International Journal of Clinical Medicine
Vol.2 No.2(2011), Article ID:5101,18 pages DOI:10.4236/ijcm.2011.22022
Glucose Metabolism in Breast Cancer and Its Implication in Cancer Therapy
![]()
1Institute of Chinese Medical Sciences, University of Macau, Macao SAR, China; 2Department of Endocrinology, Dong-Zhi-Men Hospital, Beijing University of Chinese Medicine, Beijing, China; 3Department of Pharmacology, Institute of Clinical Medical Science, China-Japan Friendship Hospital, Beijing, China; 4Faculty of Pharmacy, University of Manitoba, Winnipeg, Canada.
Email: ygong@cc.umanitoba.ca, ytwang@umac.mo
Received February 25th, 2011; revised March 8th, 2011; accepted March 22nd, 2011.
Keywords: Breast Cancer, Glucose Transport, Glycolysis, Fatty Acid Synthesis, Mitochondrial Metabolism
ABSTRACT
It is well known that malignant cells have accelerated glucose uptake and metabolism in order to maintain their fast proliferation rates. With the increased influx of glucose into cancer cells, glycolysis is facilitated through a coordinated regulation of metabolic enzymes and pyruvate consumption. Shiftting from mitochondrial oxidative phosphorylation to glycolysis and other pathways such as pentose phosphate pathway (PPP) and de novo fatty acid synthesis in the breast tumor provides not only energy but also the materials needed for cell proliferation. Glucose augmentation in tumor cells can be due to the elevated level of glucose transporter (GLUT) proteins, such as the over-expression of GLUT1 and expression of GLUT5 in breast cancers. Moreover, other factors such as hypoxia-inducible factor-1 (HIF-1), estrogen and growth factors are important modulators of glucose metabolism in the progression of breast carcinomas. Therapies targeting at the glycolytic pathway, fatty acid synthesis and GLUTs expression are currently being investigated. Restoring tumor cells to its normal glucose metabolic state would endow tumor specific and accessible treatment that targets glucose metabolism.
1. Introduction
Rapid growth and cellular proliferation are hallmarks of tumor. In order to survive, tumor cells have to adopt all possible mechanisms to meet their energy needs. The increased demand for energy also requires a higher metabolic rate of nutrients. In non-malignant cells, this is usually achieved through elevating mitochondrial oxidative phosphorylation (OXPHOS), which is the major source of cellular ATP. By contrast, in cancer cells, observations by Otto Warburg suggested that tumor cells might utilize the augmented glycolysis rather than mitochondrial phosphorylation of glucose as their main energy supply, and apply this less efficient pathway even under aerobic conditions [1]. This metabolic alternative is also closely associated with the aggressiveness of the tumor [2]. The universal ‘aerobic glycolysis’ of tumors suggests a ‘metabolic reprogramming’ of nutrients in those cells, which is characterized by facilitated glycolytic reactions, impaired OXPHOS and elevated de novo nucleotide and fatty acid biosynthesis. Since the capability of tumor cells to support their enhanced nutrient metabolism is obligatory for their growth and phenotype, mechanisms contributing to these changes may provide a biochemical basis for therapeutic strategies.
2. Glucose Metabolism in Cancer
Cells are highly organized and a constant supply of energy is required to create and maintain the biological orders that keep them alive. This energy is derived from the chemical bond stored in food molecules, which serves as fuel for cells. Glucose is a particularly important fuel molecule and dominates energy production in most animal cells. Generally, glucose metabolism provides chemical energy in the form of ATP and NADH through control of three-step continuous reactions, including glycolysis, the tricarboxylic acid (TCA) cycle and OXPHOS. The breakdown of each molecule of glucose into two smaller molecules of pyruvate is carried out through ‘glycolysis’, which involves a sequence of ten separate reactions, each of which produces different intermediates and is catalyzed by different enzymes. During glycolysis, energy is produced as activated carrier molecules (ATP and NADH) and at the end of glycolysis, there is a net gain of two molecules of ATP for each glucose molecule that is metabolized. For many anaerobic organisms, glycolysis is the principal source of cellular ATP. By contrast, in most aerobic tissues, the pyruvate derived from glycolysis directly enters mitochondria, in which each molecule of pyruvate is converted to acetyl-CoA and CO2. The complete oxidation of the acetyl groups in acetyl-CoA to CO2 occurs in the TCA cycle, starting from citrate formation by the joining of acetyl-CoA to oxaloacetate (OAA). Moreover, this cycle accounts for twothirds of total oxidation of carbon compounds in most cells and generates high-energy electrons carried by NADH and FADH2. These high-energy electrons are then passed to an electron transport chain embedded in the inner mitochondrial membrane, which eventually combine with O2 to form H2O. With the movement of electrons along the respiratory chain, energy from NADH and FADH2 is stored as an electrochemical proton gradient across the inner membrane of mitochondrial, which exerts a proton-motive force, driving the conversion of ADP and phosphate to ATP. Compared to glycolysis, mitochondrial OXPHOS produces 36 molecules of ATP per one molecule of glucose. Efficient OXPHOS relies on functional enzyme complexes of the mitochondrial respiratory chain, which are encoded by both nuclear and mitochondrial genes. Therefore, any deletions and mutations of mitochondrial DNA (mtDNA) will result in a defect of mtDNA-encoded polypeptides, which can cause a large spectrum of diseases featuring impaired mitochondrial metabolism such as cancer.
