Journal of Environmental Protection
Vol. 3 No. 8A (2012) , Article ID: 21769 , 13 pages DOI:10.4236/jep.2012.328105
Screening for Genotoxicity and Oestrogenicity of Endocrine Disrupting Chemicals in Vitro
![]()
Endocrine Disruption Group, Department of Life and Physical Sciences, School of Science, Athlone Institute of Technology, Athlone, Co. Westmeath, Ireland.
Email: *cbrougham@ait.ie
Received May 16th, 2012; revised June 18th, 2012; accepted July 20th, 2012
Keywords: Endocrine disrupting chemicals (EDCs); proliferation; mutagenicity; DNA strand breakage; comet assay; DNA repair; unscheduled DNA synthesis assay (UDS); E-SCREEN assay; YES assay; MVLN assay
ABSTRACT
A diverse range of endocrine disrupting chemicals (EDCs) was examined, using an in vitro test system, for critical events required for the onset of carcinogenesis in vivo. The initiation stage of carcinogenesis is a genotoxic process. 4-Octylphenol (alkylphenol), bisphenol A (plasticiser), coumestrol and genistein (phytoestrogens), 2,4-dichlorophenoxyacetic acid and toxaphene (pesticides) and ethinylestradiol (synthetic hormone) were investigated for potential mutagencicity, DNA strand breakage, clastogenicity and DNA repair. Significant induction in the percentage of cells containing micronuclei was observed for all the EDCs. Toxaphene and coumestrol were mutagenic in the Ames assay. They also induced significant levels of unscheduled DNA synthesis and DNA strand breakage. Bisphenol A induced low level DNA strand breakage in HepG2 cells in the comet assay. The EDCs, with the exception of toxaphene, induced transcriptional activation in the yeast estrogen screen (YES) assay. They were potently oestrogenic in the mammalian based MVLN (transactivation) and E-SCREEN (proliferation) assays. This report on the transactivational, proliferative and genotoxic ability of the EDCs suggests that these chemicals may play a role in the etiology of male and female reproductive cancers.
1. Introduction
Screening for genotoxicity and proliferative ability of endocrine disrupting chemicals (EDCs) is a reliable tactic for the evaluation of genetic hazard and to obtaining information indicating possible carcinogenic potential. The first stage of carcinogenesis is normally rapid and ireversible and is believed to involve a change in the genetic material of the cell.
Oxidation of oestradiol forms catechol oestrogens which denote intermediates in the generation of more reactive semiquinones and quinones. These quinones can serve as substrates for redox cycling with the associated generation of reactive oxygen species which can bind covalently to peptides, proteins and DNA [1]. Most carcinogenic initiators are mutagenic or genotoxic and a battery of short-term in vitro mutagenicity and genotoxicity tests have been developed to allow the detection of chemicals with potential initiating activity. While modifications in cellular DNA are essential for carcinogenesis such perturbations alone are not sufficient to bring about cancer in all cases.
After the initial transformation a pre-neoplastic cell can remain dormant and the necessary cell proliferation may be induced by a promoter. The enhanced cell turnover could lead to “fixation” of genotoxic damage. The faster the cells are dividing, the greater the chance that genotoxic damage will not be repaired prior to cell division, resulting in clonal expansion and development of neoplasia. It has been predicted that oestrogens are carcinogenic due to their ability to stimulate cell proliferation [2]. Endogenous hormones are associated with the development of specific neoplasia and are a mechanism by which epigenetic carcinogenesis arises. Hormone imbalances play a major causative role in cancers of certain hormone-sensitive tissues. Hormones are generally categorised as promoters of neoplasia. The hormonal association with cancer may relate to an increase in cell turnover among cells that already possess latent genetic damage. Alternatively, hormonal imbalance could lead to an increased proliferation of a sensitive cell population which undergoes secondary genotoxic damage from any one of several environmentally prevalent genotoxic agents.
Endogenous hormones can be replaced by chemicals in the environment that mimic their actions resulting in an agonistic or antagonistic profile [3-7]. It is possible that EDCs act in the same manner as endogenous oestrogens by increasing endometrial proliferation. Several of the most common cancers in western societies occur in hormonally responsive tissues, including breast, endometrium and ovarian in women and prostate in men. These cancers have been causally correlated to exposure to synthetic or endogenous steroidal hormones or their metabolites [8]. The reactivity of catecholoestrogens and their corresponding quinones support the proposal of a mechanism of carcinogenesis by oestrogens based on tumour initiation by covalent DNA damage and hormone-dependent growth stimulation of initiated cells [2].
The assessment of possible drug-related genotoxic potential requires the use of various short-term tests suitable for the detection of all types of genetic endpoints. The EDCs were investigated for genotoxicity, using the Comet assay, the in vitro unscheduled DNA synthesis assay (UDS) and the in vitro cytokinesis-block micronucleus technique (CBMN). The EDCs were also investigated for the potential to induce mutagenesis using the well established bacterial reverse-mutation test, the hallmark of mutagencity.
Oestrogens are defined by their ability to elicit the mitotic stimulation of the tissues of the female genital tract through genomic and non-genomic mechanisms. Measuring cell proliferation and transcriptional activity is of key importance in assessing oestrogenicity. Proliferation of oestrogen-sensitive human breast cancer cells is a simple but biological relevant in vitro model for the oestrogen-dependant tumour promotion of mammary carcinoma. The E-SCREEN assay was developed to assess the oestrogenicity of a broad spectrum of environmental chemicals using MCF-7 human breast cancer cell proliferation as an endpoint. The Yeast oestrogen screen (YES) and the MVLN (MCF-7 breast cancer cells (M) transfected with Xenopus vitellogenin A2 gene (V) controlling the firefly luciferase structural gene (L), transfected clones selected by resistance to neomycin (N)) assays are used to assess ER-dependent transcriptional expression of the EDCs.
The EDCs tested for their ability to induce initiation and proliferation were 4-octylphenol (4-OP), bisphenol A (BIS A), toxaphene (TOX), 2,4-dichlorophenoxyacetic acid (2,4-D), genistein (GEN), coumestrol (COUM) and ethinylestradiol (EE2).
2. Materials and Methods
2.1. Materials
Dimethyl sulphoxide, diisononylphthalate (≥99%), diethylhexylphthalate (≥99%), diisododecylphthalate (≥99%), dibutylphthalate (≥99%), L-glutamine, sodium bicarbonate, D-glucose, sulphorhodamine B, TCA, tris base, phenol red-free Dulbecco’s modified eagle medium (DMEM), potassium phosphate monobasic anhydrous, ammonium sulphate, potassium hydroxide pellets, magnesium sulphate anhydrous, iron(III) sulphate pentahydrate, L-leucine, L-histidine free base, adenine free base, L-arginine hydrochloride, L-methionine, L-tyrosine free base, L-isoleucine, L-lysine monohydrochloride, L-phenylalanine, L-glutamic acid free acid, L-valine, L-serine, thiamine hydrochloride, pyridoxine, D-pantothenic acid hemicalcium salt, inosital, d-biotin, D-(+)-glucose anhydrous, L-aspartic acid free acid, L-threonine, copper(II) sulphate anhydrous, sodium hydroxide pellets, sodium chloride, EDTA dihyate, triton X-100, sigma 7-9R tris, ethidium Bromide, electrophoresis film, hydrogen peroxide, Hams F-12 nutrient mixture, cytochalasin B, acridine orange, formaldehyde, methanol, hydroxyurea crystalline, nitroquinoline-N-oxide, acetic acid, non-essential amino acids and glycerol purchased from Sigma Aldrich (Ireland). 4-onylphenol (≥98%), 2-aminoanthracene and sodium azide purchased from Lennox (Ireland). 2-Nitrofluorene purased from Merck (Ireland). 17bOestradiol (≥97%) purhased from Merck (Germany). Hylcone foetal bovine serum, sodium pyruvate, L.M.P. agarose, scintillation cocktail-Ecoscint, Nunc 96 microwell plates and white solid 96 microwell plates purchased from Bio Sciences (Ireland). Linbro 24 well tissue culture plates and DMEM purchased from MP Biomedicals (UK). 3H Thymidine purchased from Amersham (UK). CPRG purchased from Fannin Healthcare (Ireland). Luciferase cell culture lysis reagent and Bright glo luciferase assay system purchased from Medical Supply (Ireland). Quadriperm plus dishes purchased from Sartorius (UK). Petri dishes, 6 well TC plate and cell culture discs purchased from Sarstedt (Ireland). Corning 12 well TC plates purchased from Fannin (Ireland). Nutrient agar and nutrient broth Oxoid No.2 purchased from Fannin (Ireland). NADPH reagent “A”, NADPH reagent “B” and S9 fraction purchased from Mol.Tox.Inc. (USA).
