Advances in Bioscience and Biotechnology
Vol. 3 No. 7A (2012) , Article ID: 25170 , 8 pages DOI:10.4236/abb.2012.327127
Oxidative stress and free radicals related diseases of the newborn
![]()
Department of Pediatrics, Obstetrics and Reproductive Medicine, University of Siena, Siena, Italy
Email: alnatour@just.edu.jo
Received 13 August 2012; revised 21 September 2012; accepted 11 October 2012
Keywords: Newborn Infant; Free Radicals; Perinatal Diseases; Oxidative Stress
ABSTRACT
Free radicals (FRs) generation is an unavoidable consequence of the life in an oxygen-rich atmosphere. FRs can be considered a double-edged sword. Beneficial effects of FRs occur at moderate concentrations and involve physiological roles in cellular responses to noxia, as in defense against infectious agents, in the function of a number of cellular signaling pathways and the induction of a mitogenic response. The overproduction of FRs and the insufficiency of an antioxidant mechanism result in oxidative stress (OS), a deleterious process and important mediator of damage to cell structures and tissues. It occurs at birth in all newborns as a consequence of the hyperoxic challenge after the transition from the hypoxic intrauterine environment to extrauterine life. During the perinatal period, OS can be magnified by others predisposing conditions such as hyperoxia, hypoxia, ischemia, hypoxia-reperfusion, inflam mation and high levels of non-protein bound iron. Epidemiological studies linked OS occurring during fetal stages and early infancy with adverse health outcomes later in life, indicating that OS is an early event in the etiology of these chronic diseases. Newborns, especially if preterm, are particularly susceptible to OS and damage due to the increased generation of FRs, the lack of adequate antioxidant protection, and the inability to induce antioxidant defenses during the hyperoxic challenge at birth. This impairment of the oxidative balance has been thought to be the common factor of pathologies grouped together as “free radical disease in the neonate” that include retinopathy of prematurity (which may lead to blindness in severe cases), bronchopulmonary dysplasia (a particularly debilitating pulmonary lesion of the preterm infant), periventricular leukomalacia (an important cause of severe neurodisability) and necrotizing enterocolitis. In this review we discuss in detail these perinatal diseases. Particularly, we analyze the current knowledge about the role of OS in their pathogenesis.
1. INTRODUCTION
Oxidative stress (OS) occurs when the production of free radicals (FRs) exceeds the capacity of antioxidant defenses [1]. It represents an imbalance between the production of reactive species and the biological system’s ability to readily detoxify the reactive intermediates or to repair the resulting damage. Each cell is characterized by a particular concentration of electrons stored in many cellular constituents and the redox state of a cell with its oscillation determines cellular functioning [2]. Disturbances in the normal redox state of tissues can cause toxic effects through the production of peroxides and FRs that damage all components of the cell, including proteins, lipids and DNA. Some reactive oxidative species can even act as messengers: at low levels, they are signaling molecules, and at high levels, they can damage organelles, particularly the mitochondria. Oxidative damage and the associated mitochondrial dysfunction may result in energy depletion, accumulation of cytotoxic mediators and cell death. Understanding the interface between stress adaptation and cell death is important for clarify redox biology and disease pathogenesis [3]. OS is an unavoidable consequence of life in an oxygen-rich atmosphere. It occurs at birth in all newborns as a consequence of the hyperoxic challenge due to the transition from the hypoxic intrauterine environment to extrauterine life. FRs sources such as hyperoxia, hypoxia, ischemia, hypoxia-reperfusion, inflammation and high levels of non-protein bound iron (NPBI) increase OS during the perinatal period. In addition preterm babies have reduced antioxidant defenses that are not able to counteract the harmful effects of FRs, leading to cellular, tissue and organ damages (kidney, retina, lung and bowel injury) [4].
2. OXIDATIVE STRESS
The increased generation of FRs in the cells relates directly to the fact that each oxygen atom has one unpaired electron in its outer valence shell, and molecular oxygen has two unpaired electrons. If the oxygen atom does not find a twin atom, it can accept hydrogen to form water (H2O), but there may be one electron less or more, resulting in no stability. This complex represents a FR. FRs are capable of giving rise to chain reactions, i.e. reactions that involve a number of steps, each of which forms a FR that triggers the next step. There are three phases: initiation, propagation and termination, and there are different free radical species: oxygen-centred radicals (ROS), nitrogen-centred radicals (RNS), carbon-centred radicals and sulphur-centred radicals [4]. Superoxide anion (O2–.) is considered the primary ROS and can generate secondary ROS. It is generated primarily within the mitochondrial respiratory chain, that is fundamental for the ATP production in mammalian cells. During the respiratory process, oxygen is utilized as an electron acceptor and completely reduced to water through the acquisition of four electrons:
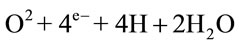
Radical formation is possible, when this process is realized through subsequent steps:
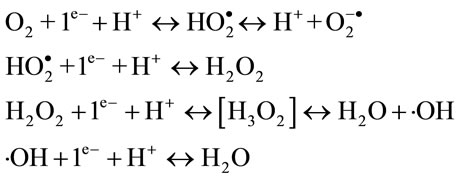
Three intermediate products are generated from oxygen reduction: superoxide anion (O2–.), hydrogen peroxide (H2O2) and hydroxyl radical (.OH). The second one is the most stable and it can be accumulated in a large quantity.