Cancer cells, characterized by uncontrolled proliferation in the hypoxic and poorly vascularized environment, require a significant amount of glucose and accelerated glucose metabolism to meet their need for energy and biosynthetic elements. However, a metabolic alteration observed by Otto Warburg (Warburg effect) in 1924, suggested that in contrast to non-malignant cells, which utilize mitochondrial OXPHOS to produce ATP in the presence of adequate oxygen and anaerobic glycolysis when adequate oxygen supply is not available, malignant cells preferentially depend on converting glucose into lactate rather than mitochondrial glucose metabolism even under high oxygen environments [1]. Though the exact molecular mechanisms underlying the ‘Warburg effect’ are unknown, the ‘aerobic glycolysis’, which presents elevated glucose transport, increased glycolytic activity and reduced mitochondrial glucose metabolism, has been one of the important tumor metabolic phenotypes. The advantage of this shifted energy conversion is likely to provide tumor cells adequate microenvironment for multiple bioenergetic and biosynthetic pathways [3] .
2.1. Facilitated Glycolysis
Glycolytic enzymes in malignant cells were distinctive from their counterparts in normal cells [4-6] . For instance, phosphoglycerate mutase (PGM) and enolase, catalyzing glucose to pyruvate—the end-product of glycolysis, were phosphorylated at the tyrosine residues in the cells transformed with Rous sarcoma virus or stimulated by growth factors [7]. The same was observed with lactate dehydrogenase (LDH)-A, which is responsible for the conversion of pyruvate to lactate—a main product of anaerobic pathway. The phosphorylation level of these three enzymes correlated with the increased rate of glycolysis [7]. Typically in malignant human breast tissue, the overexpression of LDH-A was associated with tumor invasiveness. Accordingly, LDH-A has been proposed as a marker of tumor progression [8] .
In addition to the phosphorylation of glycolytic enzymes, the enzyme activities were significantly increased in the malignant breast tissues compared to the normal and benign breast tissues, such as hexokinase (HK), phosphofructokinase (PFK), aldolase and pyruvate kinase (PK) (see Glycolysis in the Figure 1) [<9] . For example, HK activation was related to the suppression of apoptosis since the promoter region of the HK gene contains the DNA responsible elements of both p53 and hypoxia-inducible factor (HIF)-1, and both mutated p53 and HIF-1 increased the HK expression [10] . HIF-1 is considered as an oxygen sensor and is stabilized under hypoxic stress. It transcriptionally modulates a number of genes as a metabolic adaptation in cancer cells [11,12] . HIF-1-induced activation of HK triggers the transloca-tion of HK from the cytoplasm to mitochondrial membranes with which it interacts and suppresses several key components of mitochondria-dependent apoptotic proteins [13]. Moreover, the activity of PK, which irreversibly catalyzes the last step of pyruvate formation, has been shown to have a strong connection with the malignancy of breast carcinomas [14].
The glycolytic end-product pyruvate is consumed differently in tumors. Pyruvate metabolism stands at the crossroad between oxidative and glycolytic pathways. Transportation of pyruvate into mitochondria results in its oxidation, whereas its maintenance in cytosol leads to the reduction of pyruvate. The inhibition of pyruvate oxidation in mitochondria is considered as a metabolic deviation that could explain Warburg effect of tumor cells [15]: down-regulation of pyruvate oxidation by phosphorylated pyruvate dehydrogenase (PDH) complex would redirect pyruvate into the cytoplasm, which will then be metabolized into lactate. Moreover, the mitochondrial metabolic flux of pyruvate through the lefthand side of the TCA cycle (from α-ketoglutarate to OAA) is much greater than that through the right-hand side (from acetyl-CoA to α-ketoglutarate), which may be due to the higher rate of glutaminolysis in tumor [16] .