2.2. Propagation of MCF-7 BOS and MVLN Cells
MCF-7 BOS cell and MVLN cells were cultivated in DMEM supplemented with sodium bicarbonate and 5% Hyclone foetal bovine serum. The cell lines were maintained in a cell incubator with humidified air and a CO2 concentration of 5%.
2.3. E-SCREEN Assay
Preparation and storage of media, charcoal-dextran stripped serum and assay procedure for the E-SCREEN assay was carried out according to Soto et al., 1995 [9] with the following deviation. The bioassay was terminated on day six by carrying out a sulphorhodamine B (SRB) Protein/biomass estimation assay.
2.4. MVLN Assay
Assay procedure for the MVLN assay was carried out according to Pons et al., 1990 [10] with the following deviations. Cells were seeded at 5 × 105 cells/ml in growth medium for 24 h. Cells were washed and resuspended in experimental medium for 48 h. Test-compounds were added in experimental medium for 24 h. Firefly luminescence was measured immediately.
2.5. YES Assay
The yeast oestrogen screen, previously described by Routledge and Sumpter, 1996 [11] was used to test the environmental samples for oestrogenic activity.
2.6. Ames Standard Plate Incorporation Assay
The procedure of bacterial cultivation, verification of genetic markers and incubation with microsome fraction from rat liver were preformed following standard procedures [12].
2.7. The CBMN Assay
Chinese Hamster Ovary (CHO) cells (ECACC, UK) (2 × 104/ml) were seeded onto cell culture discs in 6 well tissue culture plates. Test chemical was added for 24 h. The slides were washed twice with 0.1 M phosphate buffer pH 6.45 and cytochalasin B (3 µg/ml) was added for 12 h. The slides were fixed in ice cold methanol: acetic acid (3:1) containing 0.74% formaldehyde. The fixed cells were washed in 0.1 M phosphate buffer pH 6.45 and stained in acridine orange (12.5 mg/100ml in 0.1 M phosphate buffer pH 6.45) for 1 min. The discs were rinsed in phosphate buffer for 10 min and rinsed in fresh phosphate buffer for 15 min. Cells were examined for micronuclei using fluorescent microscopy. The criteria for identifying micronuclei were performed according to Fenech, 1993 [13].
2.8. UDS Assay
HepG2 cells (5 × 105 ml) were seeded in 12 well cell culture dishes for 24 h in low serum medium (0.5%) for 4 days. 1 ml medium containing 10 mM hydroxyurea was added and incubated for 1 h. Test chemical was added in fresh culture containing 0.005 mCi/ml 3H thymidine in the presence of 10 mM hydroxyurea and incubated for 3 h. The cells were collected onto glass microfibre filter discs using a cell scraper and were washed with 6 ml PBS, 10 ml of 5% TCA and 5 ml of absolute ethanol. The filters were placed in 10 ml of scintillation cocktail and analysed in the liquid scintillation counter for radioactivity due to thymidine uptake in the cells.
2.9. Comet Assay
Assay procedure for the comet assay was carried out according to Singh et al., 1988 [14]. Tail moment [15] was chosen as a measure of DNA damage and was obtained using a computerized image analysis system—Comet assay IV perceptive instruments.
2.10. Statistical Analysis
The EC20 values were calculated using the LevenbergMarquardt fit model (Xlfit2, Microsoft Excel, ID Business Solutions, UK). The EEq of each sample was calculated using the EC20 value of the sample.
Each experiment was tested for normality using the Anderson-Darling test. Differences between the equality of population medians and diverse treatment groups were assessed using the Kruskal-Wallis test and the MannWhitney test. One-way analysis of variance (ANOVA) was used for normally distributed data and the 2-sample t test was used to compute the difference between the means of the diverse treatment groups. A p value of ≤0.05 was regarded as significant.
3. Results
3.1. Mutagenic and Genotoxic Potential of EDCs Using the Ames, CBMN, UDS and Comet Assays
TOX induced significant levels of frameshift mutations in TA98 (Tables 1, 4 and 5), TA100, TA1538 and TA97a in the absence of metabolic activation. A mutagenic index of 128.7 was achieved at 1000 µg/plate in TA97a. The lowest observed effect concentration (LOEC) for TOX occurred at 0.01 µg/plate for TA97a. A decrease in TOX mutagenicity was observed in the presence of metabolic activation. COUM induced a dose-related increase in TA97a with a mutagenic index of 15 at 10 µg/ plate (Tables 1, 4 and 5). A dose-related increase was also observed in TA102 in the absence of metabolic activation. COUM was not mutagenic following exposure to the mixed function oxidase enzymes of the S9 fraction. Bioactivation of TOX and COUM resulted in reduced or lack of mutagenicity, indicating the parent compound being more mutagenic that the metabolites.
All of the EDCs tested induced statistically significant clastogenicity in the CBMN assay (Tables 2, 4 and 5). Significant induction in the percentage of cells containing UDS was observed after treatment with 1 × 10−7 M TOX

Table 1. Mutagenicity of EDCs in the Ames assay.
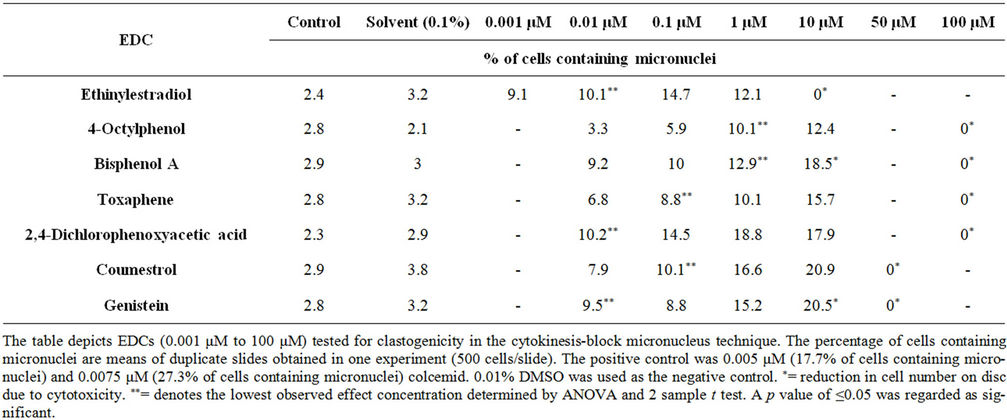
Table 2. Response of micronucleus assay to EDCs using Chinese Hamster ovary (CHO) cell line.
while COUM did not induce DNA repair at the doses tested (Figure 1, Tables 4 and 5). Statistically significant induction in DNA damage was observed in the comet assay after treatment with 1 × 10−8 M BIS A, TOX and COUM (Figure 2, Tables 4 and 5) in the HepG-2 cell line. BIS A induced low level DNA strand breakage (Table 4).
3.2. Oestrogenic Potential of EDCs in the E-SCREEN Assay
MCF-7 BOS cells exhibited a good response after treatment with the EDCs inducing statistically significant proliferation (Tables 4 and 5). The maximum amplitudes obtained with some of these chemicals were close to 17β-oestradiol used as a positive control (Figures 3 and 4). A comparison of lowest observed effect concentration (LOEC), the relative proliferative effect (RPE) and the relative proliferative potency (RPP), obtained with the test compounds in the E-SCREEN assay showed considerable differences in potency between the chemicals tested (Table 3). 17β-Oestradiol induced the maximal oestrogenic response at the lowest concentrations. The detection limit of the assay for the endogenous oestrogen 17β-oestradiol was at 1 × 10−4 nM.