Besides mitochondrial function, there are many sources, both endogenous and exogenous, of FRs generation. In cells, one-electron abstraction of molecules can yield sulfur-, oxygen-, carbon-, and nitrogen-centered FRs. For example, removal of one electron (and a proton, H+) from a -SH group of a protein by a radical species (R.) yields a sulfur-centered free radical. If R. had just one unpaired electron, the reaction would convert it to a nonradical species. Another example is the one-electron removal from bis-allylic C-H bonds of polyunsaturated fatty acids (PUFAs) that yields a carbon-centered FR.
Nitric oxide (NO) can be also a FR source because it contains an unpaired electron in the last orbital, resulting uncommonly stable. NO interacts with the heme prosthetic group of the soluble guanylate cyclase, prompting cyclic guanosine-monophosphate (cGMP) formation and activating cGMP-dependent ion channels and kinases. Nitric oxide synthase (NOS) catalyzes the formation of NO. It reacts relatively slowly with O2 producing the orangebrown gas nitrogen dioxide (.NO2), a very reactive radical species [5].
Other potential endogenous sources include inflammatory cell activation (through NADPH oxidase of phagocytes and some endothelial cells), monoxygenase system, nitric oxide synthase and several other enzymes involved in the inflammatory process [6]. The burden of ROS can be further amplified by the presence of ‘free’ metals, such as iron, copper and manganese released from metalloprotein complexes [7]. Iron, when unbound to plasma proteins, can damage tissues by catalyzing the conversion of superoxide and hydrogen peroxide to FR species [8] through the Haber-Weiss and Fenton reactions.
Additional endogenous sources of cellular ROS are activated neutrophils, eosinophils and macrophages [9]. FRs can also be produced by a host of exogenous processes such as environmental agents and xenobiotics (metal ions, radiations, barbiturates) [10]. Stress factors such as tumour necrosis factor alpha (TNFa) can induce increase in ROS production with a redox cascade which leads to the activation of both pro-survival and pro-celldeath factors [4]. Whatever the source of FRs, they are really dangerous because of their toxic effects able to damage all components of the cell, including proteins, lipids and DNA. OS also causes a very wide spectrum of genetic, metabolic, and cellular responses. Most oxidative conditions will modulate gene expression, stimulate cell growth or cause a protective temporary growth-arrest [11]. Necrosis is the most extreme outcome and involves direct cell destruction.
3. OXIDATIVE STRESS AND FREE RADICALS RELATED DISEASES
Newborns are particularly susceptible to OS; this is in part due to the rapid environment change from a poor (womb) to a relatively rich oxygen habitat. There are also others predisposing conditions such as the relative antioxidant systems deficiency, the increased release of NPBI, the exposure to hyperoxia, to hypoxic-ischemic conditions and to inflammation events. The term “oxygen free radicals disease” was used to pull together all neonatal pathologies with a probable oxidative etiology [12]. It includes many diseases such as intraventricular haemorrhage, retinopathy of prematurity, bronchopulmonary dysplasia and necrotizing enterocolitis.
4. INTRAVENTRICULAR HAEMORRHAGE
Intraventricular hemorrhage (IVH) in very preterm infants is a common disease associated with long-term consequences [13]. In Italy 20% - 25% of all very low birth weight (VLBW) infants suffer from IVH. Importantly, 10% - 15% of neonates of <1500 g birth weight experiment with the more severe grades of hemorrhage, and over three-quarters of these develop mental retardation and/or cerebral palsy [14].
IVH has been attributed to changes in cerebral blood flow to the immature germinal matrix microvasculature and secondary periventricular venous infarction [14]. Recently, more detailed analyses have demonstrated the role of OS in this context [4]. Important modulators of cerebral blood flow in the developing brain include the cyclo-oxygenase 2 (COX-2) system and prostaglandins (PGs). COX-2 expression is induced by hypoxia, hypotension, growth factors such as epidermal growth factor receptor, transforming growth factor β (TGFβ), inflammatory modulators including IL-6, IL-1β, TNF-α, and NFkappaB [15]. The resultant prostanoids promote the production and release of vascular endothelial growth factor (VEGF), a potent angiogenic factor [16].
