International Journal of Clinical Medicine
Vol. 3 No. 7 (2012) , Article ID: 26173 , 4 pages DOI:10.4236/ijcm.2012.37113
Co-Inheritance of Beta & Delta-Globin Gene (HbYialousa) Mutations in an Iranian β-Thalassemia Carrier
![]()
1Molecular Medicine Department, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran; 2Kawsar Human Genetics Research Center, Tehran, Iran.
Email: *siruszeinali@yahoo.com
Received September 7th, 2012; revised October 24th, 2012; accepted November 25th, 2012
Keywords: δ-Globin Gene; β-Thalassemia; HbYialousa; β-Globin Gene; CD39
ABSTRACT
Introduction: Beta-thalassemia is characterized by absence or reduced synthesis of the β-globin. Carriers of β-thalassemia, typically have microcytic hypochromic anemia and elevated hemoglobin HbA2 and normal HbF level. On the other hand carriers of severe alpha-thalassemia also have similar CBC parameters to that of β-thalassemia with normal HbA2 level. Co-presence of mutations in the β-globin and delta-globin genes (point mutations or deletions) usually give normal HbA2 and elevated HbF level. We report a β-thal carrier with normal level of HbA2 and increased level of HbF who had a point mutation in CD39 on the beta-globin gene and a point mutation in CD27 on the δ-globin gene named Hb-Yialousa. Materials & Methods: An individual with low hematological indices, normal HbA2 and elevated HbF was referred to our center as routine premarital screening program. Mutations in the β-globin and δ-globin genes were screened using ARMS and sequencing methods. Results: The mutation in β- and δ-globin genes were identified as CD39 and CD27 (HbYialousa) respectively. No point mutation or deletion in α-globin gene was identified. Discussion: We showed that normal HBA2 with elevated HbF level is due to co-inheritance of delta-globin gene mutation with mutation in the β-globin gene. When screening for β-thalassemia, one has to either rule out presence of α-globin gene mutation of mutation in the delta-globin gene.
1. Introduction
β-Thalassemia is one of the most common single-gene disorders with more than 400,000 new born affected per year worldwide [1]. In Iran, premarital screening has been in effect since 1997 to curve the birth of affected children [2]. Couples are primarily screed for MCV, MCH and HbA2 levels.
β-globin gene cluster is located at the short arm of the chromosome 11. The cluster contains five functional genes (i.e., ε, Aγ, Gγ, δ and β-globin). They are expressed in different stages of human development. Thalassemia is an inherited disorder in which there is an abnormality in one or more of the globin genes. β-thalassemia is the result of decreased (β+ thalassemia) or complete absence (β0 thalassemia) of normal β-globin chain [3], depending on the mutation in HBB gene.
More than 200 mutations in the beta-globin gene have been identified to date [4]. The reduced β-globin output leads to an excess of α-chains that precipitate in the red cell precursors, inhibiting their normal function and survival.
Usually carriers of β-thal have microcytic hypochromic anemia (reduced mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) and elevated hemoglobin HbA2 level. The HbA2 level is important to distinguish between carriers of α-thal from the β-thal. The human delta globin gene, HBD is one of the β-like globin genes, expressed in adults at low level (usually less than 3.5% of total Hb).
Coinheritance of δ-globin gene (mutated δ-globin gene) with β-globin can reduce the HbA2 level to that of normal level but MCV and MCH level are usually similar to those of beta-thal carriers (personal observation). However, the HbF values are usually elevated above the normal level.
HbF is largely replaced by adult hemoglobin shortly after birth with small amounts of the fetal molecule continuing to be produced through adulthood (less than 2% of total hemoglobin on average).
Normal HbA2 with reduced MCV and MCH are hallmarks of α-thalassemia trait. Therefore, the above status should be differentiated from α-thalassemia trait. This combination should be considered in couple when one partner is carrier of β-thalassemia.
The HbF level is a quantitative trait influenced by many loci inside or outside the β-globin gene cluster [5]. The switch from fetal to adult hemoglobin is usually incomplete. There is residual fetal hemoglobin in adults, which is restricted to a subset of erythrocytes called F cells. F-cell levels are influenced by a sequence variant (C > T) at position -158 upstream of the gamma-globin gene, termed as XmnI-Ggamma (GγXmnI) polymerphism [6].