3.3. Oestrogenic Potential of EDCs in the MVLN and YES Assays
The maximum amplitudes obtained with some of the EDCs in the MVLN assay were close to 17β-oestradiol (Figures 5 and 6, Table 4). Statistically significant induction in reporter gene activity was observed after treatment with all of the EDCs indicating that these chemicals are capable of binding to and activating the oestrogen receptor (Table 5).
Maximal induction of luciferse in the MVLN assay was recorded as that achieved for 10 nM 17β-oestradiol however, 50 µM GEN produced a luminescence response approximately 2.5 times the maximal induction achieved

Figure 1. DNA repair induced by EDCs measured by the unscheduled DNA synthesis assay using the Human Hepatoma cell line, HepG2. Values represent the mean nanogram level of thymidine incorporated per 5 × 105 cells, where n = 3. Graph depicts DNA repair symbolic of DNA damage induced by toxaphene (TOX) and coumestrol (COUM) (0.1 μM to 100 μM). The positive control was nitroquinoline-N-oxide which gave 0.9 ng thymidine incorporated at 0.1 μM. 0.01% DMSO was used as the negative control (0.06 ng thymidine incorporation).
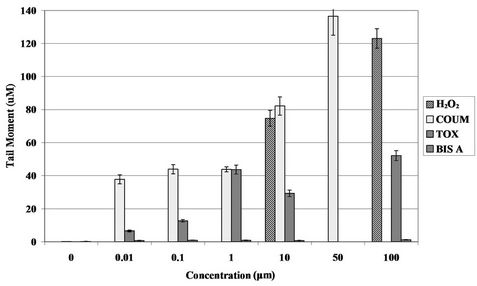
Figure 2. DNA strand breakage induced by EDCs detected by the comet assay using a Human Hepatoma cell line (HepG2). Values represent the mean tail moment, where n = 50. Graph depicts DNA strand breakage induced by hydrogen peroxide (H2O2), the positive control (10 μM and 100 μM), coumestrol (COUM), toxaphene (TOX) and bisphenol A (BIS A) (0.01 μM to 100 μM). 10 μM and 100 µM H2O2 gave a tail moment values of 75 µM and 123 µM respectively. Tail moment is the equivalent to the integrated value of density multiplied by migration distance. 0.01% DMSO was used as the negative control inducing inducing tail moment values between 0.1 μM and 0.2 µM.
for 17β-oestradiol (Figure 6 and Table 4).
The potency of the EDCs in the YES assay was characterised by the lowest observed effect concentration (LOEC) determined by statistical analysis (Table 5). Potency of the test chemical is related to the effective concentrations i.e. the lower the effective concentration the higher the potency. A comparison of these values obtained with the test compounds in the YES assay showed considerable differences in potency between the EDCs tested (Table 4). Statistically significant induction in

Figure 3. Proliferative ability of EDCs demonstrated by the E-SCREEN cell proliferation assay. Values represent the mean +/− S.E.M., where n = 4. Graph depicts proliferation induced by the positive control, 17β-oestradiol (E2) (1 × 10−5 nM to 1 × 102 nM), ethinylestradiol (EE2), bisphenol A (BIS A) and 4-octylphenol (4-OP) (1 × 10−3 nM to 1 × 105 nM) (1.E−03 = 1 × 10−3). 0.01% DMSO was used as the negative control inducing proliferation between 3 × 104 and 5 × 104 cells per well.
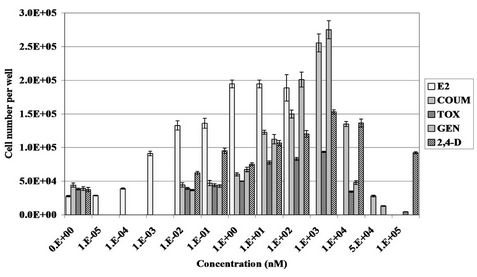
Figure 4. Proliferative ability of EDCs demonstrated by the E-SCREEN cell proliferation assay. Values represent the mean +/− S.E.M., where n = 4. Graph depicts proliferation induced by the positive control, 17β-oestradiol (E2) (1 × 10−5 nM to 1 × 102 nM), coumestrol (COUM), toxaphene (TOX), genistein (GEN) and 2,4-dichlorophenoxyacetic acid (2,4-D) (1 × 10−3 nM to 1 × 105 nM) (1.E−03 = 1 × 10−3). 0.01% DMSO was used as the negative control inducing proliferation between 3 × 104 and 5 × 104 cells per well.
reporter gene activity was observed after treatment with all EDCs tested with the exception of TOX (Figures 7 and 8) indicating that these chemicals are capable of binding to and activating the oestrogen receptor.
4. Discussion
The chemical stability of DNA is one of the fundamentals of life. Alterations in the chemical structure of DNA occur frequently, interfere with transcription and replication, and result in cell death or carcinogenesis if allowed to accumulate. Contaminants released into the aquatic environment have the potential to damage the DNA of exposed organisms resulting in genetic disorders and
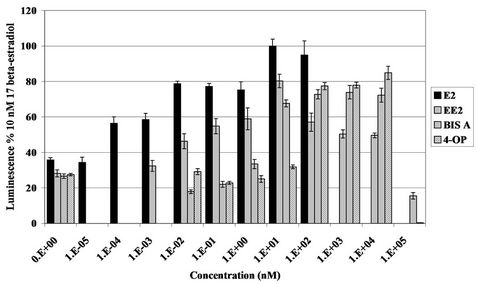
Figure 5. Transcriptional activation induced by EDC demonstrated by the MVLN reporter gene assay. Values represent the mean +/− S.E.M., where n = 4. Graph depicts oestrogenic effect of ethinylestradiol (EE2) (1 × 10−3 nM {1.E− 03 = 1 × 10−3} to 1 × 104 nM), bisphenol A (BIS A) and 4-octylphenol (4-OP) (1 × 10−2 nM to 1 × 105 nM) compared to 17β-oestradiol, the positive control (1 × 10−4 nM to 1 × 102 nM). Chemical concentrations were plotted against luciferase activity achieved as a percentage of the positive control, 10 nM 17β-oestradiol. 0.01% DMSO was used as the negative control.

Figure 6. Transcriptional activation induced by EDC demonstrated by the MVLN reporter gene assay. Values represent the mean +/− S.E.M., where n = 4. Graph depicts oestrogenic effect of toxaphene (TOX), 2,4-dichlorophenoxyacetic acid (2,4-D), genistein (GEN) and coumeatrol (COUM), bisphenol A (BIS A) and 4-octylphenol (4-OP) (1 × 10−2 nM {1.E−02 = 1 × 10−2} to 1 × 105 nM) compared to 17β-oestradiol, the positive control (1 × 10−4 nM to 1 × 102 nM). Chemical concentrations were plotted against luciferase activity achieved as a percentage of the positive control, 10 nM 17β- oestradiol. 0.01% DMSO was used as the negative control.
genotoxic effects. DNA contains a number of reactive sites and its structure can be modified in numerous ways by genotoxic agents. Scrutinising the genotoxic effects of pollutants is of fundamental relevance in the aquatic environment in the assessment of pollution-associated strain on living organisms. The most obvious reason for measuring the level of exposure to genotoxic substances in the environment relates to carcinogenesis. Many carcinogenic initiators are genotoxic and genotoxic chemicals may also contribute to other toxic responses including
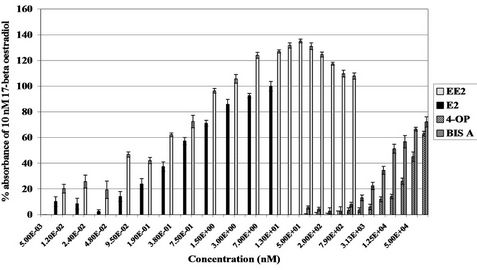
Figure 7. Transcriptional activation induced by EDCs demonstrated by the YES reporter gene assay. Values respresent the mean +/− S.E.M., where n = 4. Graph depicts oestrogenicity induced by ethinylestradiol (EE2) (1.2 × 10−2 nM to 1.57 × 103 nM), 4-octylphenol (4-OP) and bisphenol A (BIS A) (50 nM to 1 × 105 nM {5.0E+01 = 50 nM}). Chemical concentrations were plotted against absorbance at 560 nM achieved as a percentage of the positive control, 10 nM 17β-oestradiol. Ethanol was used as the negative control.

