Advances in Bioscience and Biotechnology
Vol.4 No.2(2013), Article ID:28430,13 pages DOI:10.4236/abb.2013.42034
Autophagic cell death: A new frontier in cancer research
![]()
1Department of Biochemistry, Biophysics and General Pathology, Second University of Naples, Naples, Italy
2Istituto Auxologico Italiano IRCCS, Milan, Italy
3Dipartimento di Scienze Mediche, Università degli Studi di Milano, Milan, Italy
Email: michele.caraglia@unina2.it, michele.caraglia@alice.it
Received 30 August 2012; revised 30 September 2012; accepted 8 October 2012
Keywords: Autophagy; Cancer; Cellular Death; mTOR; PPAR-γ
ABSTRACT
Autophagy is a conserved and tightly regulated cellular catabolic process that involves the lysosomal degradation pathway. Intracellular recycling of macromolecules and organelles provided by autophagy is an integral part of normal cellular function and permits cells survival under starvation conditions, maintaining cell growth and the homeostasis of organisms. In addition to its normal role in cell physiology, autophagy is closely linked to both tumorigenesis and cancer cell response to treatments. In fact, anticancer drugs can induce autophagy but it remains controversial whether this process leads to cancer cell death or protects cancer cells from cellular stress. The role of autophagy in cancer is complex and is likely dependent on tumor type, stage, and genetic context. However, recent evidences demonstrate a tight interconnection of autophagy with several cell death pathways and reveal an active contribution of autophagy to cell death. When autophagy is directly involved in the death process, the cell death process is designated “autophagic cell death” (ACD). In this review, we will give a comprehensive overview of the autophagic signaling pathway, its role and regulation in cancer cells; moreover, we will try to define the molecular mechanisms at the basis of the autophagic cell death showing that PPAR-γ activation plays a role in the induction of autophagy in cancer cells.
1. INTRODUCTION
In multicellular organisms, the number of cells is determined by a tightly regulated balance between their proliferation and their death. Maintaining this balance is a prerequisite for the development and growth as well as for adult tissue homeostasis [1]. In contrast with the necrosis (mechanism of cell death resulting from excessive cell trauma that follows to a pathological external stimulus), apoptosis represents a physiological mechanism of programmed cell death, which ensures the elimination of cells that have completed their life cycle, or that, as a result of genetic damage, have become useless or harmful to the organism [2]. Cell death is a process that can be triggered by many physiological stimuli or pathological mechanisms that trigger cell suicide. Alternatively, exogenous highly destructive stimuli can induce a passive mechanism of cell death and fragmentation called necrosis.
Necrosis is a passive pathological process triggered in response to an acute injury such as hypoxia, hypothermia, radiation, viral infections, immune response, chemicals and pharmaceuticals [3].
Thus, the term programmed cell death has been coined to distinguish between physiological cell death and destruction of necrotic cells. Programmed cell death is an integral part of normal development and it is involved not only in processes such as limb formation and remodeling of the nervous system but also in the removal of abnormal cells during development [4]. Morphological studies have led to the definition of three types of physiological cell death [5]. The first type, known as apoptosis, is characterized by the condensation of the nucleus and of the cytoplasm, followed by DNA fragmentation and formation of apoptotic bodies [2]. The second type, known as autophagy, is characterized by the formation of autophagic vacuoles in the cytoplasm of cells that incorporate and degrade the cellular components. The third type, known as non-lysosomal cell death, is less common, and is characterized by degradation of cellular components due to non-lysosomal activity. In particular: autophagy, or lysosomal cell death, is an important process in the preservation of cytoplasmic homeostasis, which gives form to the selective elimination of cytoplasmic material and organelles sequestered in autophagic vacuoles of lysosomal nature. It is possible this mechanism is able to modulate the intracellular stores of functional mitochondria, and thus that it is able to influence events dependent on the mitochondrial activity, such as apoptosis [6].
Apoptosis or programmed cell death, is an active physiological process, usually caspase-dependent (but can also be caspase-independent). Programmed cell death provides for two different activation pathways, the extrinsic or receptor-dependent in which the death signal is provided by oligomerization of the death receptor, expressed on the cell surface, by binding to its ligand (in the case is induced by cytokines), and death induced by the intrinsic pathway, independent of extracellular receptors but closely related to the presence in the cytosol of pro-apoptotic molecules, otherwise confined and sequestered in the cell compartments. The first way presents the caspase-8 as initiator of the execution phase of apoptotic process, while the intrinsic pathway using the caspase-9. It is known that apoptosis plays a fundamental role in the development and homeostasis of multicellular organisms. Alteration of the physiological balance of these events can lead to pathological situations: a low apoptotic machinery to carry out its function is often the cause of autoimmune disorders and of the development of tumors; on the contrary, an excess of activity is evident in acute and chronic degenerative diseases, including some affecting the nervous system (Figure 1).
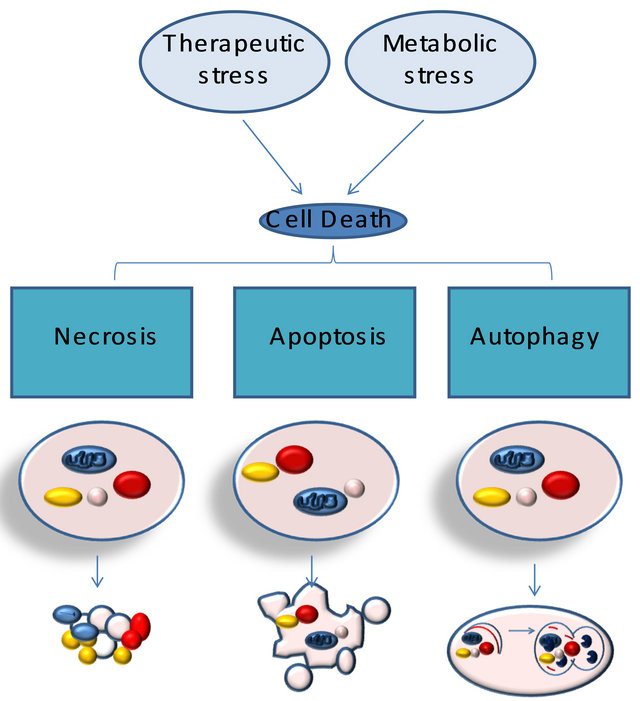
Relationship between necrosis, autophagy, and apoptosis. Metabolic and therapeutic stresses lead to acute NAD+ and ATP depletion accompanied by increased intracellular calcium and ROS. Cells that do not adapt to these changes undergo necrotic cell death. The activation of stress regulators, such as AMPK, allows cells to acutely survive these changes. AMPK-dependent phosphorylation results in the inhibition of mammalian target of rapamycin, which inhibits autophagy. AMPK-dependent phosphorylation also activates p53, which can lead to autophagy or apoptosis, through the activation of Bax and Bak, the cytoplasmic release of cytochrome c (Cyt. C), and the activation of caspases. Unlike apoptosis or necrosis, stress-induced autophagy can lead to autophagic cell death or to cell survival.
Figure 1. Typologies of cellular death.
Other types of cell death include:
-Senescence, caspase-independent mechanism, is due to an arrest of metabolic cellular functions with an intracellular accumulation of non-functional proteins which form aggregates of histamine in the cytoplasm and precipitation of lipids. Senescence is a mechanism different from apoptosis, a senescent cell doesn’t proliferate anymore, and it stops its replication cycle and cannot go toward to apoptosis.
-Mitotic catastrophe is characterized by multiple nuclear fragmentation; is a process independent of caspases and leads to the activation of certain molecules involved in cell cycle regulation as cdk 1. Therefore, it is characterized by aberrant activation of mitosis; the cell rather than to divide itself runs into death, it fragments and dies.
2. AUTOPHAGY
Autophagy is a cellular process complex, ATP-dependent, highly conserved and regulated, which occurs in all eukaryotic cells.