We report a β-thal carrier with normal level of HbA2 and increased level of HbF who had a point mutation in CD39 on the beta-globin gene and a point mutation in CD27 on the δ-globin gene named Hb-Yialousa.
2. Materials and Method
A young couple suspected of being carrier of beta-thalassemia, with abnormal hematological indices, was referred to our center for further characterization. A written informed consent was signed by both individuals. The hematological parameters of the couple and their parents are shown in Table 1.
After obtaining 10 ml of peripheral blood in tubes containing EDTA, genomic DNA was extracted from leukocyte by salting out method [7]. Complete Blood Count (CBC) was performed on an automated cell counter (Sysmex k12; Sysmex Corporation, Kobe, Japan).
β-globin gene mutation was analyzed by ARMS method for common HBB mutations in Iran [8].
MLPA (multiplex ligation probe amplification) [9] was performed using SALSA MLPA P102-B1 HBB and HBA kits (MRC Holland, Amsterdam, the Netherlands) according to manufacture protocol. Results were analyzed on ABI 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Primers used to sequences the δ-globin gene in two fragments were as follow:
δ1 F: 5’CTGAGTCAAGACACACATGACAG3’, δ2 R: 5’TGGTATGCATAATTTGAGTTGTTG3’δ3 F: 5’AATATCCTGTCTTTCTCTCCCAAC3’, δ4 R: 5’TAATTTCTGCTCTTTGGAGGTAG3’.
The Gγ and Aγ regulatory regions were sequenced using the following primers Aγ5’CACAGTACCTGCCAAAGAACATTC3’, Gγ5’GGCCTAAAACCACAGAGAGT- 3’ and common primer as 5’TTGTCCTCCTCTGTGAAATGACC3’.
The δ-globin gene was sequenced according to our pervious method [10]. For investigating the coinheritance of δand β-globin gene mutations, both genes were sequenced by KBC (Kawsar Biotech Co., Tehran, Iran). Alpha-globin genes mutation analysis was performed by either gap-PCR for the detection of α(−α3.7, −α4.2, −α20.5)- Med deletions [11] or by DNA sequencing. The α-globin genes were sequenced using the following primers:
αseq-F: 5’GCTCCGCGCCAGCCAATGAG3’,
αseq1-R: 5’CATGTGTGTCCCAGCTGCTGTC3’.
αseq2-R: 5’GAGAGGTCCTTGGTCTGAGACAG3’.
3. Results
To investigate the reason for normal HbA2 and raised HbF in the proposita, MLPA technique was used to check for presence of any deletion covering both the δ- and β-globin genes (complete or partial). No deletion was observed. The β-globin gene was sequenced and codon 39 (CD39) mutation was seen which also indicated presence of all or most of β-globin gene. Deletions in the α-globin genes were checked using gap-PCR [11] and MLPA. No deletion was seen. The α-globin genes were sequenced. No point mutation was seen either. The promoter regions for the Aγ and Gγ genes were sequenced for finding any nucleotide changes which can explain raised HbF level. No mutation was seen.
We then investigated mutation in the δ-globin gene by sequencing. A mutation was found at codon 27 (GCC > TCC). This mutation had already been reported as HbYialousa (Figure 1). Parental CBC parameters indicated a trans inheritance in which the CD39 has been inherited by her mother and the HbYialousashould have been inherited by the father since the mother had normal HbF. We then sequenced maternal DNA for the δ-globin gene and it was shown that he was carrier of this mutation.
4. Discussion
Carriers of beta-thalassemia are usually characterized by raised HbA2 and low MCV and MCH. Borderline HbA2 and even normal HbA2 beta-thal carriers have been reported. Beta-thal carriers with normal HbA2 could be due to δβ, γδβ or εγδβt halassemia [12]. The γδβ and
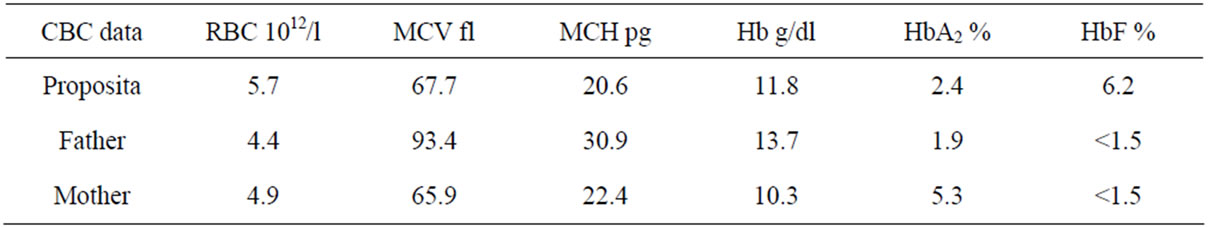
Table 1. Summary of hematological data in the proposita and her parents.