Figure 8. Transcriptional activation induced by EDCs demonstrated by the YES reporter gene assay. Values respresent the mean +/− S.E.M., where n = 4. Graph depicts oestrogenicity induced by coumestrol (COUM) (7.5 × 10−1 nM to 1 × 105 nM), genistein (GEN) and 2,4-dichlorophenoxyacetic acid (2,4-D) (50 nM to 1 × 105 nM {5.0E+01 = 50 nM}). Chemical concentrations were plotted against absorbance at 560 nM achieved as a percentage of the positive control, 10 nM 17β-oestradiol. Ethanol was used as the negative control.
teratogenic effects and hereditary defects through mutations in germ line cells.
The Ames assay was used as a first step in defining the potential mutagenicity of the EDCs. Most established carcinogens are mutagens with 85% of the carcinogens tested been detected as mutagens. TOX and COUM induced frameshift mutations in one strain or more in the Ames assay. The presence of micronuclei in somatic cells is recognised as a cytogenetic indicator of genotoxicity. Chromosome breakage and aneuploidy is an indication of exposure to genotoxic compounds which may increase the risk of cancer [16]. The in vitro micronucleus test with human lymphocytes is also used for human monitoring. An increase in the percentage of cells
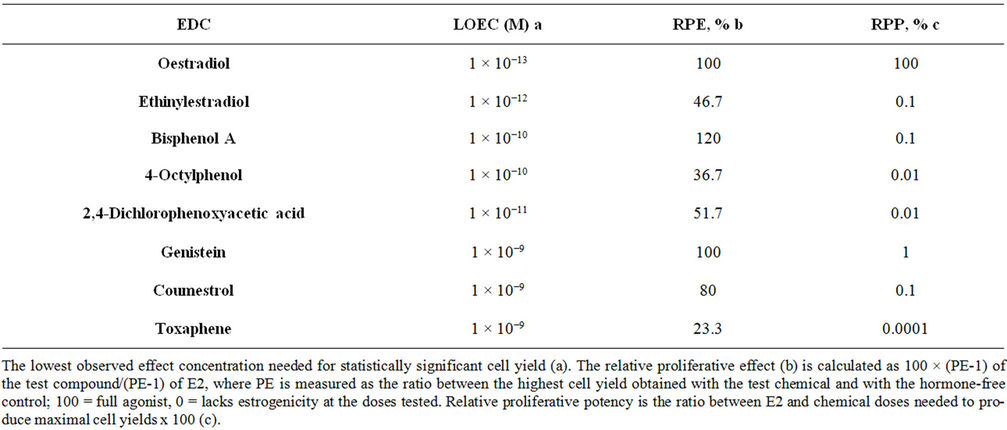
Table 3. Oestrogenic effect of various EDCs measured by the E-SCREEN assay.
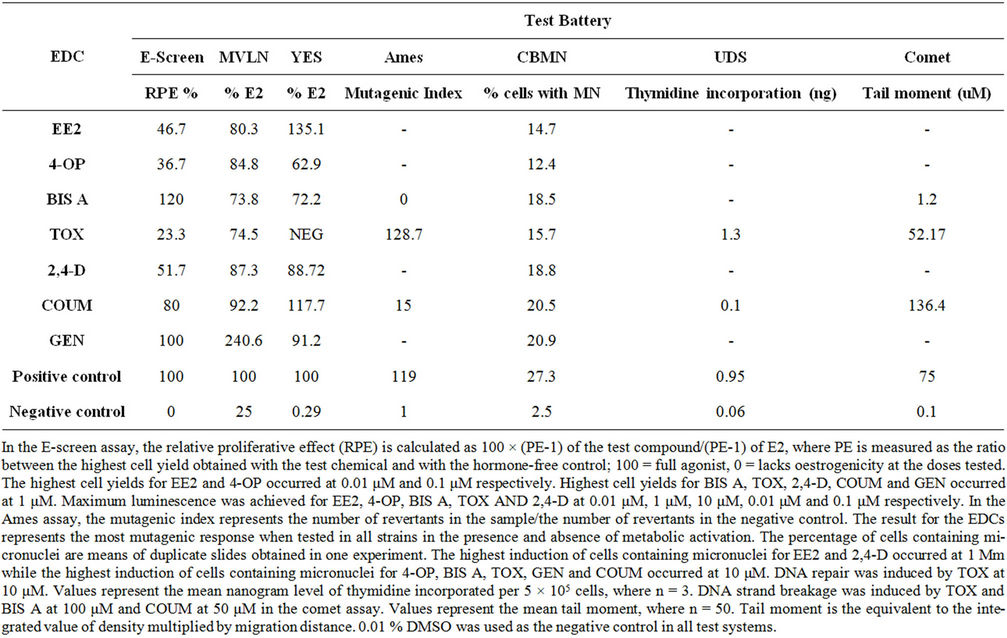
Table 4. The oestrogenic and genotoxic potential of EDCs.
containing micronuclei was observed after exposure to all of the EDCs tested indicating that these chemicals are capable of DNA damage. Some of the EDCs induced DNA damage at similar levels to colcemid, while others were weakly genotoxic. BIS A induces kinetochorepositive micronuclei formation in V79 cells [17] while Lehmann and Metzler, 2004 [18] have reported that two congeners of BIS A induce mitotic arrest and kinetochore-positive micronuclei in human fibroblasts. It has been reported that BIS A is not likely to be carcinogenic to humans based on experimental animal and metabolism studies. However in the same study it did induce micronuclei formation in Chinese hamster V79 cells [19].
COUM is a strong inducer of structural chromosomal
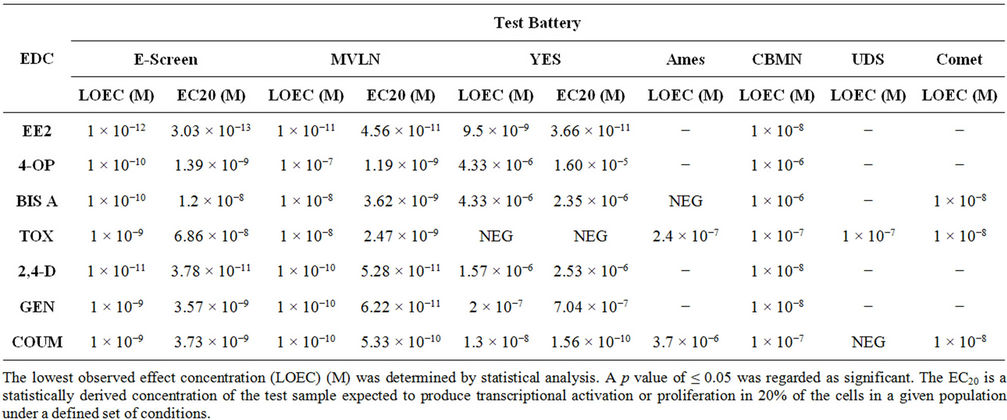
Table 5. Summary table of proliferative, transactivational and genotoxic ability of EDCs.
aberrations in cultured human peripheral blood lymphocytes [20]. The ability of the EDCs to induce micronucleus frequencies does not necessarily correlate with their hormonal activity. For example, TOX was one of the most potent of the EDCs tested in the microncleus assay and the comet assay but proved to be one of the least oestrogenic of the EDCs tested in both the MVLN and E-SCREEN assays.
As most chemical carcinogens in their ultimate reactive form are electrophiles that react with DNA and thereby may result in DNA repair, the measurement of DNA repair is a reliable determination of carcinogenic potential. Berwick and Vineis, 2000 [21] have shown a positive correlation between defects in DNA repair capacity and development of cancer in humans.
