NO under oxidative stress.
Based on our observations in the current study, we postulate that light may induce post-translational modifications of target proteins, including vimentin and PP2Ac. We speculate that PP2Ac phosphorylation may control its subunit binding that determines its phosphatase activity and substrate specificity under stress conditions. A stabilized vimentin may act as an anti-apoptotic agent when cells are under stress. Our previous results demonstrated that PP2A and vimentin are modulated by constant light, and are key elements involved in cytoskeletal signaling in rd1 mutation model [8]. A positive correlation between the levels of PP2A and vimentin under light-induced stress suggests that cytoskeletal dynamics are regulated by phosphorylation of vimentin.
PP2A is a heterotrimeric serine/threonine phosphatase, which ubiquitously performs dephosphory-lation reactions in cell signaling cascades, including Raf, MEK, and AKT. The regulatory subunit PP2Ab enhances the formation of stable complexes between PP2Aa-c. PP2A positively regulates apoptosis by controlling pro-apoptotic molecules (Bad, Bak, Bax) and anti-apoptotic Bcl-2 family members (Bcl-xL) [20]. PP2A dephosphorylates phosducin, a phosphoprotein which modulates the light activated phototransduction cascade in vertebrate retina [19, 20]. Suppression of endogenous PP2Aa by 50% leads to cell transformation; further suppression results in cell cycle arrest and apoptosis. This suggests that the suppression of PP2Aa subunit or PP2Ac induces apoptosis and that a minimal level of PP2Aa/c is required for cell survival [41]. Under light condition, efficient dephosphorylation of phosducin by PP2A is required to quench high sensitivity so that more intense light can be sensed.
PP2Ac tyrosine residue undergoes nitration by peroxynitrite that correlates with decreased tyrosine phosphorylation and increased dephosphorylation activity [42].
Although the potential importance of protein dephosphorylation as a therapeutic target has been appreciated, no detailed approach to date has been made on PP2A, especially to treat retinal degeneration [43,44]. Abnormalities in PP2A activity and concentration have been linked to neurodegenerative diseases, including Alzheimer’s and Parkinson’s. However, the mechanism by which PP2A activity is regulated under oxidative stress remains largely elusive. Recent advances in the structural investigation and the light-driven switch mechanism have elucidated PP2A-induced dephosphorylation signaling in the retina [45-47]. Modulating light-induced post-translational modifications of PP2A and vimentin through NO signaling may assist in maintaining the proper filament network in Müller cells, which subsequently supports cell survival and architecture in the retina and RPE.
Our results demonstrate that NO concentration and duration may influence cytoskeletal dynamics and apoptotic pathway via phosphorylation signaling under stress conditions. As progressive loss of neuronal structure precedes cell death, our data elucidates the regulatory mechanism of cytoskeletal organization governed by NO and PP2A/vimentin phosphorylation. These outcomes are expected to have an important positive impact, because modulation of NO might be assigned as a therapeutic intervention to serve as the pathological target for the balanced phosphorylated cytoskeleton [48,49].
5. ACKNOWLEDGEMENTS
We thank Drs. Ramakrishna Wusirika, Mike Gibson, Haiying Liu, and Mi Hye Song for insightful discussions and sharing equipment. We thank Parrisha Louis, Rissa McDonough, Lu Song, and Alex Keim for their excellent technical assistance. Matthew Durocher is acknowledged for his critical reading and suggestion. This study was supported by NSF (MCF), NIH (JG), the Century II Equipment Fund (MCF, WJJ) and the Research Excellence Fund (MCF, WJJ) of Michigan Technological University.
REFERENCES
- Noell, W.K., Walker, V.S., Kang, B.S. and Berman, S. (1966) Retinal damage by light in rats. Investigative Ophthalmology, 5, 450-473.
- Adrian, W., Everson, R.W. and Schmidt, I. (1977) Protection against photic damage in retinitis pigmentosa. Advances in Experimental Medicine and Biology, 77, 233- 247.