It is characterized by the rearrangement of subcellular membranes in order to sequester components of the cytoplasm and cellular organelles to be exposed to the action of degradative enzymes present in particular organelles, such as the vacuole in yeast and lysosomes in mammalian cells, where the seized material is degraded and recycled [7,8]. Autophagy has multiple functions. In fact, this process is involved: 1) in remodeling during development and cell differentiation; 2) in the degradation of intracellular proteins; 3) in the production of amino acids when there is a decrease of nutrients; 4) in the mechanism of exchange of citomembrane and organelles such as mitochondria and peroxisomes; 5) in the control of the mechanism of programmed cell death of type II (non-apoptotic). Differentiation and development both require cells to undergo significant phenotypic changes and, presumably, must entail a mechanism for the breakdown and recycling of obsolete cellular components. In the developing organism, autophagy plays an essential role in the cellular and tissue remodeling that occurs during morphogenesis. In many tissues in the adult organism this function of autophagy is largely obsolete; however, protein and organelle turnover by autophagy plays an essential cellular homeostatic or housekeeping function, removing damaged or unwanted organelles and proteins. One of the most fundamental and evolutionarily conserved functions of autophagy is its role in the response to starvation. During early starvetion, glycogen is used for glucose homeostasis. However, in mammals, glycogen stores are consumed within 1 day. Subsequently, blood glucose is maintained by gluconeogenesis from pyruvate or tricarboxylic acid (TCA) cycle intermediates such oxaloacetate in the liver. Amino acids produced by autophagy in the liver and other tissues could be utilized for this aim. In addition, amino acids can be directly oxidized to produce energy in some tissues, such as muscle, or used for synthesis of proteins that are required for proper starvation adaptation. Autophagy could account for the majority of the so-called protein catabolism during starvation. Autophagy can also have a role in the control of cell growth, even in that tumoral. This process has been proposed as anti-aging mechanism because autophagy is involved in the elimination of organelles and citomembrane damaged due to age-dependent peroxidation of different molecules [9]. Autophagy is stimulated in response to various stress situations such as fasting, changes in cell volume, oxidative stress, accumulation of damaged proteins, hormonal signals, irradiation and treatment with xenobiotics [10]. Role of autophagy extends beyond the general maintenance of homeostasis, degradation, and recycling of damaged proteins and specific organelles for many physiological and pathological processes, such as development, immunity, energy homeostasis, cell death, tumorigenesis, etc. [11]. The involvement of autophagy in tumor development is out of question, but it is little known at the time (Figure 2).
Types of Autophagy: Macroautophagy, Microautofagia and Autophagy Chaperone-Mediated
Types of autophagy better characterized are macro-autophagy, microautofagia and autophagy chaperone-mediated [12]. They are differentiated: the way in which the material to be degraded is transported inside the lumen lysosomal, for the type of conveyed material and for their regulation. The macroautophagy is the major form of autophagy responsible for degradation both of soluble proteins and of organelles under stress conditions. It involves the formation of double-membrane vacuoles which seize portions of cytoplasm transporting them to the lysosomes. The macroautophagy consists of several steps: initially an membranous organelle, enveloping a portion of cytoplasm and then shutting form a vacuole, usually delimited by two or more membranes, called autophagosome [13]. Numerous membranes appear involved in the origin of this vacuole, in particular the membranes of the endoplasmic reticulum (ER) and Golgi cisternae. It is proposed that in some cases the autophagosomes may originate from a pre-existing structure called phagophore [14]: it would represent a membranous structure condensed, localized in the cytosol and rich of osmolipidic material. In a second step the autophagosome acquires lysosomal membrane proteins as LGP10 and the ATPase proton pump, responsible for acidification of the vacuole [15]. Use of nocodazole is a possible cause of the accumulation of autophagosomes acids, but devoid of hydrolytic activity, morphologically
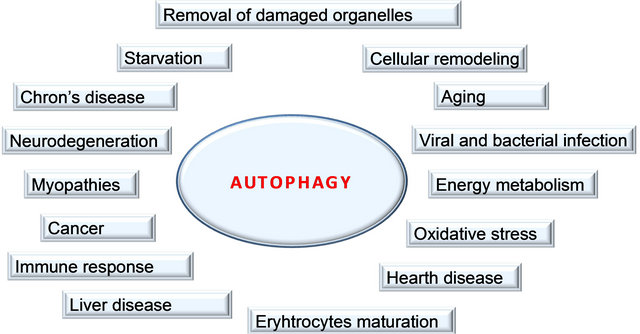
Macroautophagy is extensively involved in cellular homeostasis. The initial sequestering compartment, the phagophore, expands into the doublemembrane autophagosome. Fusion with an endosome generates the singlemembrane amphisome, which subsequently fuses with a lysosome. The degraded cytoplasm is released back into the cytosol through permeases. Some of the physiological connections between macroautophagy and human health and disease are indicated by the surrounding terms
Figure 2. Functions of autophagy.
indistinguishable from autophagosomes [16]. It is believed these vacuoles, called late autophagosomes, are formed from the merger of autophagosomes with vesicles derived both from the Golgi apparatus and from late endosomes. Other studies have identified the anphisome as autophagic vacuoles which are formed subsequently to initial autophagosomes [17]. Anphisome is a structure capable of receiving material both from endocytosis and from autophagy suggesting that it is an autophagic vacuole with auto functional properties [17]. In third step the autophagic anphysoma is fused with lysosomes. Fusion brings the auto phagocytosed material in contact with lysosomal enzymes, causing intralysosomal degradation. The volume of the vacuole is reduced and the final appearance is that of an electro-dense structure composed of amorphous material; at this stage the vacuole is defined dense body [18]. Once degraded simple molecular substances contained in the vacuole are absorbed or used by cells or available to the body. The undigested material is usually discharged from the cells by exocytosis, or remains inside the vacuole going to acquire with the time the biochemical characteristics of the pigments by wear or lipofuscin [19] (Figure 3).
Microautophagy is involved in assimilation and degradation of complete regions of the cytosol, including proteins and cytoplasmic organelles, directly by the lysosomes, without requiring the formation of intermediate autophagic vacuoles [20]. Lysosome, in fact, for invagination or protrusion, surrounds the cytoplasm and then closes itself to form an interior vesicle that contains the material to degraded. The microautophagy traditionally has been regarded as a form of active autophagy that ensures the turnover protein in long half-life in basal conditions, but recent studies [20] believe that it is also responsible, in part, of the degradation of peroxisomes.
Many types of cells can degrade cytosolic proteins
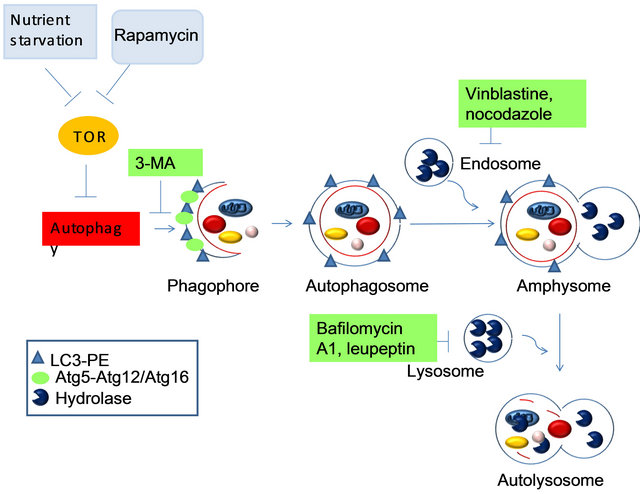
The markers that are present at each morphological step are indicated in the legend, as there are several known inhibitors and the steps at which they are thought to act. Caution must be used in interpreting the results obtained using all of these inhibitors, due to their pleiotropic effects. 3-MA, 3-methyladenine; LC3, microtubule-associated-protein light-chain 3; PE, phosphatidylethanolamine.
Figure 3. The known steps of induction, execution and maturation of autophagosomes and autolysosomes.
through chaperone-mediated autophagy (CMA) [21]. Distinctive characteristic of CMA is its selectivity for a particular group of cytoplasmic proteins and these substrates are directly translocated across the lysosomal membrane without requiring the formation of vacuoles or the deformation of the membrane. All substrates of CMA contain a sequence biochemically related to the pentapeptide KFERQ. This motif is recognized by chaperonins, including the heat shock protein of 70 kDa (hsp70) [22], that the target proteins bind. The complex chaperonin-protein interacts with the receptor LAMP-2a at the lysosomal membrane and the protein is pulled into the lysosomal lumen with the help of lysosomal hsc70 (lys-hsp70). Levels of LAMP-2a, which are in part regulated by changes in its degradation [23], may be important in the modulation of CMA. During stress conditions, such as the lack of nutrients, the activity of CMA increases when macroautophagy, which is also activated, begins to decrease [21]. This sequential activation of first a non-selective and then a selective autophagy pathway may be intended to avoid the degradation of essential cellular components after extended fasting [24] (Figure 4).