TOX induced significant levels of DNA repair in the HepG2 cell line. TOX is a complex mixture containing hundreds of polychlorinated bornane isomers which is formed by chlorination of technical camphene. The use of TOX has been limited in the United States since 1982 and banned by the EPA in 1990 following concerns over bioaccumulation and toxicity [22]. TOX and toxaphenelike preparations are still utilised in South America, India, Africa, Mexico, Romania, Hungary, Germany, Poland, and The USSR. It has been shown to bioaccumulate by biota inhabiting regions hundreds and thousands of kilometres away from location of use. Although most isomers persevere in the environment, the human body burden of TOX consists of two to three particular congeners that preferentially accumulate in liver and adipose tissue, and become concentrated in the food chain [22]. Very high concentrations of TOX have been found in the lipidrich tissues of crocodiles (Crocodylus porosus) from the Ord River in Western Australia almost thirty years after its use ceased in the area [23]. A case study analysing two mountain lakes for TOX residues in the water, aquatic plants, aquatic invertebrates and fish, following treatment with TOX for fish-eradication demonstrated the highly persistent nature of TOX and its potential bioaccumulation in higher trophic levels [24]. The initial residues, in one particular lake, declined sharply and remained at 2 p.p.b. (4.83 × 10−9 M) for approximately five years. This concentration of TOX had the ability to induce cell proliferation in the E-SCREEN in this report. In the same study [24] levels in aquatic plants were as high as 17 p.p.m. (41 μM) and accumulation of whole body concentrations of TOX in trout inhabitants of the shallow lake occurred up to 14 p.p.m (34 µM). It is im- portant to note that in this study TOX had the ability to stimulate the oestrogen receptor and induce mutagenicity and genotoxicity in the test battery at these micromolar levels. This signifies the critical aspect of bioaccumulation and persistence of pesticides in the environment.
It has been noted that the amount of DNA repair induced in a given time is sometimes greater with weak carcinogens than with potent ones. Certain carcinogens may have different effects on repair enzyme activity or stimulation of different repair processes depending on the lesion. The relative average amount of UDS elicited by carcinogens in this system is probably more a function of the type of DNA damage and repair provoked than of the potency of the carcinogen. COUM did not induce DNA repair in the UDS assay. It is important to note that the phytoestrogen was positive in the Ames assay, the comet assay and the micronucleus assay.
The comet assay was used to ascertain the genotoxic potency of the EDCs through induction of DNA strand breaks in human hepatoma cells (HepG2). The use of the HepG2 cell line in the comet assay demonstrates toxicity in vitro from a mammalian perspective. HepG2 cells retain the activities of phase I and phase II enzymes during in vitro cultivation. These enzymes play key roles in the activation and detoxification of DNA reactive carcinogens. The comet assay detects strand breaks with high sensitivity for carcinogens [25]. It provides a useful tool for simultaneous comparison of effects of environmental contaminants. The EDCs analysed in the comet assay all induced DNA strand breaks. BIS A did induce single strand breakage albeit at low levels.
The genotoxicity assays outlined here can be used as indicators for chromosomal and DNA damaging effects of environmental contaminants. Aberrations in the genome can ultimately lead to the development of cancer therefore exposure to EDCs reported here may have serious implications for reproductive integrity and tumourigenesis.
The oestrogen induced increase in the number of MCF-7 breast cancer cells is recognised as biologically equivalent to the increase of mitotic activity in the rodent endometrium [26]. The results reported here for 4-OP, BIS A, TOX, 2,4-D, GEN, COUM and EE2 in the ESCREEN assay suggest that they have the ability to mimic oestrogen action in breast cancer cells indicated here by proliferation and therefore may also act as promoters in the carcinogenic process by inducing proliferation and possibly permitting uncontrolled growth of spontaneous or carcinogen-induced mutations. TOX has extremely toxic endocrine disrupting abilities in female zebrafish with a decrease in oviposition, number of eggs spawned and an increase in severely deformed embryos exposed to the pesticide [27]. It has been suggested that toxaphene is a hepatocarcinogen exerting its effect through a non-genotoxic or promotional mechanism rather than a genetic mechanism [28]. Carcinogenic effects of toxaphene exposure have been encountered in vivo in both male and female rats, with an increase in benign and malignant neoplasm’s of the reproductive organs in females, the mammary gland in males and endocrine system and liver in both [29]. White et al., 1994 [30] demonstrated that a number of alkylphenolic compounds found in river water were capable of stimulating growth of breast cancer cells while laboratory trials have shown that BIS A induces expression of oestrogen controlled genes in various assays [31]. Shimizu et al., 2002 [32] demonstrated that sulphation of BIS A abolished its oestrogenicity based on proliferation and gene expression in the ESCREEN assay. The phytoestrogen COUM was potently oestrogenic producing a statistically significant proliferation response (1 µM) approximately 1.5 times the maximal induction achieved for 17β-oestradiol (10 nM).
High sensitivity is an essential prerequisite for screening assays to facilitate detection of compounds of low potency that might be of biological significance through chronic exposure and high profusion in the environment. The determination of whether EDCs interact directly with the oestrogen receptor is essential for understanding the risk associated with exposure to them. Luciferase expression in the MVLN cell line mimics a human natural hormonal response. The ability of a chemical to activate oestrogen-responsive genes through the ER may indicate that the chemical will be oestrogenic in vivo. It is possible that some EDCs may show preferential binding to one or the other oestrogen receptors. EDCs may exert a spectrum of activity different from that caused by endogenous oestrogen because of different affinities of these compounds for oestrogen receptor α and β. This perhaps explains the difference in potency for the chemicals tested here in the MVLN assay. It has been shown that 17β-oestradiol binds preferentially to oestrogen receptor-α while xenoestrogens and phytoestrogens showed an almost equal binding affinity for both receptor subtypes [33].
The potency of some of the EDCs in the MVLN assay was slightly lower than those achieved for the ESCREEN assay however the MVLN assay appeared to be more sensitive to phytoestrogen exposure than the E-SCREEN assay. This phenomenon of super-induction by GEN in the MVLN assay was not achieved by any of the other chemicals tested. This may be explained by the high affinity of GEN for the oestrogen receptor β but the fundamental mechanism remains unknown and it is unclear whether cell processes other than the oestrogen receptor are involved. GEN has shown differential transactivation of the oestrogen receptor α and oestrogen receptor β with up to 100-fold stronger activation of the oestrogen receptor β in breast cancer cells [34]. Metabolites of isoflavones i.e. isoflavone glycosides bind weakly to both oestrogen receptors and subsequently transcriptional expression is poor however the GEN glycoside can stimulate proliferation more strongly than the parent compound, suggesting bioactivation is necessary for its oestrogenic activity [35]. GEN can be found in herbal remedies for the treatment of menopausal symptoms and is administered up to levels of 10 mg daily.
The relative potencies of EDCs in the YES assay when compared to 17β-oestradiol are perhaps underestimated by the in vitro test due to the fact that circulating endogenous oestrogen is bound to plasma proteins and only a small percentage has the ability to infiltrate cells and activate the oestrogen receptor. EDCs have a much lower affinity for plasma proteins and as a result these chemicals are unbound in the blood and possibly will be available for oestrogen receptor activation [36]. It has also been suggested that EDCs may displace endogenous sex steroid hormones from human sex hormone-binding globulin binding sites and disrupt the androgen-to-oestrogen balance leading to elevated levels of endogenous hormones [37].
The activities of EE2, BIS A, 4-OP, 2,4-D, COUM and GEN in the YES assay were reproduced in the mammalian assays, MVLN and E-SCREEN, thus implying that these are real oestrogenic effects. Both the E-SCREEN and MVLN assays appear to be more sensitive in detecting weak oestrogenic activities than the YES assay.
For some of the chemicals tested, only a partial dose response was observed after the incubation time. Bioassays like the E-SCREEN assay, the MVLN assay and the YES assay are preferential methods when assessing whether a chemical is oestrogenic and when trying to ascertain the cause of suspected oestrogen contamination. The oestrogenicity of chemicals cannot be predicted on a structural basis alone therefore, it is unknown how many EDCs are present in the environment. Molecular identification methods are valuable to evaluate the presence and concentration of particular EDCs previously known to be oestrogenic but this precludes the use of chemical analysis as a tool for determining whether or not EDCs are present in environmental samples. The E-SCREEN assay and MVLN assay could be applied reliably to predict oestrogenic effects on human health because the target cells are of human origin. These assays also allow the screening of multiple compounds in a wide range of doses and are sensitive enough to detect weak oestrogenic activity of four orders of magnitude more concentrated than that of 17β-oestradiol without exceeding the solubility of the compounds tested.