Those same triggers, which initiate hemorrhage into the germinal matrix, promote a cascade leading to the disruption of tight junctions, increased blood brain barrier permeability and microglial activation within the developing periventricular white matter. These events are mediated by cytokines (IL-1β and TNF-α), VEGF and NO. Furthermore, hypoxia alone has been shown to alter the blood brain barrier proteins ZO-1, occluding, and ZO-2. Finally, reactive microglia release ROS, which in turn not only contribute to endothelial damage, but also alter hemostasis and increase anaerobic metabolism [17].
The preterm brain is more susceptible to ROS than adult brain because of the immaturity of detoxifying enzyme systems. In addition to their release by activated microglia, ROS are also generated following the activation of the COX-2 system. Because of their multifaceted effects on the developing vasculature, ROS are believed to play a significant role in periventricular parenchymal infarction [18]. The nature of the cells involved in OS-associated brain injury is currently unclear. In postmortem examination of premature infants, the brain oxidative damage particularly targeted the oligodendrocyte lineage, suggesting that these cells are most vulnerable to this type of injury [19]. Other vulnerable cell populations include the subplate neurons. Oxidative tissue damage was documented in the periventricular white matter of premature neonates, as well as the cerebral cortex of term infants succumbing from severe hypoxic–ischemic insults [19]. The levels of 15- F2t-IsoP in cerebrospinal fluid were significantly increased in one-third of infants with white matter injury [20]. We found increased plasma levels of total hydroperoxides (TH), advanced oxidation protein products (AOPP) and particularly NPBI in newborn who developed IVH suggesting that OS markers are direct indexes of increased production of FRs in central nervous system as a response to oxidative neuronal damage [4]. Perinatal hypoxia alters mechanisms that regulate cerebral blood flow and triggers a cascade of biochemical events that begins with a shift from oxidative to anaerobic metabolism leading to oxidative brain damage resulting from excessive production of FRs [21]. Polyunsaturated fatty acids are constituents of lipidic membrane in white matter and they are highly susceptible to FRs attack. The propensity of brain oxidative injury in the immature babies is related to deficient antioxidant defenses but also to several pro-oxidant characteristics. Basically, the developing tissues have a high metabolic rate supported almost exclusively by oxidative metabolism, an excellent source for FRs, and a relatively high concentration of NPBI [22]. The fetus accumulates iron from maternal stores during pregnancy [23] suggesting a role of iron as a trophic factor at an early developmental stage, when growth is especially active. Despite the brain’s highly regulated system for iron utilization and metabolism, iron overload is associated with saturation in protein involved in iron transport and storage, causing an increase in free ferrous iron [24]. In this case, iron is capable of causing degeneration of endothelial cells [25]. Endothelial cell injury and dysfunction may additionally contribute to the inflammatory response and alteration in coagulation, through loss of normal endothelial nitric oxide production [26]. Other potential implications of iron overload are acute impairment of endotheliumdependent flow-mediated vasodilation [27] loss of tight junction proteins and degeneration of endothelial cells, opening of the blood-brain barrier [25]. Separation of endothelial tight junctions, loss of endothelial attachment to the basement membrane, endothelial blebbing, and endothelial necrosis have been described in the cerebral vasculature following ischemic injury [28]. The progression of endothelial dysregulation can contribute to the ongoing pathogenesis of IVH.
5. RETINOPATHY OF PREMATURITY
Retinopathy of prematurity (ROP), is the major cause of visual impairment and blindness in premature neonates [29]. The overall incidence rate of the condition is about 68% among infants born with birth weight less than 1200 g and 98% among children born with birth weight less than 750 g [30]. The pathogenesis is multifactorial [30]. Normally the vascularization of the human retina is largely complete by the 4th month of gestation but peripheral retinal vascularization is still immature until the fetus is near term. ROP occurs in two phases: the vascular attenuation phase (phase I) and the fibrovascular proliferative phase (phase II). In phase I, hyperoxia causes cessation of normal retinal vascularization and in phase II, hypoxia renews vascularization. In both of these cases VEGF plays a major role [31]. It is not clear why some babies develop severe ROP whereas other babies with similar clinical characteristics do not progress to a severe stage. As multifactorial disease, genetic factors in addition to the prematurity or the environment may be responsible of the development and progression of ROP. Oxygen and overall hyperoxia are fundamental factors, with a direct relationship between a high-oxygen saturation and ROP [32]. Fluctuations in SaO2 may also be dangerous: the exposure to alternating hypoxic and hyperoxic events causes severe proliferative retinopathy in the newborn rat [33]. Very high concentrations of the FR generator, hypoxanthine, have been found in the eyes of babies at risk of developing ROP [34]. Generation of ROS and suppression of specific oxygen-regulated vascular survival factors lead to microvascular injury. We found the oxygen administration in delivery room, jointly to birth weight and prematurity, was significantly associated with the development of ROP [35]. We also, demonstrated elevated levels of TH, AOPP and NPBI, in cord blood of babies at risk for FRD and consequently of ROP [4]. Furthermore, it has been shown the retinal antioxidants and antioxyenzymes content is low in cases of ROP [36]. Some protective effects have been observed by giving the potent antioxidant D-Penicillamine and Vitamin E [37].
