Journal of Environmental Protection
Vol.07 No.03(2016), Article ID:63948,13 pages
10.4236/jep.2016.73037
Trace-Level Analysis of Hexavalent Chromium in Lake Sediment Samples Using Ion Chromatography Tandem Mass Spectrometry
Stefanie Mädler1,2, Fengrong Sun1, Cindy Tat1, Nadya Sudakova1, Peter Drouin1, Robert J. Tooley1, Eric J. Reiner1,3, Teresa A. Switzer1, Richard Dyer4, H. M. Skip Kingston5, Matt Pamuku6, Vasile I. Furdui1
1Laboratory Services Branch, Ontario Ministry of the Environment and Climate Change, Ontario, Canada
2Conservation Ontario, Ontario, Canada
3Department of Chemistry, University of Toronto, Ontario, Canada
4Ontario Geological Survey, Ontario Ministry of Northern Development and Mines, Ontario, Canada
5Department of Chemistry and Biochemistry, Duquesne University, Pittsburgh, USA
6Applied Isotope Technologies, Pittsburgh, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 18 January 2016; accepted 26 February 2016; published 29 February 2016
ABSTRACT
The analysis of hexavalent chromium, Cr(VI), in soil and sediment samples has been predominantly carried out in materials containing elevated levels. Reliable analysis of trace-level of Cr(VI) in sediment samples remains challenging. Cr(VI) analyses with multipoint calibration and speciated isotope dilution (SID) adapted from U.S. EPA method 6800 were used to measure lower- level Cr(VI) on an ion chromatograph coupled with a tandem mass spectrometer (IC-MS/MS). Lake sediment samples were collected from various locations in Northern Ontario and Cr(VI) was extracted using both alkaline digestion and ethylene diaminetetraacetic acid (EDTA) extraction. Certified reference materials were extracted and analyzed by IC-MS/MS and UV-VIS detection. The SID-MS approach allowed for the quantification of Cr(VI) in samples with concentration levels below 0.5 µg∙g−1 wet weight.
Keywords:
Trace-Level Analysis, Ion Chromatography, Tandem Mass Spectrometry, Chromium Speciation, Speciated Isotope Dilution

1. Introduction
Chromium has been heavily used in metallurgical, chemical and refractory industries, for example for the production of stainless steel, chrome-plating or leather tanning. Certain geological substrates can contribute chromium through weathering and surface and ground water flow, but atmospheric deposition and industrial waste waters are the most common and well-studied mode of movement of chromium into the aquatic environment. Association with particulate matter in waters causes deposition in sediments and poses an ecological and potentially human health risk [1] [2] .
In the environment, chromium exists predominantly in the oxidation states III (trivalent chromium) and VI (hexavalent chromium). Various processes, such as chemical and photochemical redox transformation, precipitation/dissolution and adsorption/desorption, control the presence and ratio of each species [3] . The environmental availability and fate of chromium depend strongly on the oxidation state and the geochemistry of the water/sediment system. Whereas Cr(III) tends to be bound to rather inert solid phases, such as sediment particles, with low uptake by biota, most Cr(VI) compounds are much more soluble and mobile [4] . Under oxic conditions, Cr(VI) dominates the equilibrium with low pH values favoring the presence of chromic acid (H2CrO4, pKa = −0.75) and hydrogen chromate (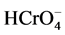 , pKa = 6.45). At physiological and high pH,
, pKa = 6.45). At physiological and high pH, 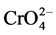 ions are the main species of Cr(VI) [5] . Anoxic conditions and the presence of reducing agents, such as iron or organic matter, tend to shift the equilibrium to Cr(III). The conditions in the location of the sediment will thus determine which oxidation state is the dominant one. Sediments close to the sediment-water interface with aerobic waters are more likely to contain Cr(VI), whereas Cr(III) is typically the most dominant species in all other sediments [4] . The pH value will also influence the Cr(III)-Cr(VI) equilibrium by favoring oxidation under alkaline and reduction under acidic conditions.
ions are the main species of Cr(VI) [5] . Anoxic conditions and the presence of reducing agents, such as iron or organic matter, tend to shift the equilibrium to Cr(III). The conditions in the location of the sediment will thus determine which oxidation state is the dominant one. Sediments close to the sediment-water interface with aerobic waters are more likely to contain Cr(VI), whereas Cr(III) is typically the most dominant species in all other sediments [4] . The pH value will also influence the Cr(III)-Cr(VI) equilibrium by favoring oxidation under alkaline and reduction under acidic conditions.
The two oxidation states differ not only in their environmental availability and fate, but also in their toxicity. Cr(VI) containing compounds are toxic with acute and chronic health effects after inhalation, ingestion and dermal contact. Numerous articles and government assessments reviewed their genotoxicity [6] - [11] and carcinogenicity [9] - [17] . In contrast, Cr(III) compounds show very low toxicity [18] and are essential for glucose, protein and fat metabolism in animals and humans [19] [20] .
Due to its heavy industrial use and toxicity, numerous methods have been developed for the quantification of Cr(VI) [3] [26] . Wet-chemistry approaches for speciation of chromium in solid matrices typically involve a digestion/extraction step to dissolve and extract Cr(III) and Cr(VI) and subsequent analysis of the solution. The quantification of Cr(VI) in solids requires the extraction of the metal ion from soluble, adsorbed, and precipitated chromium compounds from the matrix while maintaining its oxidation state [21] . U.S. EPA method 3060A recommends an alkaline digestion approach, which utilizes a Na2CO3/NaOH solvent with high pH to minimize Cr(VI) reduction during the 60 min extraction at 90˚C - 95˚C [21] . In comparison with other commonly used extractants, such as distilled water (pH 5.7), phosphate buffer (5 mM K2HPO4, 5 mM KH2PO4, pH 7.0) and hydroxide solution (0.1 M NaOH, pH 13), alkaline digestion at temperatures above 85˚C proved to be the most efficient extraction technique for soluble and insoluble Cr(VI) species in soil [22] . Although addition of MgCl2 in a phosphate buffer can reduce Cr(III) oxidation during the high pH extraction [21] , the stability of the Cr(VI)- Cr(III) oxidation system under the basic condition is determined by the nature of the sample. Thus, species interconversion in both directions cannot be completely avoided [23] . As an alternative to Na2CO3/NaOH solvent, the use of complexing agents such as ethylenediaminetetraacetic acid (EDTA) has been reported to improve Cr(VI) extraction by complexing metals that otherwise form insoluble compounds with 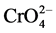 ions, such as PbCrO4, BaCrO4 or SrCrO4 [24] [25] . Simultaneous complexation of Cr(III) by EDTA further stabilizes the chromium species equilibrium as it minimizes the oxidation of the trivalent metal ion.
ions, such as PbCrO4, BaCrO4 or SrCrO4 [24] [25] . Simultaneous complexation of Cr(III) by EDTA further stabilizes the chromium species equilibrium as it minimizes the oxidation of the trivalent metal ion.