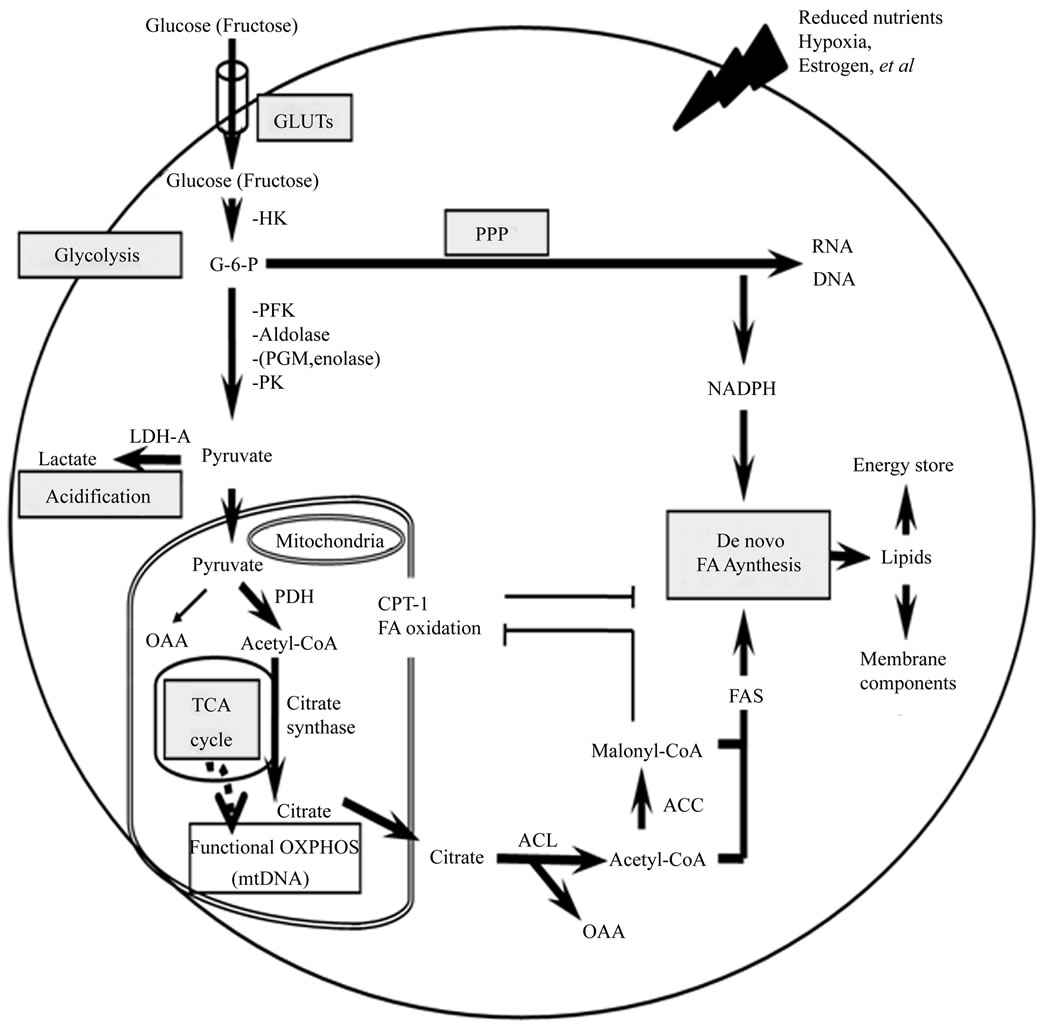
Figure 1. A brief outline from glucose uptake to mitochondrial metabolism followed by de novo nucleotide and fatty acid biosynthesis involved in this review.
Glutaminolysis is the degradation of amino acid glutamine to glutamate, pyruvate and then lactate. Therefore, the consequence of accelerated glutaminolysis in tumor cells is an increased conversion of pyruvate into lactate. In supporting this information, several studies reported that there were higher utilization of both glucose and glutamine to generate lactate in tumor cells [17,18] . As glutaminolysis is an energy producing pathway, the application of glutaminolysis in tumor cells could be an adaptive metabolic switch in order to satisfy the ATP requirement [18] . Additionally, the reduced mitochondrial pyruvate oxidation was accompanied by an increased activity of cytosolic pyruvate enzymes in tumor cells, which would reduce the amount of this substrate transported to and metabolized in the mitochondria [19, 20] .