The VEGF is produced by astrocytes in response to hypoxia [31] giving rise to retinal neovascularization at the junction between vascularized and non-vascularized retina [32]. VEGF levels are suppressed by hyperoxia with a stop of the normal retinal vascularization and loss of some developing vessels. In addition, fetal insulin like growth factor-1 (IGF-1) precipitously fall after premature birth [38]. The action of VEGF depends on IGF-1. Subsequently, with the retinal development the oxygen need increases and the result is an hypoxic environment with a consequent VEGF production. This fact, in addition to the high IGF-1 levels due to the newborn’s maturation, leads to the neo-vascularization process.
6. BRONCHOPULMONARY DYSPLASIA
Bronchopulmonary dysplasia (BPD) of the newborn is one of the most important factors influencing the perinatal mortality and morbidity. BPD occurs almost exclusively in preterm infants. Oxygen toxicity, FRs, prematurity, surfactant deficit, inflammation are major factors contributing to the pathogenesis [39]. Inadequate nutrition, and the ventilation mode participate to the increase of oxidative stress, which might trigger changes leading to permanent lung damage [40]. Phagocytic cell in the lung mediate their antimicrobial functions through the release of lysozymes, peroxidases and proteases, in addition to ROS and NO.
Activated neutrophils and pulmonary type II cells are also important inducers of the Fenton reaction and of the increase of adhesiveness to the endothelium. The release of inflammatory mediators can stimulate the endothelium to promote transendothelial migration facilitating the passage of cytokines [41]. The increase in phagocyte number and interleukin concentrations in bronchoalveolar lavage fluid of infants with BPD indicates that oxygen toxicity and inflammation are involved in the development of lung injury [42].
BPD is characterized by a tissue remodeling process, which has been divided into different phases, ending up in the chronic phase with an increased number of fibroblasts and fibrotic areas. Matrix metalloproteins (MMPs) are important in regulating fibrotic processes. They degrade extracellular matrix proteins and fibrillar collagen, the balance between MMPs and their inhibitors normally being of importance in the development and regulation of fibrosis. Their expression is regulated at the transcriptional level by cytokines, growth factors and extracellular matrix components. OS increases both MMPs and their inhibitors, so it may lead to lung damage by increasing collagenase activity, causing disruption of the extracellular matrix [40]. OS is also able to inactivate surfactant, as demonstrated in rats treated with the combination of hyperoxia and hypoxanthine infusion. As a result of inflammation and edema, transudated plasma proteins and inflammatory cells impair extracellular surfactant. Hyperoxia results in augmentation of the surfactant proteins SP-A, SP-B, and SP-C85 while oxidized SP-A loses its surfactant and immune defense functions. Hyperoxia reduces also surfactant phospholipid production [43]. It seems that glycerol-3 phosfate acyltransferase, which catalyzes the first reaction in phosphoglyceride synthesis, is a rate-limiting enzyme, extremely sensitive to oxidative damage [40].
An increased concentration of products of lipid peroxidation, pentane and ethane, in exhaled gas were demonstrated a few days after birth in infants who subsequently developed BPD [44]. A positive correlation was found between the duration of oxygen and ventilatory therapy and lipid peroxidation by Inder et al. [45].
Protein oxidation was also demonstrated by Gladstone and Levine with higher carbonyl content in lung lavage samples from preterm infants with an oxygen requirement of over 40% or who were ventilated for longer than 72 h compared with those requiring less oxygen or less time on mechanical ventilation [46]. In tracheal aspirates from premature infants requiring oxygen therapy, Varsila et al. showed that newborns who subsequently developed BPD had a significantly higher carbonyl concentration during the first 6 days of life than those who did not [47].
7. NECROTIZING ENTEROCOLITIS
Necrotizing enterocolitis (NEC) is a gastrointestinal surgical emergency in preterm neonates [48]. The overall incidence is 1 - 5 per 1000 live births, dramatically increasing to 100 per 1000 in very low birth weight and to 200 per 1000 in extremely low birth weight infants [49]. NEC has a multifactorial etiology: ischemia, infections, cytokines, enteral feeding and reactive oxygen species may contribute to the disruption of the protective gut barrier.