Several analysis methods have been applied to determine Cr(VI) in aqueous extracts. Early on, colorimetric analysis after species-specific complex formation of Cr(VI) with 1,5-diphenylcarbazide (DPC) and spectrophotometric detection at 540 nm has been described in U.S. EPA method 7196A [27] . Separation of Cr(III) and Cr(VI) species by ion chromatography (IC) and post-column DPC complex formation was added to this approach in U.S. EPA method 7199 [28] . Spectrophotometric detection, however, suffers from interferences with other metal ions that form colored complexes with DPC, such as mercury, molybdenum or vanadium, and organic matter absorbing light at the same wavelength, as well as limited sensitivity with detection limits at µg∙L−1 levels [27] .
As an alternative to the colorimetric approach, the use of inductively-coupled plasma mass spectrometry after IC separation allows chromium speciation with increased sensitivity up to ng∙L−1 and is often the method of choice for routine analysis. However, with ICP-MS, mass interferences with 40Ar12C+ and in the presence of chloride also interferences with 35Cl16O1H+ for the most abundant mass 52Cr+ and 37Cl16O+ for 53Cr+ can occur [29] . Electrospray ionization with tandem MS (ESI-MS/MS), i.e. fragmentation of an isolated precursor ion of interest and subsequent MS analysis of selected fragments, is one of the most sensitive and specific analysis tools, but to our knowledge it has not been applied for the direct analysis of Cr(VI) ions without the use of complexation agents. Hotta and Tsunoda report a detection limit of 0.7 µg∙L−1 Cr(VI) when analyzing the HCrO4- precursor at m/z 117 with ESI-MS, but did not apply tandem MS [30] . The application of tandem MS would increase the molecular selectivity and reduce interferences produced by the ICP plasma.
In most cases, the quantification of Cr(VI) requires the application of a linear calibration curve approach with the above mentioned methods. As an alternative, Kingston et al. [31] developed a direct isotope dilution mass spectrometry (DID-MS) approach for sensitive and accurate metal quantification which was incorporated into U.S. EPA method 6800 [32] : after addition of a known amount of enriched metal isotope, an analyte of interest can be quantified based on the known isotopic distribution of the unique enriched isotopic standard and natural abundance of isotopes in the natural sample. In case of the presence of several interconverting species, such as Cr(VI) and Cr(III), speciated ID-MS (SID-MS) permits evaluation of and correction for species-altering procedures by spiking several “labelled” species enriched with a different isotope, respectively.
Natural Cr has 4 stable isotopes: 50Cr (4.35%), 52Cr (83.79%), 53Cr (9.50%) and 54Cr (2.36%). When applying SID-MS to Cr speciation, known concentrations of two isotopic spikes are added to a representative sample aliquot: a Cr(III) spike enriched with 50Cr and a Cr(VI) spike enriched with 53Cr. After equilibration, four different isotope ratios are measured: 50Cr(III)/52Cr(III), 53Cr(III)/52Cr(III), 50Cr(VI)/52Cr(VI), and 53Cr(VI)/52Cr(VI) and the concentrations of Cr(III) and Cr(VI) in the sample, as well as any species interconversion after spiking determined by isotope dilution calculations. The SID-MS strategy has been shown to compensate for species interconversion during alkaline digestion based on U.S. EPA 3060A for NIST standard reference materials (SRM), aggregate materials and sediments [33] . The latter were found to contain very little to no Cr(VI).
Alonso and coworkers successfully applied the SID-MS approach in combination with IC-ICP-MS to NIST standard reference materials containing high levels of Cr(VI) and demonstrated the efficiency of the extraction solvent EDTA in complexing Cr(III) and preventing its oxidation [24] . However, the specific detection of trace-level Cr(VI) in solid matrices, such as sediments, still remains a challenge and the number of studies regarding low-level Cr(VI) in solid matrices is very limited. ESI-MS/MS enables independent chromium species measurements without interferences of polyatomic isobars produced by the ICP in the IC-ICP-MS methods. ESI ionization of these species validates and confirms more routine measurements independently. In this study, we present novel approaches to quantify Cr(VI) with highly selective IC-MS/MS for sediments and soil reference materials and show results for lake sediment samples collected in a pristine region in Northern Ontario during a 3-year period. For quantification, linear calibration curves, as well as an adapted SID-MS approach were applied.
2. Experimental
2.1. Sample and Reference Materials
Organic-rich lake sediments were collected using a gravity corer in a pristine region in Northern Ontario, Canada, as part of a regional geochemical study by the Ontario Geological Survey [34] [35] . A subset of lakes that returned high concentrations of total Cr were chosen for subsequent re-sampling for Cr(VI) quantification [35] - [37] . These samples were taken from the top 5 to 6 cm at the sediment-water interface and stored in 180 mL amber glass jars. All samples were kept refrigerated until they were delivered to the analysis lab within 7 days after collection and stored there at 5˚C ± 3˚C. Sample extraction and analysis was carried out within 30 days of sample receipt. Hexavalent chromium in soil standards were acquired from the National Institute of Standards and Technology (SRM 2701, NIST, Gaithersburg, MD, USA), Phenova (QC-CR6-SOIL, Golden, CO, USA) and Fluka (SQC012, Sigma-Aldrich, Oakville, ON, Canada) and contained the certified Cr(VI) concentrations listed in Table 1.
2.2. Sample Extraction
Extractions of Cr(VI) from soil reference materials and lake sediments were carried out for 1 h at temperatures varying between 90˚C to 95˚C using different methods. The respective amounts of sample, reference material

Table 1. Standard reference materials analyzed for Cr(VI).
aConfidence interval with a probability of 95%; bAcceptance limit, corresponding to confidence interval with a probability of 99%.
and extraction solvent are listed in Table 2. All samples and enriched standards were accurately weighed with an analytical balance and 0.0001 g precision.
Based on U.S. EPA method 3060A [21] , reference materials and samples were digested in an alkaline procedure using a 0.28 M Na2CO3/0.5 M NaOH solution and heated to 90˚C ± 5˚C on a hotplate while stirring constantly. In addition, the approach described by Fabregat-Cabello et al. [24] was followed and a 0.05 M solution of ethylenediaminetetraacetic acid tetrasodium salt (EDTA, pH 10) was used to complex extracted Cr(III) and digest Cr(VI) in solid matrices. For this method, sediment samples or the SQC012 reference material were weighed, an appropriate amount of 53Cr(VI) and 50Cr(III) internal standards (Applied Isotope Technologies, Pittsburgh, PA, USA) added and the mixture suspended in the alkaline EDTA solution. The spiked standard solutions were diluted from stock solutions with certified concentrations of 10.4616 mg∙L−1 53Cr(VI) and 10.8999 mg∙L−1 50Cr(III).
Due to the large difference in Cr(VI) concentration between sample and reference material, the concentration of the enriched chromium isotope standards was varied accordingly to maintain a concentration ratio between analyte and standard of 0.1 to 10. Standard solutions were added in a 1/100 to 1/200 (v/v) ratio of standard/extraction volume at concentrations of 0.5 and 10 mg∙L−1 for samples and reference materials, respectively. The obtained mixtures were then subjected to extraction in pre-cleaned 100 mL Teflon tubes using a Multiwave 3000 microwave system (Anton Paar, Graz, Austria) by ramping to 90˚C for 10 min and reaction for 1 h at 89.4˚C ± 0.8˚C (measured with IR sensor). In addition, a ThermoMixer® C with Thermo Top (Eppendorf, Mississauga, ON, Canada) was used with a temperature setting of 95˚C ± 4˚C (accuracy based on manufacturer’s specifications) applying 5 mL vials (Eppendorf) and 500 rpm rotation to facilitate mixing. After cooling down to room temperature, all extracts were centrifuged at 4000 rpm for 20 min in a 5810 centrifuge (Eppendorf).