The key molecule that drives glycolysis in cancer cells is HIF-1. HIF-1 is more stabilized in conditions such as nutrient depletion and reduced oxygen or hypoxic conditions. HIF-1 has been shown to induce the expression of several glucose transporters as well as most enzymes required for glycolysis [21] . Besides increasing the expression of HK in anaerobic metabolism, HIF-1 can also suppress PDH in mitochondria by inducing pyruvate dehydrogenase kinase (PDK)-1, which inhibits PDH activity by phosphorylation [22,23] . Since PDH catalyzes the mitochondrial oxidation of pyruvate to produce NADH and acetyl-CoA for ATP production, the inhibition of PDH by HIF-1 will result in a restricted entry of pyruvate into the mitochondria, a decrease of oxygen consumption, and consequently the transmission of metabolic flux to the lactate pathway [24]. Moreover, in malignant cells, there was a higher level of fructose-2, 6-bisphosphate (F-2, 6-BP), which is a powerful allosteric activator of glycolytic enzyme PFK-1. This increase could be due to a phosphorylative induction by the PFK-2 portion of the bifunctional enzyme PFK-2/fructose-2, 6-bisphosphatase in a HIF-1-dependent manner [25]. Furthermore, HIF-1 plays a transcriptional role in promoting LDH-A expression [26] , which would attenuate tumor cell respiration and viability [27] .
Diverting metabolism away from mitochondria towards cytoplasm provides malignant cells resistant to apoptosis. When cells switch from OXPHOS towards higher glycolysis, the consequent formation and accumulation of lactate can lead to cellular acidification, which would eventually facilitate tumor growth through acid-mediated extracellular matrix degradation and favour tumor invasion and metastasis (see Acidification in the Figure 1) [28] . Consequently, the persistence of this adaptive metabolic mechanism grants tumor cells a request of growth advantages. In this context, glycolytic enzymes serve as anti-apoptotic factors through inhibiting several components of mitochondria-dependent apoptosis [9, 29] . Additionally, the genetic theory of carcinogenesis regards glycolytic transformation as a consequence of activation of various oncogenes or tumor suppressor genes, including Akt/PTEN, p53 and c-myc [30]. Up-regulated Akt can be found in many cancers and could induce a glycolytic metabolic profile through augmenting HK expression and activity [31,32] . The PTEN (the antagonizer of Akt) appears to be mutated in several cancers, which fails to control the activity of Akt. Gene p53 can also influence the metabolic balance in cells between glycolysis and OXPHOS. The p53 activity can favour OXPHOS through a transcriptional regulation of F-2, 6-BP by TIGAR (a p53-induced glycolysis and apoptosis regulator). TIGAR negatively regulates glycolysis by degrading F-2, 6-BP, which activates glycolytic enzyme PFK1 [33] . Moreover, p53 contributes to the synthesis of cytochrome c oxidase (COX)—a subunit of mitochondrial electron transport chain complex IV [30,33,34] . Restoring impaired COX is able to promote the conversion of ATP production from glycolysis to OX-PHOS [34] . Accordingly, p53 mutation would result in the switch of ATP production from OXPHOS to glycolysis. Therefore, the metabolic phenotype is essential for the pathogenesis of cancer but not a ‘by-product’ of carcinogenesis.
2.2. Altered Mitochondrial Machinery
The original Warburg hypothesis was that it could be the damage of mitochondrial respiration that contributes to the enhanced glycolysis in tumor cells [1]. Mitochondria have been suggested to play an important role as an oxygen sensor to initiate the HIF-1 pathway under hypoxia [5]. Once hypoxia is initiated, reactive oxygen species (ROS) will accumulate on the site of complex III Q0 of mitochondrial respiratory chain and will stabilize rather than degrade the regulatory subunit of HIF-1 [35,36] . This results in the augmentation of HIF-1-induced gene expression and suppression of mitochondrial metabolism ADDIN EN.CITE ADDIN EN.CITE.DATA [22,23] . The role of mitochondria-derived ROS has also been implicated in a proposed vicious cycle linking mtDNA mutation in tumors. On the one hand, a burst of ROS production from mitochondria would favour oxidative dependent mitochondrial mutagenesis and hence promote tumorigenesis [37] ; While on the other hand, mutation of mtDNA in the respiratory chain components could commence a cascade of events leading to an increase in electron leakage and ROS over-generation in the process of malignant transformation and tumor progression [38,39] . Several mtDNA mutations have been found in breast tumors [40], and the presence of mtDNA mutations in breast cancer cells is consistent with the intrinsic susceptibility of mtDNA to damage and persistent oxidative stress [41] . In terms of the oncogenesis of mtDNA mutation, critical evaluation and interpretation of the literature revealed that mtDNA mutation could enhance the specificity of diagnosis, detection and prediction of breast cancer growth and/or patients’ outcomes. Therefore, the mtDNA mutation may be used as a new molecular bio-marker for breast cancer [42].