- Young, R.W. (1988) Solar radiation and age-related macular degeneration. Survey of Ophthalmology, 32, 252-269. doi:10.1016/0039-6257(88)90174-9
- Grimm, C., Wenzel, A., Hafezi, F., Yu, S., Redmond, T.M. and Reme, C.E. (2000) Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nature Genetics, 25, 63-66. doi:10.1038/75614
- Grimm, C., Wenzel, A., Groszer, M., Mayser, H., Seeliger, M., Samardzija, M., Bauer, C., Gassmann, M. and Reme, C.E. (2002) HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nature Medicine, 8, 718-724. doi:10.1038/nm723
- Chung, H., Lee, L., Lamoke, F., Hrushesky, W.J.M., Wood, P.A. and Jahng, W.J. (2009) Neuroprotective role of erythropoietin by antiapoptosis in the retina. Journal of Neuroscience Research, 87, 2365-2374. doi:10.1002/jnr.22046
- Lee, H., Chung, H., Arnouk, H., Lamoke, F., Hunt, R.C., Hrushesky, W.J.M., Wood, P.A., Lee, S.H. and Jahng, W.J. (2010) Cleavage of the retinal pigment epitheliumspecific protein RPE65 under oxidative stress. International Journal of Biological Macromolecules, 47, 104- 108. doi:10.1016/j.ijbiomac.2010.05.014
- Zhang, R., Hrushesky, W.J.M., Wood, P.A., Lee, S.H., Hunt, R.C. and Jahng, W.J. (2010) Melatonin reprogrammes proteomic profile in light-exposed retina in vivo. International Journal of Biological Macromolecules, 47, 255-260. doi:10.1016/j.ijbiomac.2010.04.013
- Arnouk, H., Lee, H., Zhang, R., Chung, H., Hunt, R.C. and Jahng, W.J. (2011) Early biosignature of oxidative stress in the retinal pigment epithelium. Journal of Proteomics, 74, 254-261. doi:10.1016/j.jprot.2010.11.004
- Sripathi, S.R., He, W., Atkinson, C.L., Smith, J.J., Liu, Z., Elledge, B.M. and Jahng, W.J. (2011) Mitochondrial-nuclear communication by prohibitin shuttling under oxidative stress. Biochemistry 50, 8342-8351. doi:10.1021/bi2008933
- Genc, S., Kuralay, F., Genc, K., Akhisaroglu, M., Fadiloglu, S., Yorukoglu, K., Fadiloglu, M. and Gure, A. (2001) Erythropoietin exerts neuroprotection in 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine-treated C57/BL mice via increasing nitric oxide production. Neuroscience Letters, 298, 139-141. doi:10.1016/S0304-3940(00)01716-X
- Chen, S.J., Wang, Y.L., Lo, W.T., Wu, C.C., Hsieh, C.W., Huang, C.F., Lan, Y.H., Wang, C.C., Chang, D.M. and Sytwu, H.K. (2010) Erythropoietin enhances endogenous haem oxygenase-1 and represses immune responses to ameliorate experimental autoimmune encephalomyelitis. Clinical & Experimental Immunology, 162, 210-223. doi:10.1111/j.1365-2249.2010.04238.x
- Sortino, S. (2010) Light-controlled nitric oxide delivering molecular assemblies. Chemical Society Review, 39, 2903- 2913. doi:10.1039/b908663n
- Souza, J.M., Peluffo, G. and Radi, R. (2008) Protein tyrosine nitration—Functional alteration or just a biomarker? Free Radical Biology and Medicine, 45, 357-366. doi:10.1016/j.freeradbiomed.2008.04.010
- Sugimoto, K., Fujii, S., Takemasa, T. and Yamashita, K. (2000) Detection of intracellular nitric oxide using a combination of aldehyde fixatives with 4,5-iaminofluorescein diacetate. Histochemistry and Cell Biology, 113, 341-347.
- Karnovsky, M.J. (1965) A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron-microscopy. Journal of Cell Biology, 27, 137-138A.
- Bailey, T.A., Kanuga, N., Romero, I.A., Greenwood, J., Luthert, P.J. and Cheetham, M.E. (2004) Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Investigative Ophthalmology & Visual Sciences, 45, 675-684. doi:10.1167/iovs.03-0351
- Ando, S., Tanabe, K., Gonda, Y., Sato, C. and Inagaki, M. (1989) Domain-specific and sequence-specific phosphorylation of vimentin induces disassembly of the filament structure. Biochemistry, 28, 2974-2979. doi:10.1021/bi00433a035