2.3. IC-MS/MS Analysis
Prior to analysis, all extracts were diluted 10 to 5000-fold (v/v) with Milli-Q water (Millipore (Canada), Etobicoke, ON, Canada) and filtered using Mini-UniPrep™ syringeless filter devices having 0.2 μm polypropylene (PP) filter media and PP housings (Whatman, Forham Park, NJ, USA). Analysis was carried out using an ICS 5000+ ion chromatograph consisting of autosampler, 2 dual pumps, auxiliary pump (0.5 mL water∙min−1 to flush the ion suppressor), eluent generator equipped with EluGen potassium hydroxide cartridge and ASRS-300 2 mm ion suppressor operated at 45 mA (all Thermo Scientific Dionex, Sunnyvale, CA, USA). Separation was achieved by flushing 200 μL of analyte through guard column (Dionex, AG20, 2 mm i.D. × 50 mm) and analytical separation column (Dionex, AS20, 2 mm i.D. × 250 mm) at 0.3 mL∙min−1 using the following gradient: 0 - 6 min: 30 mM KOH, 8 - 13 min: 60 mM KOH, 16 - 20 min: 30 mM KOH. After passing through the ion suppressor at 0.3 mL∙min−1, the eluate flow was combined with 0.2 mL∙min−1 methanol (Thermo Fisher Scientific) using a Tee connection. The sample analysis was carried out on an API 3200 triple-quadrupole MS (SCIEX, Concord, ON, Canada) operated in negative ion mode using multiple reaction monitoring (MRM). The eluate was ionized by a TurboSpray electrospray source (SCIEX) with the following settings: curtain gas: 50.0, spray voltage: −3500 V, gas temperature: 450˚C, nebulizer gas 50, and turbo gas: 60. The MRM settings are displayed in Table 3. For data acquisition and handling, Analyst software version 1.6.1 and 1.6.2 (SCIEX) and DCMS Link version 2.12 (Dionex) were applied.
2.4. Quantification Using a Linear Multipoint Calibration Curve
A classical linear multipoint calibration (MPC) quantification was carried out for selected samples. A 1000
Table 2. Extraction parameters.
Table 3. MRM parameters for quantification of Cr(VI) on the API 3200 triple-quadrupole system.
aUsed only during MPC quantification; bUsed only during SID quantification.
mg∙L−1 CrO4 standard (Sigma-Aldrich) was diluted to 9 concentrations between 0.010 to 1.0 μg∙L−1 Cr(VI) and analyzed by IC-MS/MS along with alkaline-digested sediment and reference material extracts. Prior to IC-MS/MS analysis, the samples and calibration standards were spiked with an internal standard in a 50:1 (v/v) ratio containing 10 ng∙mL−1 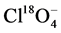 (≥98%, Sigma-Aldrich, Isotec Stable Isotopes) and 100 ng∙mL−1
(≥98%, Sigma-Aldrich, Isotec Stable Isotopes) and 100 ng∙mL−1  (98%, Cambridge Isotope Laboratories, Tewksbury, MA). Two quality control (QC) samples containing 0.800 and 0.200 μg∙L−1 Cr(VI), respectively were analyzed with each batch of samples. The QC samples were prepared by diluting a 1000 mg/L CrO4 standard (Inorganic Ventures, Christiansburg, Virginia, USA) with Milli-Q water. Sample concentrations were calculated relative to the peak areas of the internal standard
(98%, Cambridge Isotope Laboratories, Tewksbury, MA). Two quality control (QC) samples containing 0.800 and 0.200 μg∙L−1 Cr(VI), respectively were analyzed with each batch of samples. The QC samples were prepared by diluting a 1000 mg/L CrO4 standard (Inorganic Ventures, Christiansburg, Virginia, USA) with Milli-Q water. Sample concentrations were calculated relative to the peak areas of the internal standard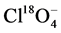 .
. 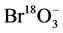 was monitored for quality control purposes.
was monitored for quality control purposes.
2.5. Quantification Using SID-MS
After double spiking with isotope-enriched 50Cr(III) and 53Cr(VI), the ratios of 52Cr/53Cr(VI) and 50Cr/52Cr(VI) were monitored for [HCrO4]−. In addition, transitions for [CrIIIEDTA]− composed of isotopes 50, 52 and 53 were investigated. Each sample was analyzed in 4 replicates on the API 3200 triple-quadrupole system. Cr(VI) concentrations were calculated relative to the internal standard peak area of 53Cr(VI) with a calculation tool supplied by Applied Isotope Technologies. The underlying mathematical equations have been published elsewhere [31] . The internal standard allowed for compensation of Cr(VI) to Cr(III) conversions occurring during extraction, as well as matrix suppression effects in the IC-MS/MS system.
2.6. Colorimetric Approach
Reference materials were also analyzed by colorimetry based on U.S. EPA method 7196A [27] . Extracts prepared by alkaline Na2CO3/NaOH digestion were pH adjusted to pH 7.5 ± 0.5 with nitric acid. Cr(VI) in the mixture was then complexed with 0.5% (w/v) 1,5-diphenylcarbazide (DPC) solution in acetone and the pH adjusted to pH 2 ± 0.5 using sulfuric acid. The absorbance of the solution was measured at a wavelength of 540 nm using a 2100 spectrophotometer (Cole-Parmer, Montréal, QC, Canada). The quantification of Cr(VI) concentrations was carried out based on a calibration curve composed of 5 standards in the range of 0.01 to 0.4 mg∙L−1.
2.7. Moisture
In order to determine the water content of the sediments, 10 g of sediment sample were dried for 16 h at 105˚C ± 5˚C. The mass difference between wet and dry weight was divided by the wet weight and defined as sample moisture content.
3. Results and Discussion
3.1. MPC-IC-MS/MS Analysis for Cr(VI) with Alkaline Digestion
A novel quantification method of low-level Cr(VI) using IC-MS/MS was developed. Anion exchange chromatography allowed for the separation of the two chromium species from each other and from most interfering inorganic and organic sample components. Electrospray tandem mass spectrometry (ESI-MS/MS) permits the detection of Cr(VI) as [HCrO4]− → [CrO3]− in negative ion mode, but cannot analyze Cr3+ directly. By applying ESI-MS/MS, we thus compromised the ability to detect both species with an immense gain in sensitivity and selectivity for Cr(VI). The method detection limit (MDL) was calculated by analyzing a 0.050 μg∙L−1 Cr(VI) standard multiple times and multiplying the resulting standard deviation of the determined Cr(VI) concentration with a one-tailed Student t-value (α = 0.01, n = 18) of 2.567. Using this approach, an MDL of 0.007 μg∙L−1 Cr(VI) was estimated. Considering an extraction volume of 0.05 L, a sample amount of 2.5 g and a dilution factor of 100 to reduce chromatographic interferences with extraction solvent used for alkaline digestion, this value translates to 0.014 μg∙g−1 wet weight Cr(VI) in solid matrices.