3. MECHANISMS OF AUTOPHAGY ACTIVATION: AUTOPHAGIC VESICLES ASSEMBLY
The molecular mechanism of autophagy is still unknown. Some years ago, genetic studies on yeast Saccharomyces cerevisiae have led to the identification of genes, in part conserved from yeast to humans, involved in autophagy, these genes were termed ATG genes (autophagy-related gene) [25]. Under favorable growth conditions autophagic degradation in yeast is inhibited by a mechanism that involves the activation of protein kinase mammalian target of rapamycin (mTOR) [26]. Activated mTOR appears to cause directly or indirectly hyperphosphorylation of Atg13. Hyperphosphorylated Atg13 has a lower affinity for the kinase Atg1 and the reduced interaction between Atg13 and Atg1 can inhibit autophagy [27]. Atg1 and Atg13 are part of a larger complex compounded also from other molecules, and the function of this complex may be required to increase the size of the autophagosome [28].
In yeast cells, the autophagosome seems to be generated from a membranous structure of unknown nature called preautophagosomal structure (PAS) [29], for the addition of other membranous components.
Many Atg gene products seem to be present in training and in elongation of the PAS. In yeast, it has been suggested that a complex called I formed by certain gene products of Atg and PI3K, to play a role in the initial formation of the autophagosomal membrane. In mammalian cells, in contrast to the yeast, there are 3 isotypes of PI3K. The isotype I of PI3K is an inhibitor of autophagy that involves the activation of the mTOR (counterpart of TOR). The isotype III of PI3K, a functional homologue of Vsp34 of yeast, is instead an enzyme constitutive of the autophagy and plays a crucial role in the early steps in the formation of the autophagosome in
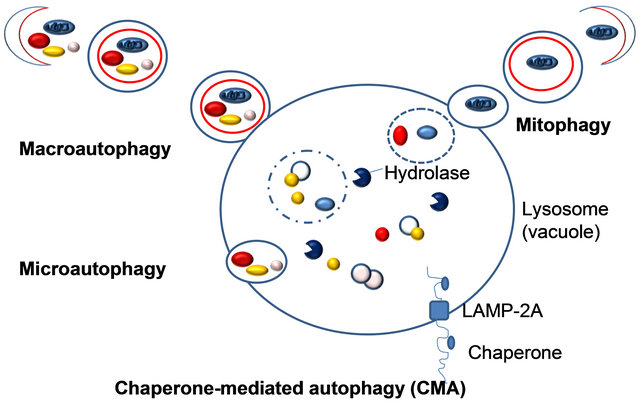
Three fundamentally different modes of autophagy are macroautophagy, microautophagy, and chaperone-mediated autophagy. Depending on the specificity of the cargos, autophagy can be a selective or a nonselective process. During nonselective autophagy, a portion of the cytoplasm is sequestered into a double-membrane autophagosome, which then fuses with the lysosome/vacuole. In contrast, the specific degradation of peroxisomes in certain conditions can be achieved by either a macroor microautophagylike mode, termed macropexophagy and micropexophagy, respectively. Piecemeal microautophagy of the nucleus allows the degradation of a portion of the nucleus. The specific degradation of mitochondria, termed mitophagy also takes place. A biosynthetic cytoplasm to vacuole targeting (Cvt) pathway in yeast also shares similar morphological features. Note that this schematic illustrates aspects of autophagy in both yeast cells and higher eukaryotes.
Figure 4. Different types of autophagy.
mammalian cells [30]. The complex I can generate phosphatidylinositol-3 phosphate in the membranes of the PAS and facilitate the recruitment of proteic components useful in the formation of the membrane of the vacuole.
In mammalian cells the isotype III of PI3K forms a complex with the protein Beclin-1, a functional homologue of Atg6 in yeast. It has been suggested this complex may play a role in the recruitment of membranes from various structures (ER, trans-Golgi, mitochondria) for the elongation of the autophagosomal membrane [31].
In the elongation phase of the membrane that will circumscribe the autophagosome, the complex Atg12- Atg5-Atg16 and the complex Atg8-phospholipid phosphatidylethanolamine (PE), two systems of conjugation similar to ubiquitin and that work in a coordinated manner [32], are activated by PI3K in yeast.
The complex Atg12-Atg5-Atg16 is the first to begin working during the expansion phase of the vacuole, forming a lining around the vacuole in formation and controlling its curvature. This conjugation system is conserved in mammalian cells where it plays a role in the formation of autophagic membranes [33].
The complex Atg8-PE, instead, mediates the expansion of the vesicle acting as a structural component. In mammalian cells different homologues of the Atg8 may be involved in the conjugation reaction [34]. One of these homologues, the microtubule Associated Protein 1 Light Chain 3 (MAP1-LC3), is involved in the formation of the autophagosome after a process similar to that which occurs in the Atg8 of yeast [35].
LC3 interacts with the membrane of the autophagosome in Atg5-dependent way and remains on the membrane after the dissociation of the complex Atg12-Atg5. For this reason LC3 can be used as a marker of the autophagosome in mammalian cells. So that the two complexes can localize on the PAS, it is requires the participation of Atg9 [36]. In yeast Atg9 is constantly exchanged between the PAS and the mitochondria. Once completed the formation of the vacuole the coat proteins, except Atg8-PE complexes, oriented toward the lumen, are dissociated from the vacuole (Figure 5).
4. PATHWAYS REGULATING AUTOPHAGIC PROCESSES (mTOR AND S6 KINASE PATHWAY)
mTOR (mammalian target of Rapamycin) [37] is a molecule of 290 kDa, so called because it is inhibited by the macrolide rapamycin, an antibiotic produced by the bacterium Streptomyces hygroscopicus. Rapamycin and its derivatives have a unique mechanism of action that involves interaction with a small protein molecule of 12 kDa, known as FKBP12 (FK506-binding protein) to form a complex that binds mTOR, inhibiting it. mTOR is a
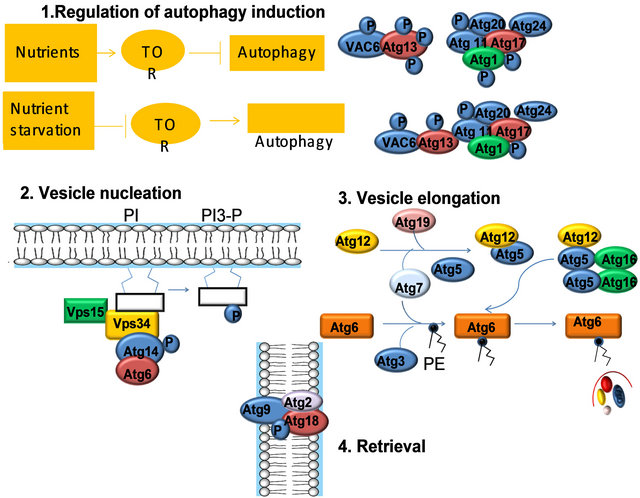
1) Regulation of induction: In yeast, the Tor kinase and its effectors regulate the induction of autophagy. UNC-51 is the C. elegans Atg1 ortholog. 2) Vesicle nucleation requires a lipid kinase complex, which includes the class III phosphatidylinositol 3-kinase (PI3K), Vps34 (in C. elegans LET-512). In yeast, Vps34 activation depends on its binding partners, Atg6, Atg14, and Vps15. In C. elegans, the ATG6 ortholog is bec-1, however, orthologs to ATG14 and VPS15 appear to be missing in the C. elegans genome. 3) Two novel ubiquitin-like conjugation pathways: the Atg12 conjugation system (Atg5, Atg12, and Atg16), and the Atg8 lipidation system (Atg8, Atg3, and Atg7) mediate vesicle expansion, and vesicle completion. 4) The retrieval of the integral membrane protein ATG-9 from the phagophore assembly site (PAS) involves ATG-2 and ATG-18, two interacting peripheral proteins.
Figure 5. Mechanism of autophagy activation.
serine/threonine protein kinase, consists of a catalytic kinase domain, a binding site for the complex FKBP12- rapamycin and a putative auto-inhibitory domain (negative regulatory domain).