In vitro studies lack the ability to detect the effects of metabolism, plasma-protein binding, and pharmacokinetics on oestrogen activity however, they are imperative for preliminary testing and prioritising chemicals for more extensive studies in vivo. Short-term methods like the E-SCREEN assay, the MVLN assay and the YES assay are necessary to detect EDCs in order to regulate the environmental burden.
Many of the chemicals shown to mimic the action of endogenous oestrogens are reaching freshwater environments and water supplies in Ireland [38-43], the United Kingdom [44,45], the Netherlands [46], Germany [47], [48], the USA [49] and China [50]. Moreover, a recent survey by Kelly et al., 2010 [51] reported levels of phthalates and alkylphenols at various river locations in the Shannon International River Basin District of Ireland. The concentrations of some of the phthalates and alkylphenols found were even higher than those required to elicit a response in the in vitro E-SCREEN and MVLN assays reported previously [52]. For example, levels of diethylhexylphthalate at Athlone Lock and the Hind River were 2 × 104 times higher than levels required to elicit a response in the E-SCREEN and 20 times higher than levels required to elicit a response in the MVLN assay. Levels of diisononylphthalate at the same locations were 4 times higher than levels required to induce a response in the in vitro E-SCREEN. 4-Nonylphenol was found at these locations 100 and 10 fold higher than levels required to elicit a response in the E-SCREEN and MVLN respectively. Intersex in populations of wild roach from rivers in the Shannon basin of Ireland have also been reported for the first time and it is hypothesised that the incidence of ovo-testis is a direct result of exposure to EDCs in waste water effluents [53].
The oestrogenicity observed for the EDCs has serious implications for human health as exposure to these chemicals occurs on a frequent basis. Increased cell proliferation, as demonstrated by the E-SCREEN assay, can have a significant input to the process of carcinogenesis. Fixation of genotoxic damage may occur due to enhanced cell turnover. The faster the cells are dividing, the greater the chance that genotoxic damage will not be repaired resulting in clonal expansion of preneoplastic cells. Hormone imbalances play a major contributory role in cancers of certain hormone-sensitive tissues. The hormonal association with cancer may relate to an increase in cell turnover among cells that already possess latent genetic damage. Therefore human exposure to these EDCs may contribute to the development of various cancers.
Some carcinogens have initiating and promoting activity and can as a consequence induce neoplasms swiftly and in high yield when exposed repetitively. The EDCs reported in this study were mutagenic, genotoxic (Comet and CBMN assays), induced proliferation of breast cancer cells in the E-SCREEN assay and transcritptional activity in the MVLN and YES assays. They possess both initiating and proliferative ability.
4. Conclusions
The second part of this century has been marked by a notable improvement of the human welfare in western countries. Although fundamental in this advancement, the chemical sciences and technologies have conversely generated apprehension in the public for unfavourable effects that may sometimes be derived from these progress endeavours. Scientists have discovered that many chemicals in the environment can mimic the action of oestrogens and are released into the environment as pollutants from agricultural spraying, industrial processes, or municipal and domestic waste.
The hypothesis that EDCs contribute to the decline in male and female reproductive health is supported by studies on wildlife and rodents. It is not acceptable to extrapolate from data that is obtained from laboratory in vitro experiments in which the magnitude and extent of xenobiotic exposure does not always represent circumstances that humans confront in their environment during a natural life.
We have established that a number of EDCs have the ability to induce mutagenesis, clastogenicity in the form of DNA strand breaks, micronuclei formation and unscheduled DNA synthesis indicating DNA repair following mutation. We have also established that the same EDCs cause proliferation in a breast carcinoma cell line and oestrogenicity in a mammalian and yeast based transactivation system. The genotoxicity and proliferative ability of the EDCs in this report is significant as these chemicals may play a role in the etiology of male and female reproductive cancers [54-59]. There is a potential risk of in vivo carcinogenicity following exposure to these EDCs as they can act as initiators and promoters in the carcinogenic process. Many of the chemicals shown to mimic the action of endogenous oestrogens are reaching freshwater environments and water supplies worldwide.
5. Acknowledgements
This work was funded by the Strand III Core Research Strengths Enhancement Scheme. The MCF-7 cells, for the detection of proliferation, were kindly provided by Dr. A.M. Soto, Tufts University, School of Medicine, Boston, Massacheusetts, U.S.A. The recombinant yeast strain for the detection of oestrogenicity was kindly provided by Prof. J.P. Sumpter, Brunel University, UK. The MVLN cell line was kindly provided by Prof. Michael Pons, INSERM, France.
REFERENCES
- J. D. Yager and J. G. Liehr, “Molecular Mechanisms of Estrogen Carcinogenesis,” Annual Review of Pharmacology and Toxicology, Vol. 36, 1996, pp. 203-232. doi:10.1146/annurev.pa.36.040196.001223
- J. Russo and I. H. Russo, “The Role of Estrogen in the Initation of Breast Cancer,” The Journal of Steroid Biochemistry and Molecular Biology, Vol. 102, No. 1-5, 2006, pp. 89-96. doi:10.1016/j.jsbmb.2006.09.004
- J. R. Karavan and M. E. Pepling, “Effects of Estrogenic Compounds on Neonatal Oocyte Development,” Reproductive Toxicology, Vol. 34, No. 1, 2012, pp. 51-56. doi:10.1016/j.reprotox.2012.02.005
- J. Matthews, T. Celius, R. Halgren and T. Zacharewski, “Differential Estrogen Receptor Binding of Estrogenic Substances: A Species Comparison,” The Journal of Steroid Biochemistry and Molecular Biology, Vol. 74, No. 4, 2000, pp. 223-234. doi:10.1016/S0960-0760(00)00126-6
- K. Morito, T. Hirose, J. Kinjo, T. Hirakawa, M. Okawa, T. Nohara, S. Ogawa, S. Inoue, M. Muramatsu and Y. Masamune, “Interaction of Phytoestrogens with Estrogen Receptors α and β,” Biological and Pharmaceutical Bulletin, Vol. 24, No. 4, 2001, pp. 351-356. doi:10.1248/bpb.24.351
- J. C. Gould, L. S. Leonard, S. C. Maness, B. L. Wagner, K. Conner, T. Zacharewski, S. Safe, D. P. McDonnell and K. W. Gaido, “Bisphenol A Interacts with the Estrogen Receptor α in a Distinct Manner from Estradiol,” Molecular and Cellular Endocrinology, Vol. 142, No. 1-2, 1998, pp. 203-214. doi:10.1016/S0303-7207(98)00084-7
- C. Wohlfahrt-Veje, H. R. Andersen, I. M. Schmidt, L. Aks-glaede, K. Sorensen, A. Juul, T. K. Jensen, P. Grandjean, N. E. Skakkebaekand and K. M. Main, “Early Breast Development in Girls after Prenatal Exposure to Non-Persistent Pesticides,” International Journal of Andrology, Vol. 35, No. 3, 2012, pp. 273-282. doi:10.1111/j.1365-2605.2011.01244.x
- C. R. Jefcoate, J. G. Liehr, R. J. Santen, T. R. Sutter, J. D. Yager, W. Yue, S. J. Santner, R. Tekmal, L. Demers, R. Pauley, F. Naftolin, G. Mor and L. Berstein, “TissueSpecific Synthesis and Oxidative Metabolism of Estrogens,” Journal of National Cancer Institute Monographs, Vol. 27, 2000, pp. 95-112.