The suppressed OXPHOS may also be interpreted as a transient adaptation in cancer cells, which disconnects glycolysis from dysfunctional OXPHOS, leaving glycolysis as the predominant ATP source, and then followed by an OXPHOS remodeling according to the growth demand of the cancer. The preference for glycolytic production of ATP in tumor cells could be due to 1) glycolysis generates ATP more rapidly although it is less efficient in long term than OXPHOS [43] ; and 2) part of the carbon intermediates from glycolysis could be used for various biosynthetic pathways to keep a constant tumor growth [44]. However, deficiencies in mitochondrial respiratory chain components such as mtDNA impairment cannot explain tumorgenesis for different kinds of tumors because in some cases the over-expression of respiratory complexes was observed [45].
2.3. Glycolysis and Mitochondrial TCA Cycle Provide Precursors for Tumor Biosynthesis
The persistent shift of malignant cells from mitochondrial OXPHOS to glycolysis for ATP production implicates that alternative pathways may be involved in maintaining cell growth. Despite of the energy production of glucose, catabolism also produces building blocks for tumor cells to divide and proliferate. Glycolytic intermediates formed in central catabolic reaction pathways can be siphoned off by other enzymes that use them as biosynthetic precursors to produce amino acids, nucleotides and lipids for cell needs. Thus, the supply of materials for biosynthesis is another primary metabolic alteration associated with proliferating tumor cells from the view of glycolysis and TCA cycle [46]. The majority of macromolecules required for proliferation are generated de novo from glucose, which is controlled through chemical modification of these large molecules in response to changes in the cellular immediate environment [47]. The presence of different glucose metabolic pathways in the cell requires a precise regulation. For long-term adaptation, glucose metabolism in tumor cells requires a balance between elimination of some particular reaction pathways and elevation of the others to meet the minimum requirements. Hence, branching pathways of glycolysis and mitochondrial TCA cycle in malignant cells are likely to function as a distinctive way for their uncontrolled growth. These are of particulate interest in understanding of the pentose phosphate pathway (PPP, also known as hexose monophosphate shunt) for de novo nucleotide biosynthesis and the de novo fatty acid synthesis in cancer cells.
2.3.1. Pentose Phosphate Pathway
Carbon derived from glycolytic flux to the PPP can be utilized to generate ribose-5-phosphate for de novo nucleotide biosynthesis. The large cellular nucleotide pool as well as the level and activity of different rate-limiting enzymes involved nucleotide synthetic pathways are the critical elements of maximal cell proliferative capacity. Compared with normal cells, tumor cells display an altered nucleotide metabolism, such as the larger nucleotide pool, higher enzymatic activity in nucleotide anabolic pathway as well as lower enzymatic activity in nucleotide catabolic pathway [48] . Understanding of the nucleotide biosynthesis in malignant cells offers possibilities to design selective therapies for human cancer. Besides the generation of ribose-5-phosphate from PPP to support nucleic acid synthesis, the reduced nicotinamide adenine dinucleotide phosphate (NADPH) was formed during PPP to provide reducing equivalents for not only lipid synthesis but also detoxification of reactive oxygen species [49]. Activities of enzymes involved in this pathway such as glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase have been reported to be increased in malignant breast tissues [50] as well as carcinomas and cancer cell lines from other origin [4,51,52] .
The remaining pyruvate from aerobic glycolysis that is not converted to lactate enters the mitochondria and is then extruded from the TCA cycle at various steps for biosynthetic pathways to satisfy the increased demand of cell proliferation. Pyruvate could be converted to acetylCoA, which will join with OAA to form citrate in the presence of citrate synthase. After translocation of citrate from mitochondria to cytosol, cleavage of citrate back to acetyl-CoA provides the precursor for the synthesis of fatty acid, which may function as essential building materials of cell plasma membrane for rapid cancer growth (see PPP and De novo FA Synthesis in the Figure 1).
2.3.2. Fatty Acid Synthesis
De novo fatty acid synthesis involves the conversion of glucose to pyruvate and then to acetyl-CoA, which will work together with OAA to generate citrate. Besides glucose metabolism, metabolites from glutaminolysis provide another carbon source to facilitate de novo fatty acid synthesis. During glutaminolysis, glutamate is produced through deamination of glutamine and can be converted to α-ketoglutarate, which is then metabolized through TCA cycle to generate citrate [53] . The citrate formed from both glucose and glutamine metabolism in mitochondria will be transported into the cytosol and broken down to acetyl-CoA and OAA by ATP citrate lyase (ACL). The acetyl-CoA generated by ACL was then carboxylated to malonyl-CoA by a rate-limiting enzyme acetyl-CoA carboxylase (ACC). In addition, malic enzyme and the PPP produce the reducing equivalence NADPH for a preparation of fatty acid synthesis. Fatty acid synthase (FAS) is the major enzyme of lipogenesis. With the provision of NADPH, FAS catalyzes the condensation of acetyl-CoA and malonyl-CoA for the de novo synthesis of 16-carbon saturated free fatty acid palmitate (see De novo FA Synthesis in the Figure 1) [54] .