In eukaryotic cells, mTOR forms two distinct multimeric complexes, mTORC1, rapamycin-sensitive, and mTORC2, which is not susceptible to inactivation in the short term by the macrolide. The complex mTORC1 is responsible for the regulation of protein synthesis and is composed of mTOR, raptor (regulatory associated protein of mTOR) and GβL [38].
Raptor is a large molecule of 150 kDa, which establishes multiple interactions with mTOR and with its substrates; GβL, instead, is a protein of 36 kDa, which binds the kinase domain of mTOR, positively regulating the catalytic activity [38]. The complex mTORC2 consists of mTOR, GβL and of a protein of about 200 kDa protein, known as rictor (rapamycin-insensitive companion of mTOR). The physiological function of mTORC2 is still not entirely clear, however to date the most accepted hypothesis is that this complex forms the organization of cytoskeleton actin [39].
The mTOR kinase promotes the synthesis of new protein molecules through phosphorylation of two different substrates: 4E-BP1 (initiation factor 4E binding protein), and p70S6k. 4EBP1 protein, also known as PHAS-1 (type 1 protein phosphorylated by heat and acid-stable), binds the translation initiation factor eIF4E making it inactive; as a result of phosphorylation by mTOR, 4EBP1 dissociates itself from the factor eIF4E that, therefore, can start the translation process [40]. p70S6k protein (70 kDa) is a member of the AGC kinases family, which also belong PKC, PKA, Akt, PDK1.
Once activated, the p70S6k kinase phosphorylates the ribosomal protein S6 resulting in transcription of the group of the m-RNA 5’-TOP (Terminal Oligo-Pyrimidine) which encode elongation factors necessary for protein synthesis [41].
The activity of the protein mTOR and its target is modulated by numerous stimuli, such as growth factors (IGF-1), nutrients, environmental stress (hypoxia), hormones (insulin, T3, GH, testosterone) and second messengers (IP3, Ca2+ ). The binding of the growth factor IGF-1 to its receptor (IGFR) triggers a intracellular enzymatic cascade along which it has the activation, by phosphorylation, of protein PI3K (phosphatidylinositol 3-kinase), which leads to the formation of IP3 (phosphatidylinositol-3,4,5-triphosphate). These, in turn, provides a binding site for the protein to the membrane Akt or PKB (Protein kinase B), which is then phosphorylated by the kinase PDK-1 and PDK-2 sequentially. After being activated, the protein Akt can phosphorylate a variety of substrates, including proteins that mediate protein synthesis, gene transcription and cell proliferation.
It’s been suggested that Akt phosphorylating TSC2 moves away it from the membrane where Rheb is localized and addresses it in the cytoplasm by binding to cytoplasmic proteins 14-3-3. TSC2 has activity that accelerates intrinsic GTPase of Rheb so it functionally inactive Rheb increasing its bound form to GDP. The move away from the membranes of TSC2 mediated by Akt, inhibits the interaction with Rheb which is normally located on the membranes through its farnesylic tail. All this leads to the activation of Rheb which in turn through the steric interaction actives mTOR pathway downstream. TSC2 can be phosphorylated on serine also by AMP kinase and Erk, but the functional role of these phosphorylations has not yet been fully defined. It is believed that the aminoacidic and insulinic adjustment may act synergistically in the control of autophagy. It seems that the signal, both of the insulin and the amino acids, can converge on the mTOR pathway [42]. At the moment, it seems clear that amino acids determine the activation of the mTOR: the enzyme is instead inhibited by conditions of lack of energy as the activation of the enzyme AMP kinase due to a decrease in the ratio ATP/ AMP. It is important to understand, however, as the amino acids signal their presence to the cell: some researchers have identified a complex localized on the cell surface that would bind itself with the leucine and they have resulted in the inhibition of autophagy with a globular peptide that binds 8 residues of leucine and not permeate the plasma membrane [43]. These results in favor of a receptor on the plasma membrane, however, have not been confirmed. In another hypothetical mechanism, the cells respond to the aminoacylation of tRNA; in fact it has been shown, in yeast, that free tRNA, which doesn’t binds amino acids, prevents the phosphorylation of p70S6k mediated by activated TOR [44]. If this was not the mechanism, since inhibition of amino acid transport system markedly inhibits the phosphorylation of p70S6k it’s favored the hypothesis that amino acids exert their effects once inside the cell [44].
As it is known, instead, for insulin there is a specific receptor on the plasma membrane (IR): the hormonereceptor bond results in an autophosphorylation of IR on the cytoplasmic side which in turn catalyzes the phosphorylation of the IR substrate (IRS). The IRS so activated probably activates PI3K [45]. Studies conducted by Brazil and Hemmings (2001) have shown that inhibition of the autophagic process mediated by insulin [46] may depend on the activation of class I PI3K, which converts PI4P and PI (4,5) P2 into PI (3,4) P2 and PI (3,4,5) P3 which are capable of binding the amino acid domains of the enzyme AKT and its activator PDK1. PDK1 phosphorylates other kinases including mTOR and p70S6k (Figure 6).
5. AUTOPHAGY IN CANCER: GOOD, BAD, OR BOTH?
Autophagy is a complex phenomenon that affects not only cancer but other diseases and studies on this subject, with its publications, are rapidly increasing.
Autophagy is defined as a genetically programmed evolutionarily conserved process that degrades longlived cellular proteins and organelles. Its role is important in the response to environmental stimuli, in the tumoral processes, in bacterial and viral infections, cardiovascular disease, and neurodegenerative disorders Countless debates have highlighted the importance of autophagy in cancer as in response to environmental stimuli. To this purpose, in fact, it was observed that autophagy allows a cell to respond to changes environmental conditions, such as nutrient deprivation, degrading proteins and organelles and thus obtaining amino acids, fatty acids and nucleotides. Thus, autophagy serves a protective role, allowing cells to survive during nutrient deprivation. Further evidence for the importance of autophagy in protecting against nutritional stress comes from studies where tumor cells are deprived of growth/survival factors, leading to an increase in autophagy that prevents the cells from dying, by inhibiting apoptosis [47]. In a tumor, this may mean that autophagy keeps tumor cells alive, when limited angiogenesis leads to nutrient deprivation and hypoxia. Therefore, we would expect that increased autophagy
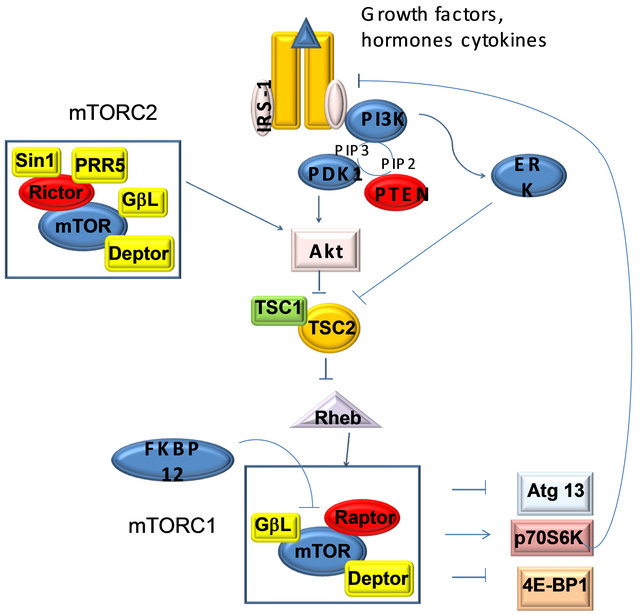
The mammalian target of rapamycin (mTOR) is an atypical serine/threonine kinase that is present in two distinct complexes. mTOR complex 1 (mTORC1) is composed of mTOR, Raptor, GβL (mLST8), and Deptor and is partially inhibited by rapamycin. Growth factors and hormones (e.g. insulin) signal to mTORC1 via Akt, which inactivates TSC2 to prevent inhibition of mTORC1. Alternatively, low ATP levels lead to the AMPKdependent activation of TSC2 to reduce mTORC1 signaling. Amino acid availability is signaled to mTORC1 via a pathway involving the Rag proteins. Active mTORC1 has a number of downstream biological effects including translation of mRNA via the phosphorylation of downstream targets (4E-BP1 and p70 S6 Kinase), suppression of autophagy, ribosome biogenesis, and activation of transcription leading to mitochondrial metabolism or adipogenesis. The mTOR complex 2 (mTORC2) is composed of mTOR, Rictor, GβL, Sin1, PRR5/Protor-1, and Deptor and promotes cellular survival by activating Akt. mTORC2 also regulates cytoskeletal dynamics by activating PKCα and regulates ion transport and growth via SGK1 phosphorylation. Aberrant mTOR signaling is involved in many disease states including cancer, cardiovascular disease, and metabolic disorders.