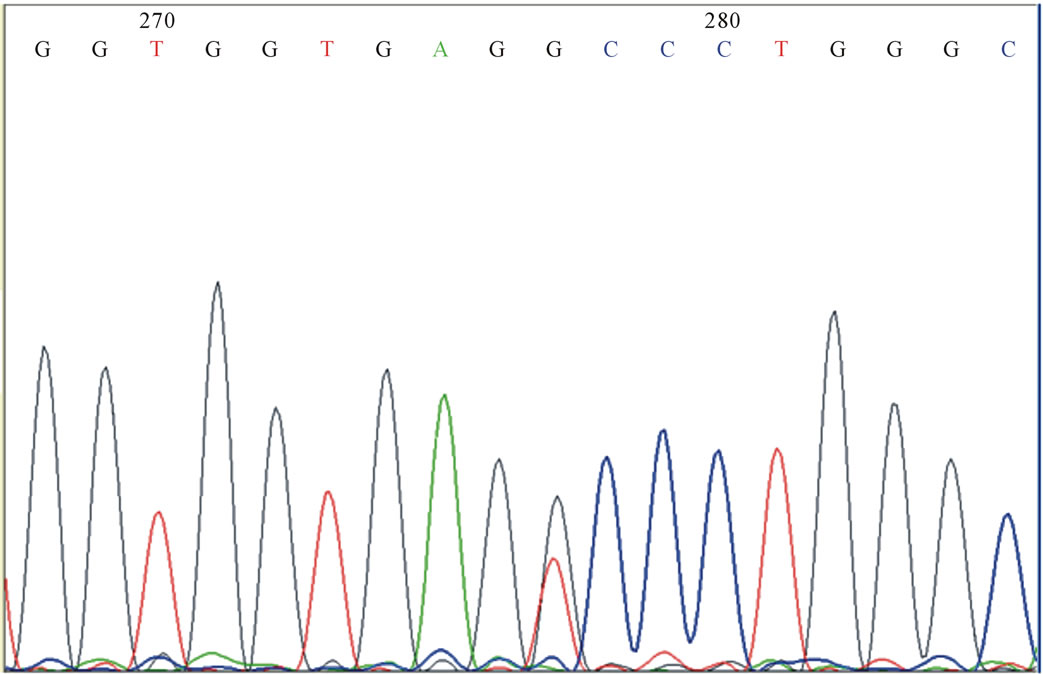
Figure 1. δ-yialousa point mutation in heterozygote form in the investigated subject.
particularly the εγδβ usually have normal HbF level due to deletion of gamma genes.
Coinheritance of δ-and β-globin mutated alleles can lower the HbA2 to that of normal range and consequently it can hinder proper diagnosis of beta-thal status. However, they usually are associated with raised HbF level. Most forms of δβ-thal are deletional forms and HbLepore being the most common in our region (personal observation). When δβ-thal is the result of point mutation then the mutated alleles can be co-inherited as trans or most rarely in cis [13]. In our case it was shown to be in trans. In these cases the fetus will inherit either the deltaglobin or the beta-gene mutation.
Our study showed that the reduced HbA2 was due to mutation in δ-globin gene in the proposita. Since her mother was beta-thal carrier and the father was normal then she should have inherited the δ-gene from the father (hence in trans).
In our experience other forms of normal HbA2 who are carriers of beta-thal are carriers of δβ-, γδβ or εγδβ. The latter two are very rare in Iran [unpublished data]). The δβ types are usually associated with raised HbF level but the other two types usually have normal HbF which makes the diagnosis even harder. In this case complete analyses of alpha and beta-globin genes have to be considered.