In particular, FRs, generated as a result of various injury to the gut, has been linked to the development of NEC in premature infants. Ischemia, hypoxia-reperfusion, infection and inflammation are mechanisms capable of produce high levels of FRs, perturbing the normal redox balance and shifting cells into a state of OS [50].
Some authors recently proposed that the underlying initial condition is the reduced ability of the neonatal gut epithelial cells to reduce OS, and that when epithelial gut cells are exposed to enteral feeding, the increased metabolic OS tips the population toward apoptosis, inflammation, bacterial activation, and eventual necrosis [51]. Recent findings suggested that mitochondria are the major source of intestinal apoptotic signaling during OS, and that modulating mitochondrial apoptotic is possible to ameliorate the NEC effects [52].
Recently we found a strong association between NEC and cord blood concentration of AOPP, NPBI and TH, showing a clear correlation between intrauterine OS events and the risk of developing NEC [53]. The immaturity of the gastrointestinal tract in preterm infants may also contribute to NEC development [54]. The decreased intestinal peristalsis expose the intestinal epithelium to noxious substances, for example, Toll-like Receptor-4 expression is down-regulated in the mature intestinal epithelium upon stimulation by gram-negative lipopolysaccharide but is increased in the immature intestinal epithelium, leading to an exaggerated pro-inflammatory response through up-regulation of the NF-kB pathway [55]. Moreover, Prolonged antibiotic exposure is associated to increased risk of NEC [54] and this association persisted in multivariate analyses excluding confounding factors (gestational age, birth weight, and sepsis) [56]. In addition, preterm infants frequently suffer from ischemic events, such as hypotension, hypothermia, anemia, and patent ductus arteriosus during the intensive care stay. Severe anemia results in insufficient oxygen for the increased requirements of the mesenteric vessels after enteral feeding. Ischemia lead to endothelial cell disfunction and alter the endothelin-1/nitric oxide balance in favor of vasoconstriction, causing expansion of ischemic intestinal lesions [57]. Other risk factors are sensitization to cow milk proteins [58] and polymorphisms, such as mutations in carbamoyl phosphate synthetase [59], vascular endothelial growth factor [60], and interleukin-10 and 12 [61].
At now, due to the established importance of perinatal oxidative insults in injured intestinal epithelial cells, more study are needed, providing a rational basis for the formulation of interventions to interrupt these mechanisms.
8. CONCLUSIONS
The existence of a redox homeostasis is essential for normal health and survival of the cell.
When an unbalance between pro-oxidant and antioxidant factors occurs, oxidative stress (OS) is produced leading to cellular and tissue damage. The newborn, especially if preterm, is highly prone to OS and to the toxic effect of FRs. The growing role of OS in the pathogenesis of preterm newborn diseases, requires careful research to better clarify this phenomenon and its impact on infants’ health.
9. ACKNOWLEDGEMENTS
Supported by grants provided by the EURAIBI Onlus Foundation.
REFERENCES
- Buonocore, G. and Groenendaal, F. (2007) Anti-oxidant strategies. Seminars in Fetal and Neonatal Medicine, 12, 287-295. doi:10.1016/j.siny.2007.01.020
- Buonocore, G., Perrone, S. And Tataranno, M.L. (2010) Oxygen toxicity: Chemistry and biology of reactive oxygen species. Seminars in Fetal and Neonatal Medicine, 15, 186-190. doi:10.1016/j.siny.2010.04.003
- Lee, J., Giordano, S. and Zhang, J. (2012) Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochemical Journal, 441, 523-540. doi:10.1042/BJ20111451
- Perrone, S., Tataranno, M.L., Negro, S., Longini, M., Marzocchi, B., Proietti, F., Iacoponi, F., Capitani, S. and Buonocore, G. (2010) Early identification of the risk for free radical-related diseases in preterm newborns. Early Human Development, 86, 241-244. doi:10.1016/j.earlhumdev.2010.03.008
- Alderton, W.K., Cooper, C.E. and Knowles, R.G. (2001) Nitric oxide synthases: Structure, function and inhibition. Biochemical Journal, 357, 593-615. doi:10.1042/0264-6021:3570593
- Auten, R.L. and Davis, J.M. (2009) Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatric Research, 66, 121-127. doi:10.1203/PDR.0b013e3181a9eafb
- McCarthy, S.M., Bove, P.F., Matthews, D.E., Akaike, T. and van der Vliet, A. (2008) Nitric oxide regulation of MMP-9 activation and its relationship to modifications of the cysteine switch. Biochemistry, 47, 5832-5840. doi:10.1021/bi702496v
- Gutteridge, J.M. (1982) The role of superoxide and hydroxyl radicals in phospholipid peroxidation catalysed by iron salts. FEBS Letters, 150, 454-458. doi:10.1016/0014-5793(82)80788-6
- Conner, E.M. and Grisham, M.B. (1996) Inflammation, free radicals, and antioxidants. Nutrition, 12, 274-277. doi:10.1016/S0899-9007(96)00000-8
- Bachowski, S., Kolaja, K.L., Xu, Y., Ketcham, C.A., Stevenson, D.E., Walborg, E.F. and Klaunig, J.E. (1997) Role of oxidative stress in the mechanism of dieldrin’s hepatotoxicity. Annals of Clinical & Laboratory Science, 27, 196-209.