Figure 6. mTOR pathway.
would promote the growth of solid tumors, whereas reduced autophagy might provide a useful way to limit tumor growth. In stark contrast to this potential cancerpromoting effect of autophagy, numerous lines of evidence indicate an anticancer role for autophagy. The autophagy gene Beclin-1 (the mammalian counterpart of the yeast Atg 6 gene), which is part of a type III phosphatidylinositol 3-kinase (PI3k) complex required for autophagic vesicle formation, is a haploinsufficient tumor suppressor in mice and is monoallelically lost in human breast, ovarian, and other tumors. Moreover, p53 and PTEN, two of the most commonly mutated tumor suppressor genes, both induce autophagy. The oncogenic protein Bcl-2 directly interacts with Beclin-1 to inhibit autophagy [48].
Because oncogenes can inhibit autophagy and tumor suppressors induce autophagy whereas that autophagy regulator is itself a tumor suppressor, these data suggest that autophagy serves an anticancer role. The mechanism through which autophagy inhibits tumor development is unclear.
Autophagy may, also, limit tumor cell growth or reduce mutagenesis or other damage caused by reactive oxygen species by removal of damaged mitochondria and other organelles. Alternatively, autophagy may kill developing tumor cells. Taken together, these data suggest that autophagy can both stimulate and prevent cancer depending on the context. To further test this idea, it will be interesting to examine mice with defects in other Atg genes, to see if they too have a cancer predisposition phenotype similar to the Beclin 1+/− mice.
Contrasting roles for autophagy during cancer development, progression, and treatment. Stimulation of autophagy suppresses cancer development, but may promote tumor growth and survival under conditions of nutrient deprivation in poorly angiogenic tumors. Autophagy may protect tumor cells from undergoing apoptosis in response to treatment with anticancer agents. But may be a mechanism of tumor cell death in cells with defective apoptotic machinery. This fact should encourage us to better understand the nuances of how autophagy affects tumor development, progression and treatment, so that we can use this information to prevent and more effectively treat cancer (Figure 7).
6. PPARs (PEROXISOME POLIFERATOR-ACTIVATED RECEPTORS)
Peroxisome proliferator activated receptors (PPARs), are transducer proteins belonging to the nuclear receptor superfamily. To date, three major types of PPAR, encoded by separate genes, have been identified in the vertebrates; they are PPAR-α (NR1C1), PPAR-δ/β (NR1C2) and PPAR-γ (NR1C3). The three known PPAR subtypes show different tissue distributions. All three PPAR isoforms possess similar structural and functional features [49], vary in tissue distribution, in fact PPAR-α is predominantly expressed in tissues that demonstrate high catabolism for fatty acids such as liver, heart, kidney and muscles; PPAR-γ is most highly expressed in white adipose tissue and PPAR-δ/β is ubiquitously expressed and so far no specific role has been described for this isoform [50]. The exact functions of PPAR-β remain an enigma.
However, it seems involved in the regulation of energy expenditure as well as in the metabolism of glucose and of the lipids.
6.1. Structural Features of PPARs
PPARs are composed of four functional domains called A/B, C, D and E/F (Figure 8). The A/B domain is located at N-terminus and presents a AF-1 region that is responsible for independent activation by ligand of the receptor; the domain C is implicated in DNA binding; domain D is the docking region for cofactors and domain
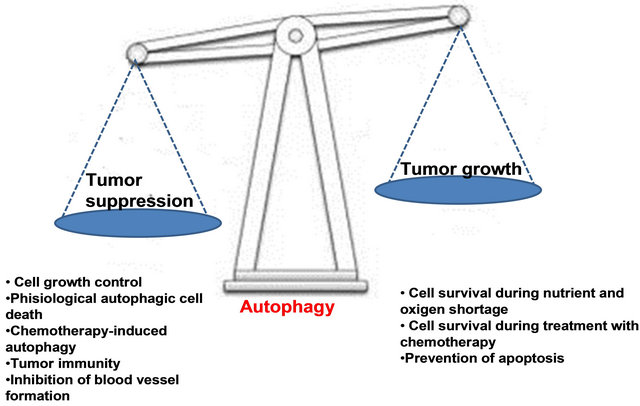
Experimental evidence supports a role for autophagy in both cancer development and suppression.
Figure 7. Autophagy in cancer: good, bad, or both?
E/F is responsible for the ligand-dependent activation containing AF-2 which promotes the recruitment of cofactors required for the gene transcription. Particularly C domain, the DNA binding domain, binds a nucleotide sequence, PPAR response element (PPRE) located in the promoter region of target genes. Before binding to PPRE, PPAR binds RXR (retinoid X receptors a subfamily of molecules within the nuclear receptor superfamily that are activated by 9-cis retinoic acid) to form a heterodimer that can activate or repress gene transcription depending on its interaction with coactivators or corepressors factors respectively [49].
γ isoform is the best known and the heterodimer inactive PPAR-γ/RXR is localized in cytoplasm and it is strongly tied to corepressors that prevent it from binding to DNA. Following binding with the ligand, the heterodimer is activated and the corepressor is detached; in this way the PPARγ/RXR is free to enter the nucleus and interact at the level of PPRE in the promoter region of the target gene, causing activation or repression of the gene [51]. The transcriptional interaction of the heterodimer with the response element in the promoter region requires the recruitment of coactivator proteins (Figure 9).
6.2. PPAR-γ
In humans, the gene PPAR-γ is located on chromosome 3 in position 3p25. In humans, four different subtypes of PPAR-γ mRNA have been identified (-γ1, γ2-, -γ3, and γ4-) which are transcribed from four different promoters [52] (Figure 10).
PPAR-γ is mainly expressed in adipose tissue, plays an

Schematic representation of the functional domains of PPARs. PPARs are composed of four distinct functional regions. The A/B domain located at N-terminal with AF-1 is responsible for phosphorylation, the domain C is implicated in DNA binding, domain D is the docking region for cofactors and domain E/F is the ligand specific domain, containing AF-2, which promotes the recruitment of cofactors required for the gene transcription.
Figure 8. Functional domains of PPARs.
active role in many biological processes such as adipocyte differentiation, lipid metabolism and glucose homeostasis. In recent years, also an extensive involvement in many aspects of inflammation it has been attributed to it. In recent years, various ligands of PPAR-γ have been studied, natural and synthetic, such as PPAR-γ agonists, PPAR-γ partial agonists, and (PPAR-α/γ dual agonist).
The known endogenous agonists of PPAR-γ are polyunsaturated fatty acids (linoleic acid and arachidonic acid), and oxidized fatty acids (9- and 13-Hode, 15- HETE) (Figure 11). The synthetic agonists of PPAR-γ are considered determinants of adipocyte differentiation as well as potential antidiabetic drugs [53]. TZDs compounds are used clinically as insulin sensitizers. Many drugs belonging to the TZD class shows high selectivity for PPAR-γ and little or no activity toward other sub-α and-β. For example, troglitazone (Rezulin) is highly specific affinity for the PPAR-γ ligand.
Thiazolidinediones (TZDs) are selective ligands for the nuclear transcription factor peroxisome proliferatoractivated receptor PPAR-γ [54]. These TZDs improve insulin sensitivity by transcriptional activation of insulinsensitive genes involved in glucose homeostasis, fatty
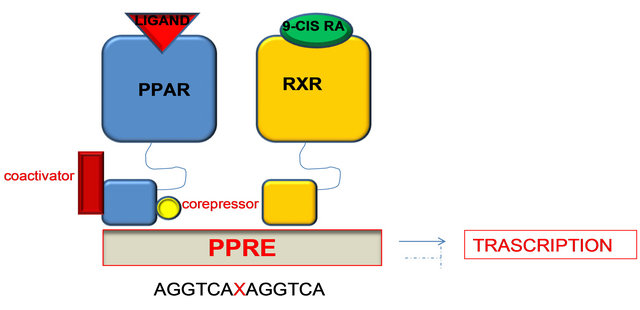
Figure 9. PPAR-γ/RXR inactive heterodimer is localized in the cytoplasm. Following binding ligand, the heterodimer is activated and the corepressor is detached; in this way the PPARγ/RXR is free to enter the nucleus and interact at the level of PPRE in the promoter region of target genes, resulting in activation or repression of the gene.