In a first attempt to develop a quantification method for Cr(VI) in lake sediments, three different reference materials were extracted in 15 replicates, respectively, using alkaline digestion. Due to the lack of sediment SRMs certified for Cr(VI), soil reference material was used. The extracts were analyzed by IC-MS/MS with a linear multipoint calibration curve (MPC) quantification method and compared with colorimetric measurements after formation of a DPC-Cr(VI) complex. The average Cr(VI) concentrations (n = 15) are listed in Table 1. The colorimetric approach generated values between 73% and 90% of the certified concentration, whereas the IC-MS/MS method resulted in recoveries of 95% and 111% for the Phenova and NIST reference materials, respectively. When looking at the individual concentrations shown in Figure 1, an excellent correlation between colorimetric and IC-MS/MS data was observed. This suggests that the extraction itself caused a large portion of the data variance for each set. Colorimetric data were typically 20% lower than Cr(VI) concentrations determined by IC-MS/MS, with both methods showing slight biases in opposite directions when compared with the certified concentration. However, for NIST 2701, one third of the samples were quantified within the 95% confidence interval of the certified concentration for both methods, respectively. For the Phenova standard, all samples were detected within the 99% confidence acceptance limits. For the colorimetric method, a bias towards
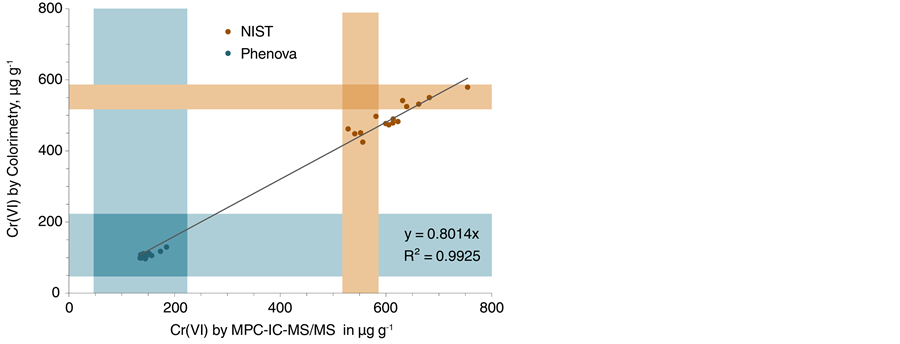
Figure 1. Comparison of IC-MS/MS and colorimetric data for two reference materials. The shaded areas correspond to the confidence intervals of the certified values with 99% confidence for the Phenova standard and 95% confidence for the NIST 2701 reference material. The linear regression curve was calculated based on data for both reference materials.
lower Cr(VI) concentrations can occur in the presence of reducing agents present in soils, such as Fe(II), sulfide or organic matter and is amplified by the acidic conditions required for the DPC-Cr(VI) complex formation [23] . A multi-laboratory study for NIST 2701 found even lower average Cr(VI) concentrations of 384.3 ± 73.5 µg∙g−1 and only 70% recovery when analyzed by DPC-UV-VIS [38] .
As shown by Kingston and coworkers [33] , alkaline digestion can cause oxidation of soluble Cr(III) of up to 60%, which could not be accounted for using a traditional MPC approach with IC-MS/MS and resulted in overestimation of the Cr(VI) concentrations for one out of two reference materials in this study. The addition of Mg(II) ions and a phosphate buffer as recommended in U.S. EPA 3060A in the presence of oxidizing conditions was not applied for this data set in order to reduce interferences with the IC separation and MS/MS detection method. Additionally, matrix matching between samples and calibration standards proved to be difficult considering the complexity of each individual solid matrix. The internal standard 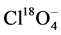 counteracted matrix suppression/enhancement effects and elevated background signals to some degree, since data analysis without its consideration resulted in even higher Cr(VI) recoveries between 113% and 118%. However, the slightly stronger retention of the standard on the anion exchange column and different ionization efficiency during electrospray ionization made accurate compensation challenging. Both methods were also used to analyze 38 lake sediment samples collected in a pristine area in Northern Ontario. For the colorimetric approach, all sample results were below the method detection limit of 0.2 µg∙g−1 Cr(VI). In addition, the dark-brown color of the extracts interfered with the analysis by UV-VIS. When applying the IC-MS/MS method to the environmental sample extracts, the results for 8 samples were below the MDL of 0.01 µg∙g−1 wet weight Cr(VI).
counteracted matrix suppression/enhancement effects and elevated background signals to some degree, since data analysis without its consideration resulted in even higher Cr(VI) recoveries between 113% and 118%. However, the slightly stronger retention of the standard on the anion exchange column and different ionization efficiency during electrospray ionization made accurate compensation challenging. Both methods were also used to analyze 38 lake sediment samples collected in a pristine area in Northern Ontario. For the colorimetric approach, all sample results were below the method detection limit of 0.2 µg∙g−1 Cr(VI). In addition, the dark-brown color of the extracts interfered with the analysis by UV-VIS. When applying the IC-MS/MS method to the environmental sample extracts, the results for 8 samples were below the MDL of 0.01 µg∙g−1 wet weight Cr(VI).
3.2. SID-IC-MS/MS Analysis with EDTA Extraction
In order to compensate for any oxidation and reduction processes altering the Cr(VI) concentration during the extraction, a speciated isotope dilution approach [24] [31] - [33] [39] was applied for the quantification of Cr(VI) in reference material and samples. An alkaline EDTA extraction [24] was chosen over the alkaline digestion based on U.S. EPA 3060A [21] for the SID-MS experiments for several reasons: Alkaline EDTA extractioncauses presumably less to no Cr(III) oxidation than U.S. EPA 3060A [24] [39] , reduces precipitation of Cr(III) and allows direct formation of the [CrIIIEDTA]− complex [39] . Theoretically, conversion of Cr3+ to [CrIIIEDTA]− improves not only its retention on the anion exchange column, but also allows indirect detection by ESI-MS/MS of the otherwise inaccessible ion. Since electrospray ionization suffers from severe ion suppression in the presence of ionic eluent compounds, the IC conditions used herein had to be modified from the elution parameters suggested by Fabregat-Cabello et al. [24] : The isocratic elution with 5 mmol∙L−1 EDTA, pH 10 as mobile phase was replaced with a 30 to 60 mM hydroxide gradient. The Dionex system used in this study offers online post-column electrochemical removal of hydroxide ions in the mobile phase and is thus ideally suited for ESI-MS/MS analysis. The extraction time was kept at 1 h to facilitate comparison between samples analyzed by alkaline and EDTA extraction. The presence of 50 mmol∙L−1 EDTA in the sample extracts required at least 1/10 dilution prior to IC-MS/MS analysis in order to retain the chromate ion on the anion exchange column.