- Davies, K.J. (2000) Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life, 50, 279-289. doi:10.1080/15216540051081010
- Saugstad, O.D. (1989) The oxygen radical disease in neonatology. Indian Journal of Pediatrics, 56, 585-593. doi:10.1007/BF02722373
- Volpe, J.J. (2001) Perinatal brain injury: From pathogenesis to neuroprotection. Mental Retardation and Developmental Disabilities Research Reviews, 7, 56-64. doi:10.1002/1098-2779(200102)7:1<56::AID-MRDD1008>3.0.CO;2-A
- McCrea, H.J. and Ment, L.R. (2008) The diagnosis, management, and postnatal prevention of intraventricular hemorrhage in the preterm neonate. Clinics in Perinatology, 35, 777-792. doi:10.1016/j.clp.2008.07.014
- Ackerman, W.E. 4th, Rovin, B.H. and Kniss, D.A. (2004) Epidermal growth factor and interleukin-1beta utilize divergent signaling pathways to synergistically upregulate cyclooxygenase-2 gene expression in human amnion-derived WISH cells. Biology of Reproduction, 71, 2079- 2086. doi:10.1095/biolreprod.104.030841
- Kuwano, T., Nakao, S., Yamamoto, H., Tsuneyoshi, M., Kuwano, M. and Ono, M. (2004) Cyclooxygenase 2 is a key enzyme for inflammatory cytokine-induced angiogenesis. FASEB Journal, 18, 300-310. doi:10.1096/fj.03-0473com
- Ulfig, N., Bohl, J., Neudörfer, F. and Rezaie, P. (2004) Brain macrophages and microglia in human fetal hydrocephalus. Brain & Development, 26, 307-315. doi:10.1016/S0387-7604(03)00172-4
- Folkerth, R.D., Keefe, R.J., Haynes, R.L., Trachtenberg, F.L., Volpe, J.J. and Kinney, H.C. (2004) Interferongamma expression in periventricular leukomalacia in the human brain. Brain Pathology, 14, 265-274. doi:10.1111/j.1750-3639.2004.tb00063.x
- Back, S.A., Luo, N.L., Mallinson, R.A., O’Malley, J.P., Wallen, L.D., Frei, B., Morrow, J.D., Petito, C.K., Roberts, C.T. Jr., Murdoch, G.H. and Montine, T.J. (2005) Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Annals of Neurology, 58, 108-120. doi:10.1002/ana.20530
- Inder, T., Mocatta, T., Darlow, B., Spencer, C., Volpe, J.J. and Winterbourn, C. (2002) Elevated free radical products in the cerebrospinal fluid of VLBW infants with cerebral white matter injury. Pediatric Research, 52, 213- 218. doi:10.1203/00006450-200208000-00013
- Perrone, S., Bracci, R. and Buonocore G. (2002) New biomarkers of fetal-neonatal hypoxic stress. Acta Paediatrica, 91, 135-138. doi:10.1111/j.1651-2227.2002.tb02919.x
- McQuillen, P.S. and Ferriero, D.M. (2004) Selective vulnerability in the developing central nervous system. Pediatric Neurology, 30, 227-235. doi:10.1016/j.pediatrneurol.2003.10.001
- Gazzolo, D., Perrone, S., Paffetti, P., Longini, M., Vezzosi, P., Bruschettini, M., Lituania, M. and Buonocore, G. (2005) Non protein bound iron concentrations in amniotic fluid. Clinical Biochemistry, 38, 674-677. doi:10.1016/j.clinbiochem.2005.03.010
- Gaasch, J.A., Lockman, P.R., Geldenhuys, W.J., Allen, D.D. and Van der Schyf, C.J. (2007) Brain iron toxicity: Differential responses of astrocytes, neurons, and endothelial cells. Neurochemical Research, 32, 1196-1208. doi:10.1007/s11064-007-9290-4
- Won, S.M., Lee, J.H., Park, U.J., Gwag, J., Gwag, B.J. and Lee, Y.B. (2011) Iron mediates endothelial cell damage and blood-brain barrier opening in the hippocampus after transient forebrain ischemia in rats. Experimental and Molecular Medicine, 43, 121-128. doi:10.3858/emm.2011.43.2.020
- Yang, Y. and Loscalzo, J. (2000) Regulation of tissue factor expression in human microvascular endothelial cells by nitric oxide. Circulation, 101, 2144-2148. doi:10.1161/01.CIR.101.18.2144
- Van Buren, P., Velez, R.L., Vaziri, N.D. and Zhou, X.J. (2012) Iron overdose: A contributor to adverse outcomes in randomized trials of anemia correction in CKD. International Urology and Nephrology, 44, 499-507. doi:10.1007/s11255-011-0028-5
- Pomfy, M. and Húska, J. (1992) The state of the microcirculatory bed after total ischaemia of the brain. An experimental ultrastructural study. Functional and Developmental Morphology, 2, 253-258.