- A. M. Soto, C. Sonnenschein, K. L. Chung, M. F. Fernandez, N. Olea and F. Serrano, “The E-SCREEN Assay as a Tool to Identify Estrogens: An Update on Estrogenic Environmental Pollutants,” Environmental Health Perspect, Vol. 103, 1995, pp. 113-122. doi:10.1289/ehp.95103s7113
- M. Pons, D. Gagne, J. C. Nicolas and M. Mehtali, “A New Cellular Model of Response to Estrogens: A Bioluminescent Test to Characterise (anti) Estrogen Molecules,” Biotechniques, Vol. 9, 1990, pp. 450-459. doi:10.1234/12345678
- E. J. Routledge and J. P. Sumpter, “Estrogenic Activity of Surfactants and Some of Their Degradation Products Assessed Using a Recombinant Yeast Screen,” Environmental Toxicology and Chemistry, Vol. 15, No. 3, 1996, pp. 241-248. doi:10.1002/etc.5620150303
- K. Mortelmans and E. Zeiger, “The Ames Salmo-Nella/ Microsome Mutagenicity Assay,” Mutation Research, Vol. 455, No. 1-2, 2000, pp. 29-60. doi:10.1016/S0027-5107(00)00064-6
- M. Fenech, “The Cytokinesis-Block Micronucleus Technique: A Detailed Description of the Method and Its Application to Genotoxicity Studies in Human Populations,” Mutation Research, Vol. 285, No. 1, 1993, pp. 35-44. doi:10.1016/0027-5107(93)90049-L
- N. P. Singh, M. T. McCoy, R. R. Tice and E. L. Schneider, “A Simple Technique for Quantitation of Low Levels of DNA Damage in Individual Cells,” Experimental Cell Research, Vol. 175, No. 1, 1988, pp. 184-191. doi:10.1016/0014-4827(88)90265-0
- C. R. Kent, J. J. Eady, G. M. Ross and G. G. Steel, “The Comet Moment as a Measure of DNA Damage in the Comet Assay,” International Journal of Radiation Biology, Vol. 67, 1995, pp. 655-660. doi:10.1080/09553009514550771
- N. J. Duker, “Chromosome Breakage Syndromes and Cancer,” American Journal of Medical Genetics, Vol. 115, No. 3, 2002, pp. 125-129. doi:10.1002/ajmg.10688
- E. Pfeiffer, B. Rosenberg, S. Deuschel and M. Metzler, “Interference with Microtubules and Induction of Micronuclei in Vitro by Various Bisphenols,” Mutation Research, Vol. 390, No. 1, 1997, pp. 21-31.
- L. Lehmann and M. Metzler, “Bisphenol A and Its Methylated Congeners Inhibit Growth and Interfere with Microtubules in Human Fibroblasts in Vitro,” ChemicoBiological Interactions, Vol. 147, 2004, pp. 273-285.
- L. A. Haighton, J. J. Hlywka, J. Doull, R. Kroes, B. S. Lynch and I. C. Munro, “An Evaluation of the Possible Carcinogenicity of Bisphenol A to Humans,” Regulatory Toxicology and Pharmamcology, Vol. 35, No. 2, 2002, pp. 238-254. doi:10.1006/rtph.2001.1525
- S. E. Kulling, B. Rosenberg, E. Jacobs and M. Metzler, “The Phytoestrogens Coumestrol and Genistein Induce Structural Chromomsomal Aberrations in Cultured Human Peripheral Blood Lymphocytes,” Archives in Toxicology, Vol. 73, 1999, pp. 50-54.
- M. Berwick and P. Vineis, “Markers of DNA Repair and Susceptibility to Cancer in Humans: An Epidemiologic Review,” Journal National Cancer Institute, Vol. 92, No. 11, 2000, pp. 874-897. doi:10.1093/jnci/92.11.874
- H. J. De Geus, H. Besselink, A. Brouwer, J. Klungsoyr, B. McHugh, E. Nixon, G. G. Rimkus, P. G. Wester and J. de Boer, “Environmental Occurrence, Analysis, and Toxicology of Toxaphene Compounds,” Environmental Health Perspectives, Vol. 107, 1999, pp. 115-144.
- M. Yoshikane, W. R. Kay, Y. Shibata, M. Inoue, T. Yanai, R. Kamata, J. S. Edmonds and M. Morita, “Very High Concentrations of DDE and Toxaphene Residues in Crocodiles from the Ord River, Western Australia: An Investigation into Possible Endocrine Disruption,” Journal Environmental Monitoring, Vol. 8, No. 6, 2006, pp. 649-661. doi:10.1039/b518059g
- L. C. Terriere, U. Kiigemagi, A. R. Gerlach and R. L. Borovicka, “The Persistence of Toxaphene in Lake Water and Its Uptake by Aquatic Plants and Animals,” Journal Agricultural and Food Chemistry, Vol. 14, No. 1, 1966, pp. 66-69. doi:10.1021/jf60143a021
- W. Liao, M. A. McNutt and W. G. Zhu, “The Comet Assay: A Sensitive Method for Detecting DNA Damage in Individual Cells,” Methods, Vol. 48, No. 1, 2009, pp. 46-53. doi:10.1016/j.ymeth.2009.02.016
- A. M. Soto, T.-M. Lin, H. Justicia, R. M. Silvia and C. Sonnenschein, “An ‘in Culture’ Bioassay to Assess the Estrogenicity of Xenobiotics,” In: T. Colborn and C. Clement, Eds., Chemically Induced Alterations in Sexual Development: The Wildlife/Human Connection, Princeton Scientific Publishing, Princeton, 1992, pp. 295-309.
- G. E. Fahraeus-van Ree and J. F. Payne, “Effect of Toxaphene on Reproduction of Fish,” Chemosphere, Vol. 34, 1997, pp. 855-867. doi:10.1016/S0045-6535(97)00016-7
- C. C. Hedli, R. Snyder, F. K. Kinoshita and M. Steinberg, “Investigation of Hepatic Cytochrome P-450 Enzyme Induction and DNA Adduct Formation in Male CD/1 Mice Following Oral Administration of Toxaphen,” Journal of Applied Toxicology, Vol. 18, No. 3, 1998, pp. 173-178. doi:10.1002/(SICI)1099-1263(199805/06)18:3<173::AID-JAT488>3.0.CO;2-2
- M. D. Reuber, “Carcinogenicity of Toxaphene: A Review,” Toxicology and Environmental Health, Vol. 5, No. 4, 1979, pp. 729-748. doi:10.1080/15287397909529783
- R. White, S. Jobling, S. A. Hoare, J. P. Sumpter and M.G. Parker, “Environmentally Persistent Alkylphenolic Compounds Are Estrogenic,” Endocrinology, Vol. 135, No. 1, 1994, pp. 175-182. doi:10.1210/en.135.1.175
- A. Rivas, M. Lacroix, F. Olea-Serrano, I. Laios, G. Leclercq and N. Olea, “Estrogenic Effect of a Series of Bisphenol Analogues on Gene and Protein Expression in MCF-7 Breast Cancer Cells,” Journal of Steroid Biochemistry and Molecular Biology, Vol. 82, No. 1, 2002, pp. 45-53. doi:10.1016/S0960-0760(02)00146-2
- M. Shimizu, K. Ohta, Y. Matsumoto, M. Fukuoka, Y. Ohno and S. Ozawa, “Sulphation of Bisphenol A Abolished Its Estrogenicity Based on Proliferation and Gene Expression in Human Breast Cancer MCF-7 Cells,” Toxicology in Vitro, Vol. 16, 2002, pp. 549-556. doi:10.1016/S0887-2333(02)00055-3
- B. Gutendorf and J. Westendorf, “Comparison of an Array of in Vitro Assays for the Assessment of the Oestrogenic Potential of Natural and Synthetic Oestrogens, Phytoestrogens and Xenoestrogens,” Toxicology, Vol. 166, No. 1-2, 2001, pp. 79-89. doi:10.1016/S0300-483X(01)00437-1
- D. M. Harries, E. Besselink, S. M. Henning, V. L. W. Go and D. Heber, “Phytoestrogens Induce Differential Estrogen Receptor Alphaor Beta-Mediated Responses in Transfected Breast Cancer Cells,” Experimental. Biology and Medicine, Vol. 230, 2005, pp. 558-568.