EDTA extraction facilitated by microwave and ThermoMixer was applied to 36 and 47 lake sediment samples, respectively, as well as the SQC012 soil reference material. All extracts were analyzed by SID-IC-MS/MS. None of the analyzed lake sediment or SQC012 samples showed any formation of [CrEDTA]− for the 50Cr(III) standard or natCr(III). It has to be noted that the sensitivity of [CrEDTA]− by ESI-MS/MS is significantly lower than for [HCrO4]− with a detection limit above 2 µg∙L−1. Although the lack of a [CrEDTA]− signal did not allow the simultaneous quantification of Cr(VI) and Cr(III), accurate Cr(VI) results were obtained for the certified reference material without consideration of any Cr(III) to Cr(VI) species interconversion. As pointed out before [24] , the complexation of Cr(III) with EDTA and the consequential absence of oxidation processes thus enables the practice of a single spike DID-MS procedure for Cr(VI) analysis in solid matrices. In addition, the detection of oxidized 50Cr spike as [H50CrO4]− allowed for a control tool to estimate the extent of potential oxidation.
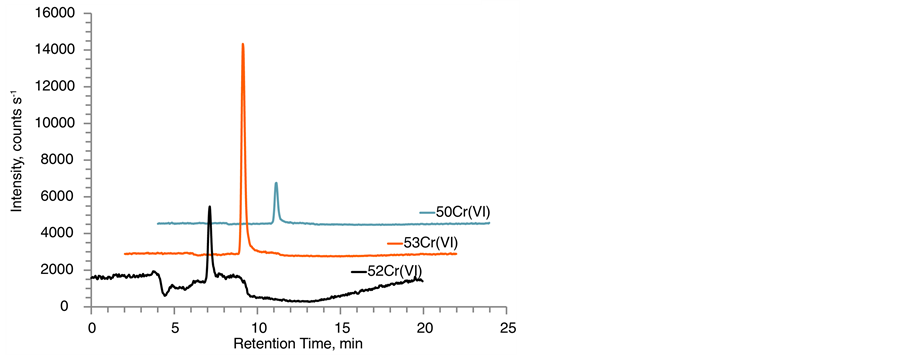
Figure 2 contains typical chromatograms for the three chromium isotopes analyzed as chromate ions for a lake sediment sample and SQC012. The three chromatographic traces reflect the abundance of several species: natCr(VI), Cr(VI) spike enriched for 53Cr, as well as potentially oxidized natCr(III) and oxidized Cr(III) spike enriched for 50Cr. The main contribution for the [H53CrO4]− and [H52CrO4]− signals originated from the 53Cr(VI) spike and natural 52Cr(VI), respectively. In contrast, the relative distribution for the [H50CrO4]− trace was heavily dependent on the presence of oxidation reactions. SID-IC-MS/MS analysis on all SQC012 extracts amounted in
 (a)
(a)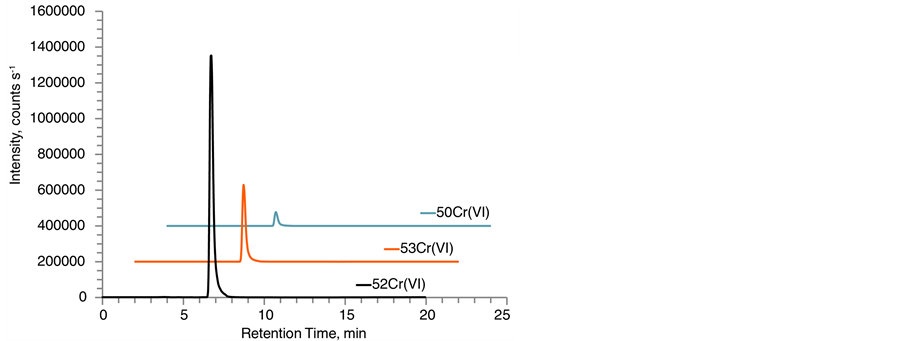 (b)
(b)
Figure 2. IC-MS/MS chromatograms after EDTA/ThermoMixer extraction of (a) lake sediment extract with a Cr(VI) concentration of 0.015 ± 0.002 µg∙g−1 wet weight, 0.75 ± 0.10 µg∙g−1 dry weight and a moisture content of 98% and (b) Fluka SQC012 quantified as 152.7 ± 1.5 µg∙g−1 dry weight Cr(VI). Traces show all monitored chromium isotopes for Cr(VI) as [HCrVIO4]− → [CrVIO3]− transition. No [CrIIIEDTA]− complex was detected in the investigated samples.
Cr(VI) concentrations within the acceptance criteria with deviations between 0.2% to 18% from the certified concentration (see Table 1 and Figure 3(b)). The abundance of natural Cr(VI) was found to be very low in the environmental samples compared to the SQC012 standard. Intensities of 53Cr(VI) spiked into sediment samples were typically significantly lower than in double-spiked method blanks. Furthermore, extraction recoveries of Cr(VI) peak areas in sediment samples spiked with natural Cr(VI) pre- and post-extraction ere low in some cases and ranged from 6% to 90%. These findings suggest that the high organic content of the analyzed sediment samples triggered analyte and spike reduction processes despite the high pH of the extractant.
Even with signal losses, Cr(VI) quantification after EDTA/microwave extraction yielded results above MDL for all the 36 analyzed samples. For 9 out of 47 samples digested with the ThermoMixer, no quantifiable signals for 52Cr(VI) were detected after 60 min extraction. For 4 out of those 9 sediment samples, reducing the extraction time to 15 min allowed to increase the signal above MDL and thus quantify the Cr(VI) concentration in the sample. For SQC012, the extraction duration did not influence the intensity of any of the analyzed [HCrO4]− isotopes and the calculated Cr(VI) concentration when applying 95˚C for 15, 30, 45 and 60 min in the ThermoMixer (data not shown). For 5 out of 47 samples, no quantifiable signal for 52Cr(VI) and 53Cr(VI) could be detected despite a 4-fold increase in spike concentration and/or shorter extraction times of 15 min in additional test experiments.