- Rivera, J.C., Sapieha, P., Joyal, J.S., Duhamel, F., Shao, Z., Sitaras, N., Picard, E., Zhou, E., Lachapelle, P. and Chemtob, S. (2011) Understanding retinopathy of prematurity: Update on pathogenesis. Neonatology, 100, 343- 353. doi:10.1159/000330174
- Shastry, B.S. (2010) Genetic susceptibility to advanced retinopathy of prematurity (ROP). Journal of Biomedical Science, 17, 69. doi:10.1186/1423-0127-17-69
- Pierce, E.A., Foley, E.D. and Smith, L.E. (1996) Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Archives of Ophthalmology, 114, 1219-1228. doi:10.1001/archopht.1996.01100140419009
- Saugstad, O.D. (2006) Oxygen and retinopathy of prematurity. Journal of Perinatology, 26, S46-S50.
- Penn, J.S., Henry, M.M. and Tolman, B.L. (1994) Exposure to alternating hypoxia and hyperoxia causes severe proliferative retinopathy in the newborn rat. Pediatric Research, 36, 724-731. doi:10.1203/00006450-199412000-00007
- Saugstad, O.D. and Rognum, T.O. (1988) High postmortem levels of hypoxanthine in the vitreous humor of premature babies with respiratory distress syndrome. Pediatrics, 81, 395-398.
- Perrone, S., Vezzosi, P., Longini, M., Marzocchi, B., Paffetti, P., Bellieni, C.V., Martinelli, S. and Buonocore, G. (2009) Biomarkers of oxidative stress in babies at high risk for retinopathy of prematurity. Frontiers in Bioscience, 1, 547- 552.
- Chen, W., Hunt, D.M., Lu, H. and Hunt, R.C. (1999) Expression of antioxidant protective proteins in the rat retina during prenatal and postnatal development. Investigative Ophthalmology & Visual Science, 40, 744-751.
- Raju, T.N., Langenberg, P., Bhutani, V. and Quinn, G.E. (1997) Vitamin E prophylaxis to reduce retinopathy of prematurity: A reappraisal of published trials. Journal of Pediatrics, 131, 844-850. doi:10.1016/S0022-3476(97)70031-3
- Lineham, J.D., Smith, R.M., Dahlenburg, G.W., King, R.A., Haslam, R.R., Stuart, M.C. and Faull, L. (1986) Circulating insulin-like growth factor I levels in newborn premature and full-term infants followed longitudinally. Early Human Development, 13, 37-46. doi:10.1016/0378-3782(86)90096-4
- Joung, K.E., Kim, H.S., Lee ,J., Shim, G.H., Choi, C.W., Kim, E.K., Kim, B.I. and Choi, J.H. (2011) Correlation of urinary inflammatory and oxidative stress markers in very low birth weight infants with subsequent development of bronchopulmonary dysplasia. Free Radical Research, 45, 1024-1032. doi:10.3109/10715762.2011.588229
- Saugstad, O.D. (2003) Bronchopulmonary dysplasiaoxidative stress and antioxidants. Seminars in Neonatology, 8, 39-49. doi:10.1016/S1084-2756(02)00194-X
- Fellman, V. (1997) Respiratory distress syndrome of neonates today. Duodecim, 113, 1024-1031.
- Groneck, P. and Speer, C.P. (1993) Interleukin-8 in pulmonary effluent fluid of preterm infants. Journal of Pediatrics, 123, 839-840. doi:10.1016/S0022-3476(05)80884-4
- Haagsman, H.P. (1998) Interactions of surfactant protein A with pathogens. Biochimica et Biophysica Acta, 1408, 264-277. doi:10.1016/S0925-4439(98)00072-6
- Varsila, E., Hallman, M. and Andersson, S. (1994) Freeradical-induced lipid peroxidation during the early neonatal period. Acta Paediatrica, 83, 692-695. doi:10.1111/j.1651-2227.1994.tb13120.x
- Inder, T.E., Graham, P., Sanderson, K. and Taylor, B.J. (1994) Lipid peroxidation as a measure of oxygen free radical damage in the very low birthweight infant. Archives of Disease in Childhood Fetal Neonatal Edition, 70, F107-F111. doi:10.1136/fn.70.2.F107
- Gladstone Jr., I.M. and Levine, R.L. (1994) Oxidation of proteins in neonatal lungs. Pediatrics, 93, 764-768.