Fatty acids are the source of membrane components such as phospholipids and glycolipids. It also provides precursors of critical signaling molecules for proliferation and differentiation [55] . Fatty acids are generally acquired from diet and high level of fat in the Western food has been implicated in the development of human malignancies including carcinomas of colon, breast and ovarian [56-60] . In fact, except for some organs such as the liver, adipose tissue and lactating mammary gland, other normal cells rarely utilize de novo lipid synthesis pathway [61,62] ; in contrast, the de novo fatty acid synthesis was significantly elevated in malignant tumors as reported by Medes et al. [63]. Consistently, the expression of enzyme FAS is highly up-regulated in a variety of human malignancies, but FAS is hardly detected in normal tissues [54] . Through immunohistochemical and metabolic studies, FAS over-expression has been detected in cancer tissues, its surrounding non-cancer tissues and in the blood of patients with cancer [64-71] . Evidence from breast cancer, prostate cancer, ovarian cancer and thyroid cancer studies suggested an association of FAS expression and tumor aggressiveness, and indicated that the activity of FAS might serve as a malignant marker of tumor phenotype [72]. Therefore, the presence of high level of FAS is considered one of the most common molecular changes in cancer cells [64,65, 73] . Strikingly, subsequent clinical and laboratory investigations identify that human cancers have the capacity to synthesize their own fatty acid, which is independent of the inhibitory mechanisms of lipid synthesis in normal cells [72]. Moreover, studies with labeled [14C]-glucose in the Ehrlich ascites tumor model revealed that de novo fatty acid synthesis accounted for all esterified fatty acid in the tumor, and more than 93% of triacylglycerol fatty acids in the tumor cells were from fatty acid synthesis. Thus, fatty acids synthesized from the endogenous pathway could contribute a significant amount of fatty acids to tumor cell proliferation [74].
It has been documented that the expression of FAS can be regulated by fat-rich diet and hormones [56,57,59,75] . However, the mechanisms of how the FAS gene is upregulated in tumors remains largely unknown. In cancer cells, FAS was found to be increased by growth factors such as epidermal growth factor (EGF) and keratinocyte growth factor, as well as oncogene such as Her-2 (ErbB2/ neu), which is considered an EGF receptor [76-78] . It was suggested that these growth factors and receptors will activate typical mitogen-activated protein kinase (MAPK) pathway such as extracellular signal regulated kinase (ERK) 1/2, Janus Kinase (JNK), and phosphatidylinositol-3 kinase (PI3K). These kinases will then activate Akt to turn on FAS gene expression. Moreover, tumor suppressor genes like p53 and PTEN could regulate the expression of FAS [79-81] . Furthermore, HIF-1 and a key lipogenic transcriptional factor, sterol regulatory-element binding protein (SREBP), have been shown to play a role in the increased fatty acid synthesis of tumor [82,83] .
2.4. Augmented Glucose Uptake
Although glucose is the primary energy source for most cells, it is hydrophilic and does not pass through plasma membranes by diffusion; therefore, cellular utilization of glucose needs specific carrier proteins. In mammalian cells, there are two pathways for glucose uptake, namely the Na+/glucose co-transporters [84] found primarily in small intestine and kidney, and GLUTs or solute carrier family 2A (SLC2A) expressed in all cells [85]. Thirteen members of GLUTs have been identified and classified into three groups; with well-characterized GLUT1-GLUT4 belonging to Class I, fructose carrier protein GLUT5 and newly indentified GLUT7, GLUT9 and GLUT11 falling into Class II, and GLUT6, GLUT8, GLUT10, GLUT12 and the H+/myo-inositol transporter HMIT grouped into Class III, respectively [86]. Each of GLUTs presents specific tissue distribution, function and developmental regulation [87,88] . For example, GLUT1 expresses at variable levels in the early development of many tissues [89] , and is believed to be responsible for basal glucose uptake [90,<91] ; GLUT2, regarded as glucose sensor, expresses in the liver, pancreatic islets, kidney and small intestine [92] ; GLUT3 is abundant in the brain [93]; GLUT4 is restricted to adipose and skeletal tissues [94]; and GLUT5, having a high affinity to fructose instead of glucose, usually presents in small intestine and sperm cells [95,96] .