 (a)
(a)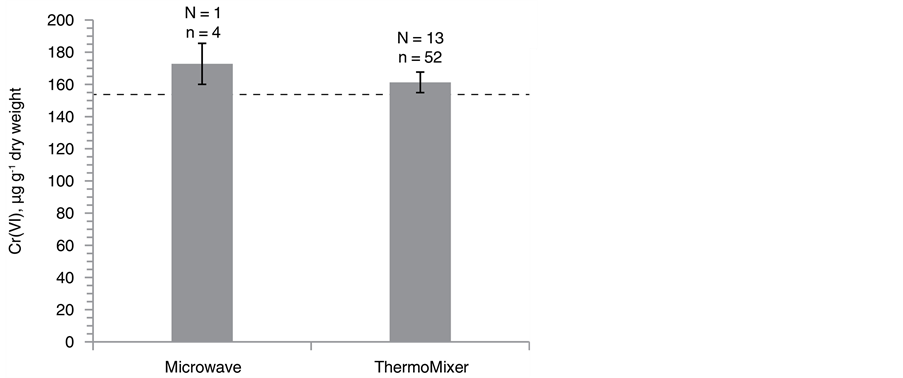 (b)
(b)
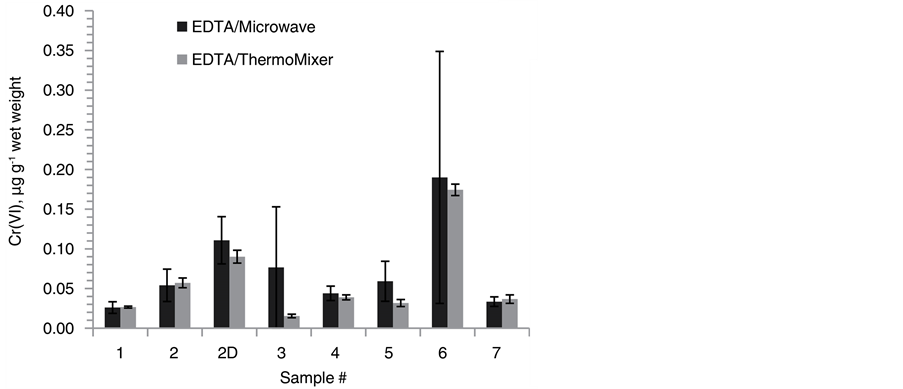
Figure 3. Direct comparison of Cr(VI) SID-IC-MS/MS concentrations between ThermoMixer- and microwave-assisted EDTA extraction for (a) lake sediments and (b) Fluka SQC012 with the respective numbers of extraction replicates (N) and total analysis replicates (n) indicated. The dashed line represents the certified Cr(VI) con- centration. Error bars illustrate the 95% confidence interval.
In order to compare the efficiency of the two different extraction protocols for microwave and ThermoMixer directly, aliquots of 8 lake sediment samples were extracted in both heating systems and analyzed by SID-IC-MS/MS. The resulting Cr(VI) concentrations are shown in Figure 3. While the concentrations correspond very well for the selected samples, standard deviations were typically larger for samples extracted in the microwave, probably mostly due to lower absolute peak areas. Lower signals could be the result of a higher reduction rate of Cr(VI) in the microwave or result from different extractant-to-sample ratios. As shown in Table 2, the ratio between sediment sample and extraction solvent was increased from 1/20 (w/w) with microwave- assisted extraction to 1/9 (w/w) when using the ThermoMixer in order to accommodate the 10-fold reduced volume capacity. As a consequence, the total concentration of natCr(VI) in the extracts was increased by a factor of 2.2. Consequentially, the decreased EDTA volume did not reduce the relative amount of extracted Cr(VI), but potentially improved the precision of the calculated concentrations. It has to be emphasized that this experiment did not allow a direct comparison between the heating systems itself, but rather between the two different extraction protocols that were commonly applied in our laboratory.
For SQC012, the sample to extractant ratio was increased from 1/500 to 1/225 when switching from micro- wave to ThermoMixer extraction with no significant changes in accuracy of Cr(VI) quantification, as shown in Figure 3(b).
In order to estimate how representative a 0.5 g subsample was for the whole sample set, 6 sediment subsamples were taken from the same jar and extracted with EDTA. Analysis with SID-IC-MS/MS yielded a Cr(VI) concentration of 0.068 ± 0.006 µg∙g−1 wet weight (N = 6, n = 24) with a coefficient of variation of 19%. Despite the difficulty of adequate mixing in a highly aqueous solid/liquid system, this experiment illustrated the adequacy of 0.5 g sample sizes for trace-level sediment samples. Single subsampling duplicates analyzed along with sample extracts resulted in average recoveries of 200% ± 58% (N = 14) for the ThermoMixer and 77% and 121% (N = 2) for the microwave extraction.
3.3. Comparison of All Lake Sediment Sample Sets
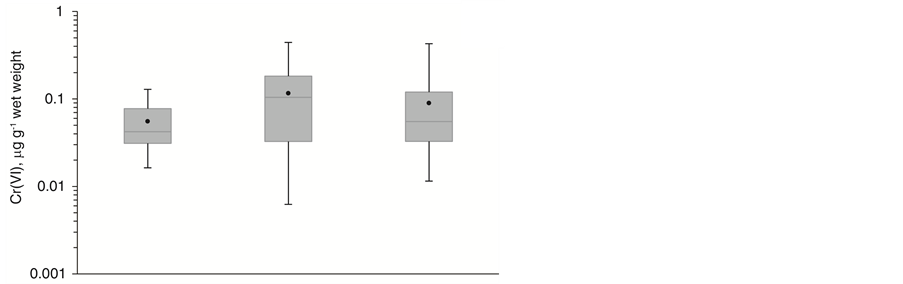
Figure 4 shows a direct comparison between one sample set digested with U.S. EPA method 3060A and analyzed using an MPC-MS approach and two sample sets processed with EDTA and an adapted SID-MS approach. All digestion/extraction and analysis methods yielded trace-level Cr(VI) concentrations. Samples analyzed without correction for Cr(VI) reduction with SID-MS yielded a lower average Cr(VI) amount in the analysis extracts and a lower maximum concentration. This finding suggests potential reduction processes even in the presence of the strongly alkaline Na2CO3/NaOH extractant. SID-MS results compensated for Cr(VI) reduction and thus provided typically higher average and maximum Cr(VI) concentrations. When comparing the wet weight data, the concentration distributions for ThermoMixer and microwave extracts aligned fairly well. However, after correction for the moisture content, the dry weight microwave data showed larger Cr(VI) concentrations than the ThermoMixer data. The moisture values have to be treated with caution since most samples contained over 90% water. Furthermore, the water content was determined in different sample aliquots than the
 (a)
(a)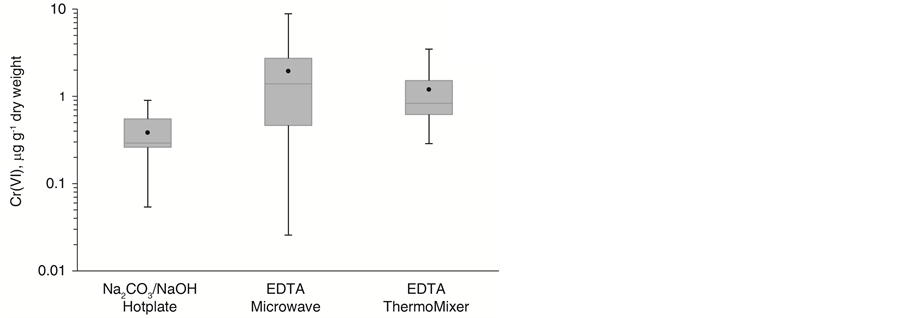 (b)
(b)
Figure 4. Boxplot comparison of IC-MS/MS data obtained for Cr(VI) quantification in lake sediment samples using threedifferent extraction protocols based on (a) wet weight and (b) dry weight of the samples after correcting for sample moisture content.
Cr(VI) analysis. A comparison between wet weight data seems thus more meaningful for sediments samples with high levels of water.