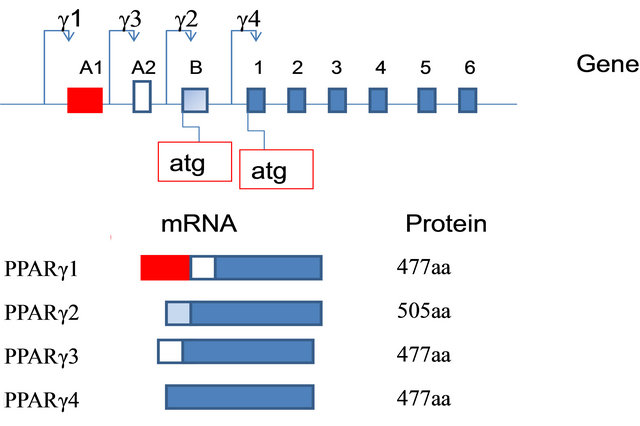
Figure 10. Diagram of the genomic structure of the PPARG gene and protein structures of the two PPAR-γ isoforms. Genomic and promoter organization, corresponding mRNAisoforms and polymorphisms of the human PPARγ gene. Exons (A1, A2, B, and 1 - 6) are represented by boxes, introns by a line. Dashed exons mean untranslated exons. P means promoter. The 4 polymorphisms are indicated with an arrow.
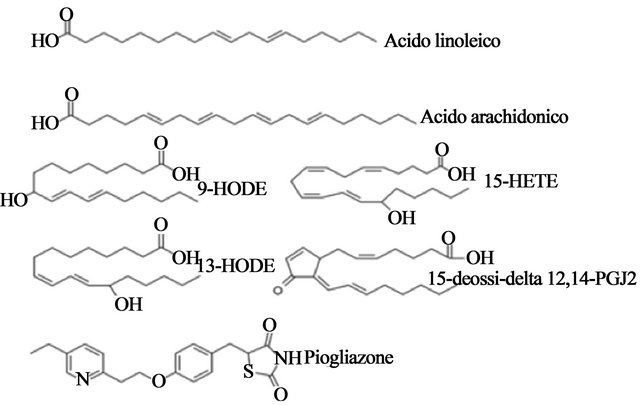
Figure 11. Chemical structures of representatives natural (linoleic acid, arachidonic acid and related compounds PG) and synthetic ligands (pioglitazone, the anti-diabetic drug TZD) of PPAR-γ.
acid metabolism, and triacylglycerol storage in adipocytes [55].
7. RELATIONSHIP BETWEEN PPAR GAMMA AND THE ADJUSTMENT OF AUTOPHAGY
The class of oral antidiabetic agents, characterized by the presence of a thiazolidinedione group (TZD) (rosiglitazone, pioglitazone, troglitazone), act as ligands of PPAR- γ. These drugs have shown considerable modulatory activity on immune and inflammatory processes. The treatment with the ligands of PPAR-γ inhibits the production of proinflammatory molecules such as TNF-α, IL-1-β and IL-6 by a mechanism that involves the interaction with transcription factors such as NF-kB, AP-1 and STAT-1.
Activation of PPAR-γ is associated to cell cycle exit and to expression of genes that promote cell differentiation. The antiproliferative and differentiative role of PPAR-γ was confirmed in cells derived from solid tumors of the colon, prostate, pancreas and breast, and although definitive data on the antitumor role of activation of PPAR-γ in vivo lacking, the expression of this recaptor in different tumor types justifies interest for potential use in oncology. PPAR ligands induce autophagy, but it is not yet clear whether this biological process takes place exclusively by activating PPAR-γ.
Troglitazone is a synthetic ligand of peroxisome proliferators activated receptor-γ (PPAR-γ) and induces apoptosis in a variety of malignant cells. However, the underlying mechanism of its regulatory role in autophagy remains largely unknown. Recent data show that troglitazone increases AMP-activated protein kinase-α (AMPKα) phosphorylation, reduces p70S6 kinase phosphorylation and stimulates autophagy, promoting autophagosome formation, that is independent of EGFR expression and transactivation. EGFR can serve as a critical transducer of intracellular signals in response to the ligands of PPAR-γ and the activated signaling pathways, may contribute to autophagy activation. Recently, it was shown that PPAR-γ may not be involved in the activation of the autophagic pathway upon TZ challenge; however, TZinduced autophagy is probably not caused by transactivation of EGFR. In fact, in cells which lack the EGFR expression or cells expressing EGFR but had been treated with AG1478, a specific EGFR kinase inhibitor, troglitazone was still able to induce autophagy. Whereas, blockage of PPAR activity by the irreversible antagonist or by overexpressing dominate-negative PPAR-γ did not affect LC3-II, a specific marker of autophagy. Several studies have attempted to define the molecular mechanisms underlying the antitumor activity of thiazolidin-ediones; a mechanism could be represented by the induction of autophagy EGFR and PPAR-γ-independent and AMPKdependent [56].
8. THE ACTIVATION OF PPAR-γ ANTAGONIZES THE RESISTANCE OF PROLIFERATIVE INHIBITION INDUCED BY INTERFERON-β IN TREATED PANCREATIC CANCER CELLS Another mechanism for the antitumor activity of thiazolidinediones may be represented by the activation of PPAR-γ and by induction of autophagy PPAR-γ-dependent. We demonstrated, for the first time, that the activation of a PPAR-γ agonist potentiates the anti-cancer effects of IFN-β through the induction of cell cycle perturbations and the occurrence of autophagy cell death in pancreatic cancer cells. In fact, PPAR-γ activation by troglitazone can counteract STAT-3-dependent escape pathways following IFN-β-induced proliferative inhibition. On these bases, we have evaluated the effects of the pharmacological interaction between IFN-β and troglitazone (TGZ), agonist of PPAR-γ. The drugs used in combination showed a strong synergism in inducing proliferative inhibition of pancreatic adenocarcinoma.
The combination between IFN-β and TGZ was not able to induce apoptosis, as evaluated by either Annexin V or propidium iodide labelling at FACS analysis. On these bases, we have studied the alternative autophagy cell death mechanism. Untreated control cells, examined by TEM after ultrathin sectioning showed their typical morphology, characterized by a compact cytoplasm with well preserved organelles and no sign of cell damage. After treatment with INF-β alone, cells did not exhibit significant changes when compared to the untreated control; the only difference was the higher number of mitochondria in treated cells. In TGZ-treated samples, many cells showed the presence of large vesicular bodies containing organelles and material of variable structure inside a well preserved cytoplasmic matrix. Such vesicles appeared to be very similar to autophagosomes, typical structures present in autophagic cells, suggesting the induction of autophagy by TGZ. Such phenomenon appeared to be extremely potentiated when the treatment with TGZ was carried out in association with INF-β. In fact, most cells treated with the combination INF-β and TGZ displayed large areas of the cytoplasm occupied by complex vacuolar structures surrounded by a well defined membrane and containing cellular components.
We have investigated the molecular mechanisms of autophagy, studying the interaction between two molecules involved in this process: Beclin-1 and Bcl-2. It is known that Bcl-2, interacting with Beclin-1, inhibits beclin-1-dependent autophagy. Beclin-1 is a tumor suppressor gene, involved in the autophagy. The combined treatment reduces the levels of the complex Beclin-1/ Bcl-2 respectively at 3, 6 and 24 h from start of treatment.