REFERENCES
- P. Lahiry, S. A. Al-Attar and R. A. Hegele, “Understanding Beta-Thalassemia with Focus on the Indian Subcontinent & the Middle East,” The Open Hematology Journal, Vol. 2, 2008, pp. 5-13. doi:10.2174/1874276900802010005
- H. Abolghasemi, A. Amid, S. Zeinali, M. Radfar, P. Eshghi, M. Rahiminejad, et al., “Thalassemia in Iran Epidemiology, Prevention, and Management,” Journal of Pediatric Hematology/Oncology, Vol. 29, No. 4, 2007, pp. 233-238. doi:10.1097/MPH.0b013e3180437e02
- D. J. Weatherall and J. B. Clegg, “The Molecular Pathology of the Thalassaemias,” 4th Edition, Wiley, Hoboken, 2008.
- N. F. Olieveri, “The β-Thalassemias,” New England Journal of Medicine, Vol. 341, 1999, pp. 99-109. doi:10.1056/NEJM199907083410207
- T. K. Nquyen, P. Joly, C. Bardel, M. Moulsma, N. Bonello-Palot and A. Francina, “The XmnI (G)Gamma Polymorphism Influences Hemoglobin F Synthesis Contrary to BCL11A and HBS1L-MYB SNPs in a Cohort of 57 Beta-Thalassemia Intermedia Patients,” Blood Cells, Molecules, and Diseases, Vol. 45, No. 2, 2010, pp. 124-127.
- C. Garner, N. Silver, S. Best, S. Menzel, C. Martin, T. D. Spector and S. L. Thein, “Quantitative Trait Locus on Chromosome 8q Influences the Switch from Fetal to Adult Hemoglobin,” Blood, Vol. 104, No. 7, 2004, pp. 2184-2186.
- S. A. Miller, D. D. Dykes and H. F. Polesky, “A Simple Salting out Procedure for Extracting DNA from Human Nucleated Cells,” Nucleic Acids Research, Vol. 16, No. 3, 1998, p. 1215. doi:10.1093/nar/16.3.1215
- J. M. Old, “Detection of Mutations by the Amplification Refractory Mutation System (ARMS),” Protocols in Human Molecular Genetics, Methods in Molecular Biology, Vol. 9, 1992, pp. 77-84. doi:10.1385/0-89603-205-1:77
- D. A. Koolen, W. M. Nillesen, M. H. A. Versteeg, G. F. M. Merkx, N. V. A. M. Knoers, M. Kets, S. Vermeer, C. M. A. van Ravenswaaij, C. G. de Kavel, H. G. Brunner, D. Smeets, B. B. A. de Vries and E. A. Sistermans, “Screening for Subtelomeric Rearrangements in 210 Patients with Unexplained Mental Retardation Using Multiplex Ligation Dependent Probe Amplification (MLPA),” Journal of Medical Genetics, Vol. 41, No. 12, 2004, pp. 892-899. doi:10.1136/jmg.2004.023671
- S. Zeinali, S. M. Eram, S. B. Azimifar, V. Lotfi, P. Foulady and M. Masrouri, “First Report on the Co-Inheritance of (b) IVS I-1(G-T) Thalassemia with the (γ) CD85 [Phe-Ser](F1) (TTT>TCT)] HbA2 Etoliain in Iran,” The Hematology Journal, Vol. 91, No. 1, 2006, p. 45.
- R. Alibakhshi, M. Arash, R. Akramipour, H. Nomani, M.-R. Farshchi, S. Fathollahi and M. Rezaei, “Molecular Analysis of Alpha Globin Gene Deletions among Patients with Microcytic Hypochromic Anemia in KermanshahIran,” Behbood Journal, Vol. 14, No. 4, 2011, pp. 336-341.
- B. H. Al-Awamy, “Thalassemia Syndromes in Saudi Arabia Meta-Analysis of Local Studies,” Saudi Medical Journal, Vol. 21, No. 1, 2000, pp. 8-17.
- A. Giambona, C. Passarello, M. Vinciguerra, R. Li Muli, P. Teresi, M. Anzà, G. Ruggeri, D. Renda and A. Maggio, “Significance of Borderline Hemoglobin A2 Values in an Italian Population with a High Prevalence of β-Thalassemia,” Haematologica, Vol. 93, No. 9, 2008, pp. 1380-1384.
NOTES
*Corresponding author.