- Wu, F. and Wilson, J.X. (2009) Peroxynitrite-dependent activation of protein phosphatase type 2A mediates microvascular endothelial barrier dysfunction. Cardiovascular Research, 81, 38-45. doi:10.1093/cvr/cvn246
- Medearis, S., Han, I.C., Huang, J.K., Yang, P. and Jaffe, G.J. (2011) The role of Bcl-xL in mouse RPE cell survival, Investigative Ophthalmology & Visual Science, 52, 6545- 6551. doi:10.1167/iovs.10-6772
- Zhang, N., Peairs, J.J., Yang, P., Tyrrell, J., Roberts, J., Kole, R. and Jaffe, G.J. (2007) The importance of Bcl-xL in the survival of human RPE cells. Investigative Ophthalmology & Visual Science, 48, 3846-3853. doi:10.1167/iovs.06-1145
- Owaribe, K., Sugino, H. and Masuda, H. (1986) Characterization of intermediate filaments and their structural organization during epithelium formation in pigmented epithelial cells of the retina in vitro. Cell and Tissue Research, 244, 87-93. doi:10.1007/BF00218385
- Chang, G.Q., Hao, Y. and Wong, F. (1993) Apoptosis: Final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron, 11, 595-605. doi:10.1016/0896-6273(93)90072-Y
- Portera-Cailliau, C., Sung, C.H., Nathans, J. and Adler, R. (1994) Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proceedings of the National Academy of Sciences USA, 91, 974-978. doi:10.1073/pnas.91.3.974
- Lee, H., Arnouk, H., Sripathi, S., Chen, P., Zhang, R., Bartoli, M., Hunt, R.C., Hrushesky, W.J.M., Chung, H., Lee, S.H. and Jahng, W.J. (2010) Prohibitin as an oxidative stress biomarker in the eye. International Journal of Biological Macromolecules, 47, 685-690. doi:10.1016/j.ijbiomac.2010.08.018
- Lee, H., Chung, H., Lee, S.H. and Jahng, W.J. (2011) Light-induced phosphorylation of crystallins in the retinal pigment epithelium. International Journal of Biological Macromolecules, 48, 194-201. doi:10.1016/j.ijbiomac.2010.11.006
- Mann, B.E. and Motterlini, R. (2007) CO and NO in medicine. Chemical Communications, 4197-4208. doi:10.1039/b704873d
- Rose, M.J. and Mascharak, P.K. (2008) Fiat lux: Selective delivery of high flux of nitric oxide (NO) to biological targets using photoactive metal nitrosyls. Current Opinion in Chemical Biology, 12, 238-244. doi:10.1016/j.cbpa.2008.02.009
- Mocellin, S., Bronte, V. and Nitti, D. (2007) Nitric oxide, a double edged sword in cancer biology: Searching for therapeutic opportunities. Medicinal Research Reviews, 27, 317-352. doi:10.1002/med.20092
- Palamalai, V., Darrow, R., Organisciak, D.T. and Miyagi, M. (2006) Light-induced changes in protein nitration in photoreceptor rod outer segments. Molecular Vision, 12, 1543-1551.
- Miyagi, M., Sakaguchi, H., Darrow, R.M., Yan, L., West, K.A., Aulak, K.S., Stuehr, D.J., Hollyfield, J.G., Organisciak, D.T. and Crabb, J.W. (2002) Evidence that light modulates protein nitration in rat retina. Molecular & Cellular Proteomics, 1, 293-303. doi:10.1074/mcp.M100034-MCP200
- Murdaugh, L.S., Wang, Z., Del Priore, L.V., Dillon, J. and Gaillard, E.R. (2010) Age-related accumulation of 3-nitrotyrosine and nitro-A2E in human Bruch’s membrane. Experimental Eye Research, 90, 564-571. doi:10.1016/j.exer.2010.01.014
- Marletta, M.A. (1993) Nitric-oxide synthase structure and mechanism. Journal of Biological Chemistry, 268, 12231- 12234.
- Knowles, R.G. and Moncada, S. (1994) Nitric-oxide synthases in mammals. Biochemical Journal, 298, 249-258.
- Becquet, F., Courtois, Y. and Goureau, O. (1997) Nitric oxide in the eye: Multifaceted roles and diverse outcomes. Survey of Ophthalmology, 42, 71-82. doi:10.1016/S0039-6257(97)84043-X
- Davidson, P.C. and Sternberg, P. (1993) Potential retinal phototoxicity. American Journal of Ophthalmology, 116, 497-501.