Moreover, we have found that the combination between IFN-β and TGZ decreased 4EBP1 phosphorylation already after 6 h, and more significantly, after 24 h; while IFN-β plus TGZ reduced eIF4E phosphorylation after 3 and 6 h, while it increased at 24 h. The inhibition of eIF4E phosphorylation, occurring at earlier time points, seemed to be independent from the interaction with 4EBP1. On this basis eIF4E is phosphorylated by the MAPK activated protein kinase (MNK1) that, in turn, is activated by Erk-1 and 2. Therefore, in order to explain the modulation of eIF4E phosphorylation induced by the different treatments, we have evaluated MNK1 expression and phosphorylation, which, upon activation by mitogenic and/or stress stimuli mediated by MAPK and p38 MAPK, phosphorylates eIF4E. We have found that the combination determined an about 60% reduction of MNK1 phosphorylation at both 3 h and 6 h, while its phosphorylation increased of about 60% at 24 h. Single agents had poor effects on the phosphorylation of MNK1 and all the treatments did not induce any significant changes in total MNK1 expression. Therefore, the combination IFN-β plus TGZ probably decreased eIF4E phosphorylation by inhibiting MNK1 activation. In conclusion, this preclinical work represents the first demonstration of a synergistic interaction between IFN-β and PPAR-y agonists in the induction of autophagy in pancreatic cancer and may represent a motivation for further investigation [57].
9. AUTOPHAGY AND CANCER PROGNOSIS
Since it is clear that autophagy has a role in the development of the tumor, it is natural to assume the selectiveity of autophagy as a realistic perspective for the therapy cancer. An immediate problem concerns the ability to target selectively the oncogenic role, played by autophagy in cancer cells subjected to stress, while not inhibiting its role, in tumor suppression. It may be that autophagy behave as oncogene in certain stages of tumor development, while in other phases, it acts as a tumor suppressive. The issue, however, can be even more complicated because the effects carried out by autophagy as an oncogene and those carried out as a tumor suppressor, can co-exist not only within a patient, but also within each individual tumor. Do not forget also that autophagy has many beneficial roles in our normal tissues and in an ideal world, it would be better if autophagy in these contexts was not affected by systemic therapy, which is finalized to strike the malignant disease. The identification of cellular signaling pathways that regulate autophagy selectively in response to specific stimuli, could be the key to the selective targeting of autophagy in human disease. It has been tried to tackle this point respect to cancer through the search of signaling pathways that regulate autophagy in response to hypoxia, a common condition in many solid tumors, but for the most part absent in normal tissue. These results show the importance of autophagic action in a condition such as that due to cancer, in which autophagy itself may assist in a way more targeted to a therapeutic gain.
10. CONCLUSION
The role that autophagy has in the pathogenesis of cancer remains controversial. The triggering of autophagic processes in tumor cells has been correlated recently with the resistance of tumors to cytostatic agents or directed against specific molecular targets. However it is possible that some agents such as agonists of PPAR-γ may act at least in part through the induction of autophagy in human cancer cells. Finally data are emerging from the literature that confirm the possibility of the cytokines such as interferon alpha to induce escape mechanisms from proliferative inhibition and from cell death through the triggering of autophagic processes in human tumor cells. In this context, the use of agents such as agonists of PPAR-γ may be useful in enhancing the antitumor activity of these cytokines antagonizing the protective antiautophagic effect. Therefore, the study of alternative models of induction of cell death could be useful to the design of innovative strategies for the treatment of human cancers.
REFERENCES
- Clarke, P.G. and Clarke, S. (1996). Nineteenth century research on naturally occurring cell death and related phenomena. Anatomy and Embryology (Berl), 193, 81- 99. doi:10.1007/BF00214700
- Kerr, J.F., Wyllie, A.H. and Curie, A.R. (1972) Apoptosis:a basic biological phenomenon with wide ranging implications in tissue kinetics. British Journal of Cancer, 26, 239-257. doi:10.1038/bjc.1972.33
- Lockshin, R.A. and Zakery, Z. (1991) Programmed cell death and apoptosis. In: Tomei, L.D. and Cope, F.O., Eds., Apoptosis: The Molecular Basis of Cell Death, Cold Spring Harbor Laboratory Press, New York, 47-60.
- Thompson, C.B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science, 267, 1456-1462. doi:10.1126/science.7878464
- Schweiichel, J.-U. and Merker, H.-J. (1973) The morphology of various types of cell death in prenatal tissues. Teratology, 7, 253-266. doi:10.1002/tera.1420070306
- Moreira, M.E. and Barcinski, M.A. (2004) Apoptotic cell and phagocyte interplay: Recognition and consequences in different cell systems. Anais da Academia Brasileira Ciencias, 76, 93-115. doi:10.1590/S0001-37652004000100009
- Kanazawa, T., Taneike, I., Akaishi, R., Yoshizawa, F., Furuya, N., Fujimura, S. and Kadowaki, M. (2003) Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mtor in isolated rat hepatocytes. Journal of Biological Chemistry, 279, 8452-8459. doi:10.1074/jbc.M306337200
- Wang, C.W. and Klionsky, D.J. (2003) The molecular mechanism of autophagy. Molecular Medicine, 9, 65-76.
- Bergamini, E., Cavallini, G., Donati, A. and Gori, Z. (2003) The anti-ageing effects of caloric restriction may involve stimulation of macroautophagy and lysosomal degradation, and can be intensified pharmacologically. Biomedicine & Pharmacotherapy, 57, 203-208. doi:10.1016/S0753-3322(03)00048-9
- Meijer, A.J. and Codogno, P. (2004) Regulation and role of autophagy in mammalian cells. International Journal of Biochemistry & Cell Biology, 36, 2445-2462. doi:10.1016/j.biocel.2004.02.002
- Mizushima, N. (2004) Methods for monitoring autophagy. International Journal of Biochemistry & Cell Biology, 36, 2491-2502. doi:10.1016/j.biocel.2004.02.005
- Klionsky, D.J. (2005) The molecular machinery of autophagy: Unanswered questions. Journal of Cell Science, 118, 7-18. doi:10.1242/jcs.01620
- Ohsumi, Y. and Mizushima, N. (2004) Two ubiquitin-like conjugation systems essential for autophagy. Seminars in Cell & Developmental Biology, 15, 231-236. doi:10.1016/j.semcdb.2003.12.004
- Seglen, P.O. (1987) Lysosomes: Their role in protein breakdown. Academic Press, New York, 369-414.
- Reggiori, F. and Klionsky, D.J. (2005) Autophagosomes: Biogenesis from scratch? Current Opinion in Cell Biology, 17, 415-422. doi:10.1016/j.ceb.2005.06.007
- Dunn Jr., W.A. (1994) Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends in Cell Biology, 4, 139-143. doi:10.1016/0962-8924(94)90069-8
- Seglen, P.O. and Bohley, P. (1992) Autophagy and other vacuolar protein degradation mechanisms. Experientia, 48, 158-172. doi:10.1007/BF01923509
- Aplin, A., Jasionowski, T., Tuttle, D.L., Lenk, S.E., Dunn Jr., W.A. (1992) Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. Journal of Cellular Physiology, 152, 458-466. doi:10.1002/jcp.1041520304
- Mortimore, G.E., Poso, A.R. and Lardeux, B.R. (1989) Mechanism and regulation of protein degradation in liver. Diabetes/Metabolism Reviews, 5, 49-70. doi:10.1002/dmr.5610050105
- Glaumann, H., Ericsson, J.L. and Marzella, L. (1981) Mechanisms of intralysosomal degradation with special reference to autophagocytosis and heterophagocytosis of cell organelles. International Review of Cytology, 73, 149- 182. doi:10.1016/S0074-7696(08)61288-7
- Cuervo, A.M. (2004) Autophagy: Many pathways to the same end. Molecular and Cellular Biochemistry, 263, 55- 72. doi:10.1023/B:MCBI.0000041848.57020.57
- Majeski, A.E. and Dice, J.F. (2004) Mechanisms of chaperone-mediated autophagy. International Journal of Biochemistry & Cell Biology, 36, 2435-2444. doi:10.1016/j.biocel.2004.02.013
- Salvador, N., Aguado, C., Horst, M. and Knecht, E. (2000) Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. Journal of Biological Chemistry, 275, 27447-27456.
- Massey, A., Kiffin, R. and Cuervo, A.M. (2004) Pathophysiology of chaperone-mediated autophagy. International Journal of Biochemistry & Cell Biology, 36, 2420- 2434. doi:10.1016/j.biocel.2004.04.010
- Klionsky, D.J. (2002) Molecular machinery required for autophagy and the cytoplasm to vacuole targeting (cvt) pathway in S. cerevisiae. Current Opinion in Cell Biology, 14, 468-475. doi:10.1016/S0955-0674(02)00343-5
- Klionsky, D.J., Cregg, J.M., Dunn, W.A.J.R., Emr, S.D., Sakai, Y., Sandoval, I.V., Sibirny, A., Subramani, S., Thumm, M., Veenhuis, M. and Ohsumi, Y. (2003) A unified nomenclature for yeast autophagy-related genes. Developmental Cell, 5, 539-545.