- Varsila, E., Pesonen, E. and Andersson, S. (1995) Early protein oxidation in the neonatal lung is related to development of chronic lung disease. Acta Paediatrica, 84, 1296-1299. doi:10.1111/j.1651-2227.1995.tb13552.x
- Baregamian, N., Song, J., Bailey, C.E., Papaconstantinou, J., Evers, B.M. and Chung, D.H. (2009) Tumor necrosis factor-alpha and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy, and c-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis. Oxidative Medicine and Cellular Longevity, 2, 297-306. doi:10.4161/oxim.2.5.9541
- Buonocore, G., Bracci, R. and Weindling, M. (2011) Neonatology. A pratical approach to neonatal management. Springer Verlag Italia, Milano.
- Aydemir, C., Dilli, D., Uras, N., Ulu, H.O., Oguz, S.S., Erdeve, O. and Dilmen, U. (2011) Total oxidant status and oxidative stress are increased in infants with necrotizing enterocolitis. Journal of Pediatric Surgery, 46, 2096-2100. doi:10.1016/j.jpedsurg.2011.06.032
- Kim, M., Christley, S., Alverdy, J.C., Liu, D. and An, G. (2012) Immature Oxidative Stress Management as a Unifying Principle in the Pathogenesis of Necrotizing Enterocolitis: Insights from an Agent-Based Model. Surgical Infections, 13, 18-32. doi:10.1089/sur.2011.057
- Baregamian, N., Song, J., Papaconstantinou, J., Hawkins, H.K., Evers, B.M. and Chung, D.H. (2011) Intestinal mitochondrial apoptotic signaling is activated during oxidative stress. Pediatric Surgery International, 27, 871-877. doi:10.1007/s00383-011-2880-x
- Perrone, S., Tataranno, M.L., Negro, S., Cornacchione, S., Longini, M., Proietti, F., Soubasi, V., Benders, M.J., Van Bel, F. and Buonocore, G. (2012) May oxidative stress biomarkers in cord blood predict the occurrence of necrotizing enterocolitis in preterm infants? Journal of Maternal-Fetal and Neonatal Medicine, 1, 128-131. doi:10.3109/14767058.2012.663197
- Lee, J.H. (2011) An update on necrotizing enterocolitis: Pathogenesis and preventive strategies. Korean Journal of Pediatrics, 54, 368-372. doi:10.3345/kjp.2011.54.9.368
- Claud, E.C., Zhang, X., Petrof, E.O. and Sun, J. (2007) Developmentally regulated tumor necrosis factor-alpha induced nuclear factor-kappaB activation in intestinal epithelium. American Journal of Physiology Gastrointestinal and Liver Physiology, 292, G1411-G1419. doi:10.1152/ajpgi.00557.2006
- Alexander, V.N., Northrup, V. and Bizzarro, M.J. (2011) Antibiotic exposure in the newborn intensive care unit and the risk of necrotizing enterocolitis. Journal of Pediatrics, 159, 392-397. doi:10.1016/j.jpeds.2011.02.035
- Nankervis, C.A., Giannone, P.J. and Reber, K.M. (2008) The neonatal intestinal vasculature: Contributing factors to necrotizing enterocolitis. Seminars in Perinatology, 32, 83-91. doi:10.1053/j.semperi.2008.01.003
- Abdelhamid, A.E., Chuang, S.L., Hayes, P. and Fell, J.M. (2011) In vitro cow’s milk protein-specific inflammatory and regulatory cytokine responses in preterm infants with necrotizing enterocolitis and sepsis. Pediatric Research, 69, 165-169. doi:10.1203/PDR.0b013e31820263e7
- Moonen, R.M., Paulussen, A.D., Souren, N.Y., Kessels, A.G., Rubio-Gozalbo, M.E. and Villamor, E. (2007) Carbamoyl phosphate synthetase polymorphisms as a risk factor for necrotizing enterocolitis. Pediatric Research, 62, 188-190. doi:10.1203/PDR.0b013e3180a0324e
- [61] Bányász, I., Bokodi, G., Vásárhelyi, B., Treszl, A., Derzbach, L., Szabó, A., Tulassay, T. and Vannay, A. (2006) Genetic polymorphisms for vascular endothelial growth factor in perinatal complications. European Cytokine Network, 17, 266-270.
- [62] Treszl, A., Kaposi, A., Hajdú, J., Szabó, M., Tulassay, T. and Vásárhelyi, B. (2007) The extent to which genotype information may add to the prediction of disturbed perinatal adaptation: None, minor, or major? Pediatric Research, 62, 610-614.