- Beckman, J.S., Chen, J., Crow, J.P. and Ye, Y.Z. (1994) Reactions of nitric-oxide, superoxide and peroxynitrite with superoxide-dismutase in neurodegeneration. Progress in Brain Research, 103, 371-380. doi:10.1016/S0079-6123(08)61151-6
- Di Stasi, A.M., Mallozzi, C., Macchia, G., Petrucci, T.C. and Minetti, M. (1999) Peroxynitrite induces tryosine nitration and modulates tyrosine phosphorylation of synaptic proteins. Journal of Neurochemistry, 73, 727-735. doi:10.1046/j.1471-4159.1999.0730727.x
- Li Calzi, S., Purich, D.L., Chang, K.H., Afzal, A., Nakagawa, T., Busik, J.V., Agarwal, A., Segal, M.S. and Grant, M.B. (2008) Carbon monoxide and nitric oxide mediate cytoskeletal reorganization in microvascular cells via vasodilator-stimulated phosphoprotein phosphorylation: Evidence for blunted responsiveness in diabetes. Diabetes, 57, 2488-2494. doi:10.2337/db08-0381
- Salceda, R., Hernandez-Espinosa, C. and Sanchez-Chavez, G. (2008) L-arginine uptake in normal and diabetic rat retina and retinal pigment epithelium. Neurochemical Research, 33, 1541-1545. doi:10.1007/s11064-008-9641-9
- Chen, W., Arroyo, J.D., Timmons, J.C., Possemato, R. and Hahn, W.C. (2005) Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Research, 65, 8183-8192. doi:10.1158/0008-5472.CAN-05-1103
- Monteiro, H.P., Arai, R.J. and Travassos, L.R. (2008) Protein tyrosine phosphorylation and protein tyrosine nitration in redox signaling. Antioxidants & Redox Signaling, 10, 843-889. doi:10.1089/ars.2007.1853
- Tanimukai, H., Kudo, T., Tanaka, T., Grundke-Iqbal, I., Iqbal, K. and Takeda, M. (2009) Novel therapeutic strategies for neurodegenerative disease. Psychogeriatrics, 9, 103-109. doi:10.1111/j.1479-8301.2009.00289.x
- Kalev, P. and Sablina, A.A. (2011) Protein phosphatase 2A as a potential target for anticancer therapy. Anti-Cancer Agents in Medicinal Chemistry, 11, 38-46. doi:10.2174/187152011794941172
- Cho, U.S. and Xu, W.Q. (2007) Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature, 445, 53-57. doi:10.1038/nature05351
- Xu, Y.H., Xing, Y.N., Chen, Y., Chao, Y., Lin, Z., Fan, E., Yu, J.W., Strack, S., Jeffrey, P.D. and Shi, Y.G. (2006) Structure of the protein phosphatase 2A holoenzyme. Cell, 127, 1239-1251. doi:10.1016/j.cell.2006.11.033
- Brown, B.M., Carlson, B.L., Zhu, X., Lolley, R.N. and Craft, C.M. (2002) Light-driven translocation of the protein phosphatase 2A complex regulates light/dark dephosphorylation of phosducin and rhodopsin. Biochemistry, 19, 13526-13538. doi:10.1021/bi0204490
- Thomas, D.D., Ridnour, L.A., Isenberg, J.S., FloresSantana, W., Switzer, C.H., Donzelli, S., Hussain, P., Vecoli, C., Paolocci, N., Ambs, C., Colton, C.A., Harris, C.C., Roberts, D.D. and Wink, D.A. (2008) The chemical biology of nitric oxide: Implications in cellular signaling. Free Radical Biology & Medicine, 45, 18-31. doi:10.1016/j.freeradbiomed.2008.03.020
- Thomas, D.D., Espey, M.G., Ridnour, L.A., Hofseth, L.J., Mancardi, D., Harris, C.C. and Wink, D.A. (2004) Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proceedings of the National Academy of Sciences USA, 101, 8894-8899. doi:10.1073/pnas.0400453101
APPENDIX

Figure S1. Western blot analysis before and after reduction of nitrotyrosine to aminotyrosine. Nitration reduction (NO2 to NH2) was performed to confirm protein nitration, using disodium dithionite (Na2S2O4) to reduce 3-nitrotyrosine to 3-aminotyrosine. Reduced tyrosine (3-aminotyrosine) showed no band on Western blotting using 3-nitrotyrosine antibody compared to control (3-nitrotyrosine). ARPE-19 cell lysates from LPS, oxidative stress, and light treatment were separated by SDS-PAGE, and proteins were visualized by Western blotting. PVDF membrane was chemically reduced by 10 mM sodium dithionite (+Na2S2O4 ) in 50 mM pyridine acetate buffer (pH 5.0) for 1 hr before probing by anti-nitrotyrosine antibody. Negative control PVDF membrane was directly probed with anti-nitrotyrosine antibody without reduction (–Na2S2O4 ).

Figure S2. Expression of p-vimentin, vimentin and BAK in RPE under oxidative stress. ARPE-19 cells were treated with oxidative stress along with the okadaic acid (100 nM) for 1hr. Proteins were separated by SDS-PAGE and visualized by Western blotting using anti-p-vimentin (S38), vimentin and BAK. Vimentin phosphorylation was downregulated in 1 hr but upregulated using okadaic acid, a PP2A inhibitor. Pro-apoptotic Bak showed a negative correlation with vimentin phosphorylation in 1 hr under oxidative stress.

Figure S3. Negative control of iNOS immunocytochemistry. iNOS expressions in RPE cells were confirmed by immunocytochemistry with Alexa-488 secondary antibody and without anti-iNOS antibody as a negative control. Absence of green fluorescence demonstrates that there are not non-specific binding proteins with Alexa-488 secondary antibody. DAPI (blue) represents counter staining for the nucleus. Scale bars represent 20 μm.
NOTES
*These authors contributed equally.
#Corresponding authors.