- Schmelzle, T. and Hall, M.N. (2000) Tor, a central controller of cell growth. Cell, 103, 253-262. doi:10.1016/S0092-8674(00)00117-3
- Kamada, Y., Funakoshi, T., Shintani, T., Nagano, K., Ohsumi, M. and Ohsumi, Y. (2000) Tor-mediated induction of autophagy via an apg1 protein kinase complex. Journal of Cell Biology, 150, 1507-1513. doi:10.1083/jcb.150.6.1507
- Abeliovich, H., Zhang, C., Dunn Jr., W.A., Shokat, K.M. and Klionsky, D.J. (2003) Chemical genetic analysis of apg1 reveals a non-kinase role in the induction of autophagy. Molecular Biology of the Cell, 14, 477-490. doi:10.1091/mbc.E02-07-0413
- Arico, S., Petiot, A., Bauvy, C., Dubbelhuis, P.F., Meijer, A.J., Codogno, P. and Ogier-denis, E. (2001) The tumor suppressor pten positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase b pathway. Journal of Biological Chemistry, 276, 35243- 35246. doi:10.1074/jbc.C100319200
- Kihara, A., Noda, T., Ishihara, N. and Ohsumi, Y. (2001) Two distinct vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase y sorting in saccharomyces cerevisiae. Journal of Cell Biology, 152, 519-530. doi:10.1083/jcb.152.3.519
- Liang, XH., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H. and Levine, B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature, 402, 672-676. doi:10.1038/45257
- Tanida, I., Tanida-miyake, E., Komatsu, M., Ueno, T. and Kominami, E. (2002) Human apg3p/aut1p homologue is an authentic e2 enzyme for multiple substrates, gate-16, gabarap, and map-lc3, and facilitates the conjugation of hapg12p to hapg5p. Journal of Biological Chemistry, 277, 13739-13744. doi:10.1074/jbc.M200385200
- Mizushima, N., Yamamoto, A., Hatano, M., Kobayashi, Y., Kabeya, Y., Suzuki, K., Tokuhisa, T., Ohsumi, Y. and Yoshimori, T. (2001) Dissection of autophagosome formation using apg5-deficient mouse embryonic stem cells. Journal of Cell Biology, 152, 657-668. doi:10.1083/jcb.152.4.657
- Yorimitsu, T. and Klionsky, D.J. (2005) Autophagy: Molecular machinery for self-eating. Cell Death and Differentiation, 12, 1542-1552. doi:10.1038/sj.cdd.4401765
- Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., Kominami, E., Ohsumi, Y. and Yoshimori, T. (2000) Lc3, a mammalian homologue of yeast apg8p, is localized in autophagosome membranes after processing. EMBO Journal, 19, 5720-5728. doi:10.1093/emboj/19.21.5720
- Yoshimori, T. (2004) Autophagy: A regulated bulk degradation process inside cells. Biochemical and Biophysical Research Communications, 313, 453-458. doi:10.1016/j.bbrc.2003.07.023
- Bjornsti, M.A. and Houghton, P.J. (2004) The tor pathway: A target for cancer therapy. Nature Reviews Cancer, 4, 335-348. doi:10.1038/nrc1362
- Hara, K., Maruki, Y., Long, X., Yoshino, K., Oshiro, N., Hidayat, S., Tokunaga, C., Avruch, J. and Yonezawa, K. (2002) Raptor, a binding partner of target of rapamycin (tor), mediates tor action. Cell, 110, 177-189. doi:10.1016/S0092-8674(02)00833-4
- Wullschleger, S., Loewith, R., Oppliger, W. and Hall, M.N. (2005) Molecular organization of target of rapamycin complex 2. Journal of Biological Chemistry, 280, 30697-30704. doi:10.1074/jbc.M505553200
- Jacinto, E., Loewith, R., Schmidt, A., Lin, S, Rüegg, M.A., Hall, A. and Hall, M.N. (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology, 6, 1122-1128. doi:10.1038/ncb1183
- Hayashi, A.A. and Proud, C.G. (2007) The rapid activation of protein synthesis by growth hormone requires signaling through mTOR. American Journal of Physiology Endocrinology and Metabolism, 292, E1647-16455. doi:10.1152/ajpendo.00674.2006
- Conus, N.M., Hemmings, B.A. and Pearson, R.B. (1998) Differential regulation by calcium reveals distinct signaling requirements for the activation of Akt and p70S6k. The Journal of Biological Chemistry, 273, 4776-4782. doi:10.1074/jbc.273.8.4776
- Hardie, D.G. (2003) Minireview: The amp-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology, 144, 5179-5183. doi:10.1210/en.2003-0982
- Miotto, G., Venerando, R., Marin, O., Siliprandi, N. and Mortimore, G.E. (1994) Inhibition of macroautophagy and proteolysis in the isolated rat hepatocyte by a nontransportable derivative of the multiple antigen peptide leu8-lys4-lys2-lys-beta ala. The Journal of Biological Chemistry, 269, 25348-25353.
- Blommaart, E.F., Luiken, J.J.F.P. and Meijer, A.J. (1997) Autophagic proteolysis: Control and specificity. Histochemical Journal, 29, 365-385. doi:10.1023/A:1026486801018
- Mortimore, G.E. and Poso, A.R. (1987) Intracellular protein catabolism and its control during nutrient deprivation and supply. Annual Review of Nutrition, 7, 539-564. doi:10.1146/annurev.nu.07.070187.002543
- Boya, P., Gonzalez-Polo, R.A., Casares, N., et al. (2005) Inhibition of macroautophagy triggers apoptosis. Molecular and Cellular Biology, 25, 1025-1040. doi:10.1128/MCB.25.3.1025-1040.2005
- Kondo, Y., Kanzawa, T., Sawaya, R. and Kondo, S. (2005) The role of autophagy in cancer development and response to therapy. Nature Reviews Cancer, 5, 726-734. doi:10.1038/nrc1692
- Pattingre, S., Tassa, A., Qu, X., et al. (2005) Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell, 122, 927-939.
- Thorburn, J., Moore, F., Rao, A., et al. (2005) Selective inactivation of a FADD-dependent apoptosis and autophagy pathway in immortal epithelial cells. Molecular Biology of the Cell, 16, 1189-1199. doi:10.1091/mbc.E04-10-0906
- Trifillieff, A., Bench, A., Hanley, M., Bayley, D., Campbell, E. and Whittaker, P. (2003) PPAR-α and -γ but not -δ agonists inhibit airway inflammation in a murine model of asthma: In vitro evidence for an NF-κB-independent model. British Journal of Pharmacology, 139, 163-171. doi:10.1038/sj.bjp.0705232
- Xu, H.E., Lambert, M.H., Montana, V.G., Plunket, K.D., Moore, L.B., Collins, J.L., Oplinger, J.A., Kliewer, S.A., Gampe, Jr., R.T., McKee, D.D., Moore, J.T. and Willson, T.M. (2001) Structural determinant of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proceedings of the National Academy of Sciences USA, 98, 13919-13924. doi:10.1073/pnas.241410198
- Nagy, L., Tontonoz, P., Alvarez, J.G., Chen, H. and Evans, R.M. (1998) Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell, 93, 229-240. doi:10.1016/S0092-8674(00)81574-3
- Lehmann, J.M., Lenhard, J.M., Oliver, B.B., Ringold, G.M. and Kliewer, S.A. (1997) Peroxisome proliferatoractivated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. Journal of Biological Chemistry, 272, 3406-3410. doi:10.1074/jbc.272.6.3406
- Kletzien, R.F., Clarke, S.D. and Ulrich, R.G. (1992) Enhancement of adipocyte differentiation by an insulinsensitizing agent. Molecular Pharmacology, 41, 393-398.
- Vitale, G., Zappavigna, S., Marra, M., Dicitore, A., Meschini, S., Condello, M., Arancia, G., Castiglioni, S., Maroni, P., Bendinelli, P., Piccoletti, R., van Koetsveld, P.M., Cavagnini, F., Abbruzzese, A., Hofland, L.J. and Caraglia, M. (2011) The PPAR-γ agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-β treated pancreatic cancer cells. Biotechnology Advances, 30, 169-184.