- K. Morito, T. Hirose, J. Kinjo, T. Hirakawa, M. Okawa, T. Nohara, S. Ogawa, S. Inoue, M. Muramatsu and Y. Masamune, Interaction of Phytoestrogens with Estrogen Receptors α and β,” Biological Pharmaceutical Bulletin, Vol. 24, No. 4, 2001, pp. 351-356. doi:10.1248/bpb.24.351
- H. H. Jury, T. R. Zacharewski and G. L. Hammond, “Interactions between Human Plasma Sex Hormone-Binding Globulin and Xenobiotic Ligands,” Journal Steroid Biochemistry and Molecular Biology, Vol. 75, 2000, pp. 167-176. doi:10.1016/S0960-0760(00)00168-0,
- H. Dechaud, C. Ravard, F. Claustrat, A. B. de la Pierriere and M. Pugeat, “Xenestrogen Interaction with Human Sex Hormone-Binding Globulin (hSHBG),” Steroids, Vol. 64, 1999, pp. 328-334. doi:10.1016/S0039-128X(98)00114-7
- A. Reid and J. Roche, “Hormone Modulating Substances in the Irish Midlands Shannon Catchment: Extraction, Analysis and Quantification,” Proceedings ESAI ENVIRON 2005.
- A. M. Reid, C. A. Brougham, A. M. Fogarty and J. J. Roche, “Approaches to Enhance the Liquid Chromatographic Determination of Phthalates in Environmental Matrices,” Proceedings ESAI ENVIRON 2006.
- A. M. Reid, C. A. Brougham, A. M. Fogarty and J. J. Roche, “Isocratic LC Methods for the Trace Analysis of Phthalates and 4-Nonylphenol in Varying Types of Landfill and Adjacent Run-Offs,” Toxicological and Environmental Chemistry, Vol. 89, No. 3, 2007, pp. 399-410. doi:10.1080/02772240601116613
- A. M. Reid, C. A. Brougham, A. M. Fogarty and J. J. Roche, “Accelerated Solvent-Based Extraction and Enrichment of Selected Plasticisers and 4-Nonylphenol, and Extraction of Tin from Organotin Sources in Sediments, Sludges and Leachate Soils,” Analytica Chimica Acta, Vol. 634, No. 2, 2008, pp. 197-204. doi:10.1016/j.aca.2008.12.032
- A. M. Reid, C. A. Brougham, A. M. Fogarty and J. J. Roche, “Analysis of Bio-Obtainable Endocrine Disrupting Metals in River Water and Sediment, Sewage Influent/Effluent, Sludge, Leachate, and Concentrated Leachate, in the Irish Midlands Shannon Catchment,” International Journal of Analytical Chemistry, Vol. 2009, 2009, Article ID: 325496, pp. 1-12. doi:10.1155/2009/325496
- H. Tarrant, N. Llewellyn, A. Lyons, N. Tattersall, S. Wylde, G. Mouzakitis, M. Maloney and C. McKenzie, “Endocrine Disruptors in the Irish Aquatic Environment. Environmental RTDI Programme 2000-2006,” Johnstown Castle, Co. Wexford, 2005.
- C. Desbrow, E. J. Routledge, G. C. Brighty, J. P. Sumpter and M. Waldock, “Identification of Estrogenic Chemicals in STW Effluent. 1. Chemical Fractionation and in Vitro Biological Screening,” Environmental Science and Technology, Vol. 32, No. 11, 1998, pp. 1549-1558. doi:10.1021/es9707973
- S. Jobling, R. Williams, A. Johnson, A. Taylor, M. GrossSorokin, M. Nolan, C. R. Tyler, R. van Aerle, E. Santos and G. Brighty, “Predicted Exposures to Steroid Estrogens in U.K. Rivers Correlate with Widespread Sexual Disruption in Wild Fish Populations,” Environmental Health Perspectives, Vol. 114, 2006, pp. 32-39. doi:10.1289/ehp.8050
- A. C. Belfroid, A. Van der Horst, A. D. Vethaak, A. J. Schafer, G. B. J. Rijs, J. Wegener and W. P. Cofino, “Analysis and Occurrence of Estrogenic Hormones and Their Glucuronides in Surface Water and Waste Water in The Netherlands,” The Science of the Total Environment, Vol. 225, No. 1-2, 1999, pp. 101-108. doi:10.1016/S0048-9697(98)00336-2
- P. Spengler, W. Korner and J. W. Metzger, “Substances with Estrogenic Activity in Effluents of Sewage Treatment Plants in South-Western Germany. 1. Chemical Analysis,” Environmental Toxicology and Chemistry, Vol. 20, No. 10, 2001, pp. 2133-2141. doi:10.1002/etc.5620201001
- W. Korner, P. Spengler, U. Bolz, W. Schuller, V. Hanf and J. W. Metzger, “Substances with Estrogenic Activity in Effluents of Sewage Treatment Plants in South-Western Germany. 2. Biological Analysis,” Environmental Toxicology and Chemistry, Vol. 20, No. 10, 2001, pp. 2142-2151. doi:10.1002/etc.5620201002
- R. A. Rudel, S. J. Melly, P. W. Geno, G. Sun and J. G. Brody, “Identification of Alkylphenols and Other Phenolic Compounds in Wastewater, Septage, and Groundwater on Cape Cod, Massachusetts,” Environmental Science Technology, Vol. 32, No. 7, 1998, pp. 861-869. doi:10.1021/es970723r
- J. H. Shen, B. Gutendorf, H. H. Vahl, L. Shen and J. Westendorf, “Toxicological Profile of Pollutants in Surface Water from an Area in Taihu Lake, Yangtze Delta,” Toxicology, Vol. 166, No. 1-2, 2001, pp. 71-78. doi:10.1016/S0300-483X(01)00439-5
- M. A. Kelly, A. M. Reid, K. M. Quinn-Hosey, A. M. Fogarty. J. J. Roche and C. A. Brougham, “Investigation of the Estrogenic Risk to Feral Male Brown Trout (Salmotrutta) in the Shannon International River Basin District of Ireland,” Ecotoxicology and Environmental Safety, Vol. 73, 2010, pp. 1658-1665. doi.org/10.1016/j.ecoenv.2010.08.018
- K. M. Quinn-Hosey, J. J. Roche, A. M. Fogarty and C. A. Brougham, “A Toxicological Assessment of Endocrine Disrupting Chemicals Found in the BMW (Border Midland and Western) Region of Ireland,” Journal of Environmental Protection, Vol. 3, No. 4, 2012, pp. 304-315. doi:10.4236/jep.2012.34039
- C. McGee, C. Brougham, J. Roche and A. Fogarty, “First Report of Intersex Roach Residing in Irish Rivers Downstream of Several Wastewater Treatment Plants,” Biology and Environment, Vol. 112, No. 1, 2012, pp. 1-9. doi:10.3318/BIOE.2011.23
- V. Pelekanou and G. Leclercg, “Recent Insights into the Effect of Natural and Environmental Estrogens on Mammary Development and Carcinogenesis,” International Journal Developmental Biology, Vol. 55, 2011, pp. 869- 878. doi:10.1387/ijdb.113369vp
- S. A. Bidgoli, R. Ahmadi and M. D. Zavarhei, “Role of Hormonal and Environmental Factors on Early Incidence of Breast Cancer in Iran,” Science Total Environment, Vol. 408, No. 19, 2010, pp. 4056-4061. doi:10.1016/j.scitotenv.2010.05.018
- A. Giwercman and Y. L. Giwercman, “Environmental Factors and Testicular Function,” Best Practice Research Clinical Endocrinology Metabolism, Vol. 25, No. 2, 2011, pp. 391-402. doi:10.1016/j.beem.2010.09.011
- K. M. Main, N. E. Skakkebaek, H. E. Virtanen and J. Toppari, “Genital Anomalies in Boys and the Environment,” Best Practice Research Clinical Endocrinology Metabolism, Vol. 24, No. 2, 2010, pp. 279-289. doi:10.1016/j.beem.2009.10.003
- D. Klingmuller and A. Allera, “Endocrine Disruptors: Hormone-Active Chemicals from the Environment: A Risk to Humans?” Dtsh Med Wochenschr, Vol. 136, 2011, pp. 967-972.
- L. F. Doherty, J. G. Bromer, Y. Zhou, T. S. Aldad and H. S. Taylor, “In Utero Exposure to Diethylstilbestrol (DES) or Bisphenol-A (BPA) Increases EZH2 Expression in the Mammary Gland: An Epigenetic Mechanism Linking Endocrine Disruptors to Breast Cancer,” Hormones Cancer, Vol. 1, No. 3, 2010, pp. 146-155. doi:10.1007/s12672-010-0015-9
NOTES
*Corresponding author.