The higher rate of glycolysis in tumor cells makes tumor cells access to glucose easier if necessary. To compete with normal cells for substrate supply, tumor cells employ this high glucose transport strategy to make them adapted in relatively poor vascularized zones. In fact, it has been shown that the efficiency of glucose entry into malignant cells was at least 20- to 30-fold higher than that in non-malignant cells [97]. The increase of glucose entry to tumor cells is initially achieved through upregulation of specific glucose transporters in the plasma membrane [98,99] .
Association between the malignancy of cells and increased glucose transportation and/or augmented GLUTs expression has been extensively investigated. Rat fibroblasts transformed by ras and src oncogenes have an increased rate of glucose uptake, which is due to activated GLUT1 expression through enhancer elements within GLUT1 promoter [100-102] . With positron emission tomography (PET) scan, clinical observations of tumor through labeled glucose analog, 18F-labeled 2- fluoro-2-deoxy-D-glucose (FDG) indentified an increase in GLUT1-mediated glucose uptake in 57 cervical carcinomas [103] , 55 breast carcinomas [104] , 34 non-small cell lung carcinomas [105] and 17 ovarian carcinomas [106] . Increased glucose uptakes as well as the overexpression of GLUT family members have been found involved in tumor aggressiveness [107,108] , therefore, GLUT family members are considered as a potential therapeutic target [86,109] . In addition, oncogenic transformation itself may be an inducer of GLUT1 [100,101, 110] . In lung and ovarian tumor, comparative increase in GLUT1 expression was observed in cancer patients with higher grade of malignancy, higher proliferative index and lower degree of differentiation [111,112] . Consistently, GLUT1 expression level reciprocally correlates with survival rate of cancer patients [112-114] . Since the expression and regulation of GLUTs are tissue and often cell-specific, expression of some isoforms may only be observed in tumor cells but absent in normal cells. For example, GLUT3, which is detected in lung, ovarian and gastric cancer tissues, was not found in adjacent nontumor tissues [<115]. Fructose transporter GLUT5 could be only found in the malignant breast tissue, but not in its counterpart normal tissues [116] .
Identification in the breast tumor by reverse transcripttase-polymerase chain reaction (RT-PCR), immunohistochemistry and immunocytochemistry, variable expression patterns of GLUTs were observed in both malignant cell lines and tumor tissues. GLUT1 is widely considered responsible for glucose transport in breast tissue [117- 120] . Existence of GLUT2 in breast was controversial and no apparent difference was observed between normal and malignant tissues [117,118,121] . GLUT3, which is more likely expressed in the brain, was not detected in either normal or tumor breast tissues [<118] , except of breast cancer cell line ZR-75 [122] . The expression of GLUT4 was found in various human malignant breast tissues, but not in human breast cancer cell lines - MCF-7, MDA-MB-435 and MDA-MB-23 ADDIN EN.CITE ADDIN EN.CITE.DATA [117,118,121] . The high-affinity fructose transporter GLUT5 was selectively expressed in human breast cancer tissues, indicating that fructose may be a good energy substrate in tumor cells [116, 119] . A novel glucose transporter protein GLUT12 has been identified in breast cancer cell line MCF-7 [123, 124] . The perinuclear sub-cellular localization of GLUT 12 suggests that GLUT12 may traffic to plasma membrane as GLUT4 does and translocation of GLUT12 may contribute to hexose supply to breast cancer cells [86,123,124] .
HIF-1 has been suggested to be one of the factors responsible for GLUT1 up-regulation in tumor cells, due to either the prevention of proteosomal degradation or the enhanced synthesis of GLUT1. HIF-1 is a transcriptional factor that can bind to the GLUT1 promoter to facilitate GLUT1 transcription [125]. Gatenby et al. demonstrated that up-regulation of GLUT1 is an adaptive process to hypoxia in breast cancer [126] . Specific disruption of the HIF-1 pathway in human breast tumor MCF-7 cells with short hairpin RNA significantly attenuated GLUT1 expression [127] . In contrast, Burgman et al. reported a divergence between increased hypoxia-induced FDG uptake and the induction of GLUT1/ GLUT3 protein levels. This observation suggested that hypoxia-induced reduction of cysteine residues in the GLUT may be the mechanism underlying the hypoxia-induced increase in FDG uptake [128]. Moreover, in mouse mammary tumor cells, the insulin receptor substrate (IRS), which was shown to be involved in breast cancer invasion, survival and metastasis, could promote tumor invasion through GLUT1-mediated sustainment of aerobic glycolysis [129] . Furthermore, cellular oncogene c-myc, protein kinase Akt, and ovarian hormone estrogen are also known to induce GLUT1 expression [130-133] . In addition, it has been demonstrated that GLUT3 expression and glucose uptake can be up-regulated through secondary messenger cAMP pathway in breast cancer cell line ZR-75 ADDIN EN.CITE ADDIN EN.CITE.DATA [122] .