4. Conclusions
Reliable Cr(VI) analysis in organic-rich lake sediments with high water contents of typically above 90% remains challenging. The geochemistry of this type of samples can favour reduction of Cr(VI) even under alkaline conditions. The application of an adapted SID-MS approach permitted compensation of Cr(VI) loss during extraction. However, the current data set does not allow to draw any conclusions whether the 53Cr(VI) spike, added as CrO42−, can adequately correct for the reduction of all chemical Cr(VI) species present in sediment samples.
IC-MS/MS is a highly sensitive analytical tool to detect Cr(VI) as [HCrO4]−. However, due to the highly reducing nature of the matrix, the abundance of natural and spiked Cr(VI) in the extracts was extremely low in some cases. Increasing sample size, while using the same or smaller volume of the extraction liquid can improve signal-to-noise ratios. In this study, two different extractants to sample ratios were used: 1/20 with 2.5 g of sample and 1/9 with 0.5 g of sample. While Cr(VI) results obtained from 0.5 g samples had the same distribution and extremes as the results obtained from 2.5 g samples, differences were observed between some replicates, although all results were found to be at trace level. These data suggest that an increase in sample-to-extractant ratio did not create additional matrix effects by suppressing the signal monitored on the mass spectrometer, while maintaining comparable extraction efficiencies.
Larger differences in Cr(VI) concentrations were observed between field replicates than between subsamples taken from the same jar. Homogeneity of the samples is a significant factor that limits the reproducibility of the low-level results. While CRMs are well homogenized and allowed obtaining reproducible results even when using a much smaller sample size (0.1 and 0.02 g), sediments must be kept moist to maintain the integrity of Cr(VI) in the sample, which excludes the possibility of homogenizing them prior to analysis. The CRMs currently available for soils have significantly higher Cr(VI) level than the samples analyzed in this study. Having a sediment CRM with a Cr(VI) level lower than 5 µg∙g−1 would be more appropriate for using U.S. EPA 6800 for trace-level measurements of Cr(VI).
Acknowledgements
S. Mädler acknowledges funding from the Ontario Ministry of Northern Development and Mines through the Southern Ontario Stream Sediment Project, a partnership between Ontario Ministry of the Environment and Climate Change, Ontario Ministry of Northern Development and Mines and Conservation Ontario. The authors thank Mizanur Rahman (Applied Isotope Technologies) and Mesay Mulugeta Wolle (Duquesne University) for their support with the SID-MS approach.
Cite this paper
Stefanie Mädler,1 1,Fengrong Sun,Cindy Tat,Nadya Sudakova,Peter Drouin,Robert J. Tooley,Eric J. Reiner,Teresa A. Switzer,Richard Dyer,H. M. Skip Kingston,Matt Pamuku,Vasile I. Furdui, (2016) Trace-Level Analysis of Hexavalent Chromium in Lake Sediment Samples Using Ion Chromatography Tandem Mass Spectrometry. Journal of Environmental Protection,07,422-434. doi: 10.4236/jep.2016.73037
References
- 1. (2001) The Incidence and Severity of Sediment Contamination in Surface Waters of the United States; EPA-823-R-01- 01. National Sediment Quality Survey, 2nd Edition, Washington DC.
https://clu-in.org/download/contaminantfocus/pcb/incidence-and-severity-2004nsqs2ed-complete.pdf - 2. Rifkin, E., Gwinn, P. and Bouwer, E. (2004) Chromium and Sediment Toxicity. Environmental Science & Technology, 38, 267A-271A. http://dx.doi.org/10.1021/es040575m
- 3. Kotas, J. and Stasicka, Z. (2000) Chromium Occurrence in the Environment and Methods of Its Speciation. Environmental Pollution, 107, 263-283. http://dx.doi.org/10.1016/S0269-7491(99)00168-2
- 4. (1994) Chromium and Its Compounds. Canadian Environmental Protection Act. Government of Canada, Ottawa.
- 5. Rai, D., Sass, B.M. and Moore, D.A. (1987) Chromium(III) Hydrolysis Constants and Solubility of Chromium(III) Hydroxide. Inorganic Chemistry, 26, 345-349. http://dx.doi.org/10.1021/ic00250a002
- 6. De Flora, S., Bagnasco, M., Serra, D. and Zanacchi, P. (1990) Genotoxicity of Chromium Compounds—A Review. Mutation Research, 238, 99-172. http://dx.doi.org/10.1016/0165-1110(90)90007-X
- 7. Codd, R., Dillon, C.T., Levina, A. and Lay, P.A. (2001) Studies on the Genotoxicity of Chromium: From the Test Tube to the Cell. Coordination Chemistry Reviews, 216, 537-582. http://dx.doi.org/10.1016/S0010-8545(00)00408-2
- 8. Zhitkovich, A. (2005) Importance of Chromium-DNA Adducts in Mutagenicity and Toxicity of Chromium(VI). Chemical Research in Toxicology, 18, 3-11. http://dx.doi.org/10.1021/tx049774+
- 9. Cohen, M.D., Kargacin, B., Klein, C.B. and Costa, M. (1993) Mechanisms of Chromium Carcinogenicity and Toxicity. Critical Reviews in Toxicology, 23, 255-281. http://dx.doi.org/10.3109/10408449309105012
- 10. Katz, S.A. and Salem, H. (1993) The Toxicology of Chromium with Respect to Its Chemical Speciation—A Review. Journal of Applied Toxicology, 13, 217-224. http://dx.doi.org/10.1002/jat.2550130314
- 11. Leonard, A. and Lauwerys, R.R. (1980) Carcinogenicity and Mutagenicity of Chromium. Mutation Research, 76, 227- 239. http://dx.doi.org/10.1016/0165-1110(80)90018-4
- 12. Langård, S. (1990) 100 Years of Chromium and Cancer—A Review of Epidemiological Evidence and Selected Case Reports. American Journal of Industrial Medicine, 17, 189-215. http://dx.doi.org/10.1002/ajim.4700170205
- 13. Langård, S. (1993) Role of Chemical Species and Exposure Characteristics in Cancer among Persons Occupationally Exposed to Chromium Products. Scandinavian Journal of Work, Environment & Health, 19, 81-89.
- 14. Costa, M. (1997) Toxicity and Carcinogenicity of Cr(VI) in Animal Models and Humans. Critical Reviews in Toxicology, 27, 431-442. http://dx.doi.org/10.3109/10408449709078442
- 15. Costa, M. and Klein, C.B. (2006) Toxicity and Carcinogenicity of Chromium Compounds in Humans. Critical Reviews in Toxicology, 36, 155-163. http://dx.doi.org/10.1080/10408440500534032
- 16. Norseth, T. (1981) The Carcinogenicity of Chromium. Environmental Health Perspectives, 40, 121-130. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1568823/# http://dx.doi.org/10.1289/ehp.8140121
- 17. Zhitkovich, A. (2011) Chromium in Drinking Water: Sources, Metabolism, and Cancer Risks. Chemical Research in Toxicology, 24, 1617-1629. http://dx.doi.org/10.1021/tx200251t
- 18. Eastmond, D.A., MacGregor, J.T. and Slesinski, R.S. (2008) Trivalent Chromium: Assessing the Genotoxic Risk of an Essential Trace Element and Widely Used Human and Animal Nutritional Supplement. Critical Reviews in Toxicology, 38, 173-190. http://dx.doi.org/10.1080/10408440701845401
- 19. Cefalu, W.T. and Hu, F.B. (2004) Role of Chromium in Human Health and in Diabetes. Diabetes Care, 27, 2741-2751. http://dx.doi.org/10.2337/diacare.27.11.2741
- 20. Barceloux, D.G. (1999) Chromium. Journal of Toxicology: Clinical Toxicology, 37, 173-194. http://dx.doi.org/10.1081/clt-100102418
- 21. (1996) U.S. EPA Method 3060A: Alkaline Digestion for Hexavalent Chromium. Test Methods for Evaluating Solid Waste, Physical/Chemical Methods SW 846. US Government Printing Office, Washington DC. http://www3.epa.gov/epawaste/hazard/testmethods/sw846/pdfs/3060a.pdf
- 22. James, B.R., Petura, J.C., Vitale, R.J. and Mussoline, G.R. (1995) Hexavalent Chromium Extraction from Soils: A Comparison of Five Methods. Environmental Science & Technology, 29, 2377-2381. http://dx.doi.org/10.1021/es00009a033
- 23. Tirez, K., Scharf, H., Calzolari, D., Cleven, R., Kisser, M. and Lück, D. (2007) Validation of a European Standard for the Determination of Hexavalent Chromium in Solid Material. Journal of Environmental Monitoring, 9, 749-759. http://dx.doi.org/10.1039/b706724k
- 24. Fabregat-Cabello, N., Rodríguez-González, P., Castillo, á., Malherbe, J., Roig-Navarro, A.F., Long, S.E. and Alonso, J.I.G. (2012) Fast and Accurate Procedure for the Determination of Cr(VI) in Solid Samples by Isotope Dilution Mass Spectrometry. Environmental Science & Technology, 46, 12542-12549. http://dx.doi.org/10.1021/es3022864
- 25. Korolczuk, M. and Grabarczyk, M. (2005) Evaluation of Ammonia Buffer Containing EDTA as an Extractant for Cr(VI) from Solid Samples. Talanta, 66, 1320-1325. http://dx.doi.org/10.1016/j.talanta.2005.01.047
- 26. Gomez, V. and Callao, M.P. (2006) Chromium Determination and Speciation since 2000. TRAC-Trends in Analytical Chemistry, 25, 1006-1015. http://dx.doi.org/10.1016/j.trac.2006.06.010
- 27. (1992) U.S. EPA Method 7196A: Chromium, Hexavalent (Colorimetric), SW-846 Third Edition, Update V. U.S. Government Printing Office, Washington DC. http://www3.epa.gov/epawaste/hazard/testmethods/sw846/pdfs/7196a.pdf
- 28. (1996) U.S. EPA Method 7199: Determination of Hexavalent Chromium in Drinking Water, Groundwater and Industrial Wasterwater Effluents by Ion Chromatography. U.S. Government Printing Office, Washington DC. http://www3.epa.gov/epawaste/hazard/testmethods/sw846/pdfs/7199.pdf
- 29. Markiewicz, B., Komorowicz, I., Sajnog, A., Belter, M. and Baralkiewicz, D. (2015) Chromium and Its Speciation in Water Samples by HPLC/ICP-MS—Technique Establishing Metrological Traceability: A Review since 2000. Talanta, 132, 814-828. http://dx.doi.org/10.1016/j.talanta.2014.10.002
- 30. Hotta, H. and Tsunoda, K. (2015) Electrospray Ionization Mass Spectrometry for the Quantification of Inorganic Cations and Anions. Analytical Sciences, 31, 7-14. http://dx.doi.org/10.2116/analsci.31.7
- 31. Kingston, H.M.S., Huo, D.W., Lu, Y.S. and Chalk, S. (1998) Accuracy in Species Analysis: Speciated Isotope Dilution Mass Spectrometry (SIDMS) Exemplified by the Evaluation of Chromium Species. Spectrochimica Acta B, 53, 299- 309. http://dx.doi.org/10.1016/S0584-8547(97)00143-2
- 32. Kingston, H.M.S. and Huo, D. (2007) U.S. EPA Method 6800: Elemental and Speciated Isotope Dilution Mass Spectrometry. Solid Waste Manual-846, Update IV. U.S. Environmental Protection Agency, Washington DC. http://www3.epa.gov/epawaste/hazard/testmethods/sw846/pdfs/6800.pdf
- 33. Rahman, G.M., Kingston, H.M.S., Towns, T.G., Vitale, R.J. and Clay, K.R. (2005) Determination of Hexavalent Chromium by Using Speciated Isotope-Dilution Mass Spectrometry after Microwave Speciated Extraction of Environmental and Other Solid Materials. Analytical and Bioanalytical Chemistry, 382, 1111-1120.http://dx.doi.org/10.1007/s00216-005-3203-7
- 34. Dyer, R.D. (2012) McFaulds Lake (“Ring of Fire”) Area Lake Sediment and Water Sampling Pilot Study, Far North, Ontario. In: Summary of Field Work and Other Activities 2012, Ontario Geological Survey, Open File Report, 6280, 128-111, 128-115. http://www.geologyontario.mndm.gov.on.ca/mndmfiles/pub/data/imaging/ofr6280//ofr6280.pdf
- 35. Dyer, R.D. and Handley, L.A. (2013) McFaulds Lake (“Ring of Fire”) Area High-Density Lake Sediment and Water Survey, Far North, Ontario. In: Summary of Field Work and Other Activities 2013, Ontario Geological Survey, Open File Report, 6290, 132-111, 132-117.http://www.geologyontario.mndm.gov.on.ca/mndmfiles/pub/data/imaging/ofr6290//OFR6290.pdf
- 36. Dyer, R.D. and Handley, L.A. (2014) McFaulds Lake (“Ring of Fire”) Area Lake, Stream and Till Geochemistry Infill Sampling, Far North, Ontario. In: Summary of Field Work and Other Activities 2014, Ontario Geological Survey, Open File Report, 6300, 28-21, 28-25. http://www.geologyontario.mndm.gov.on.ca/mndmfiles/pub/data/imaging/ofr6300//OFR6300.pdf
- 37. Dell, K.M., Dyer, R.D. and Handley, L.A. (2015) Nakina and McFaulds Lake (“Ring of Fire”) Area Lake Sediment and Till Geochemistry In-Fill Sampling, Northern Ontario. In: Summary of Field Work and Other Activities 2015, Ontario Geological Survey, Open File Report, 130-111, 130-115.
- 38. (2009) Certificate of Analysis, Standard Reference Material 2701: Hexavalent Chromium in Contaminated Soil (High Level). National Institute of Standards and Technology, Gaithersburg.
- 39. Wolle, M.M., Rahman, G.M.M., Kingston, H.M.S. and Pamuku, M. (2014) Optimization and Validation of Strategies for Quantifying Chromium Species in Soil Based on Speciated Isotope Dilution Mass Spectrometry with Mass Balance. Journal of Analytical Atomic Spectrometry, 29, 1640-1647. http://dx.doi.org/10.1039/C4JA00133H