Estrogen is a key element in the development and progression of breast cancer; therefore, anti-estrogens, such as tamoxifen, significantly improved the outcome of breast cancer patients. In human breast cancer cells, 17β- estradiol (E2) was able to induce higher rate of glucose consumption through glycolysis and TCA cycle, which lead to an increased glucose metabolism [134]. Rivenzon-Segal et al. observed that tamoxifen could suppress both estrogen-stimulated tumor growth and glycolysis. These changes were consistent with the observed downregulation of GLUT1 expression. Thus, it is proposed that in vivo monitoring of glycolysis may function as a tool to reveal hormonal regulation of GLUT1 expression in breast cancer tumors, as well as to assess tumor response to hormonal therapy [133] . In addition to estrogen, GLUTs expression correlates well with estrogen recaptors (ER) expression level, especially ER-alpha [135]. E2 could elevate the protein levels of GLUT12 after 24 h treatment in ER positive, but not ER negative breast cancer cells, suggesting an ER-dependent hormonal regulation of these GLUTs [86,135] . Epithelial growth factor (EGF) is another important factor that regulates the expression of GLUTs. It has been demonstrated that EGF could enhance not only cell proliferation but also GLUT12 protein in MCF-7 human breast cells. This observation indicates that GLUT12 may contribute to the increased glucose uptake in breast cancer tissues [86,136] .
3. Glucose Metabolism and Its Implication in Cancer Treatment
3.1. Blockage of Glycolysis
Due to the remarkable heterogeneity and adaptability of cancer cells, an effective pharmacological treatment plan is challenging. The molecular characteristics of cancers with identical histology are often dissimilar and molecular heterogeneity can even be observed within a single tumor [24]. Therefore, it is of critical importance to develop an anti-cancer drugs with a defined target and integrated molecular mechanisms participated by distinct cancers [24]. Though not all tumor cells use glycolysis as their main energy source and mitochondrial metabolism is completely functional in these cells [137,138] , they do exhibit a higher sensitivity to OXPHOS-inhibiting drugs than their normal counterparts [139] . Therefore, metabolic alterations appear to be a unique signature of diverse molecular abnormalities in tumors especially those with suppressed mitochondrial function [24]. This observation makes ‘metabolic therapy’ a promising anticancer approach [138]. Metabolic therapy may have two advantages, namely ‘specificity’ and ‘generality’. Specificity is referred to the reason why tumor cells are more sensitive to metabolic inhibitors than their normal counterparts [138,139] . Generality stands for its complete independence of specific signaling and/or epigenetic dysfunction [140]. Glycolytic suppressors have been demonstrated to be successful in experimental tumor models exhibiting that the inhibition of glycolysis and/or OXPHOS results in the delayed tumor growth and/or the impaired tumor cell viability in xenograft in nude mice and cell cultures in vitro [139,141] . As shown in the glycolysis pathway of the Figure 1, HK, PFK and PK are rate-limiting enzymes that catalyze three irreversible reactions in glycolytic pathway. Thus, identifying glycolytic inhibitors should first focus on these steps. In fact, some chemical drugs that target these reactions have been in clinical trials.
2-deoxyglucose (2-dg) is one of these drugs that is a glucose derivative where 2-hydroxyl group is replaced by hydrogen [142] . 2-deoxyglucose enters cells through the glucose transporter, therefore higher expressions of glucose transporter in tumor cells, the higher uptake of 2-deoxyglucose in those cells. Once 2-deoxyglucose enters the cells, it can be used as substrate of HK but will not go further into glycolysis like glucose [141]. Therefore, an augmented uptake of 2-deoxyglucose is likely to be used as a strategy for metabolic therapy of cancer. It has been documented that modification of 2-deoxyglucose improved its uptake in cells such as addition of hydroxyl group - esters, ethers and phosphoesters etc [143,144] , and/or replacement of hydroxyl group with fluorine, iodine, thiol or thioalkyl [145-151] . Moreover, any modification of glucose by either removal of the hydroxyl group or replacement of fluorine and/or methyl group at C-1 or C-6 positions would escape the control of HK and PFK. Furthermore, because of complexity of metabolic interactions, it will be better for a drug that can target one or more glycolytic steps and/or the potential paralleled pathways than just disrupt the rate-limiting step such as HK, PFK and PK. For example, modifications of glucose at C-3 and C-4 could block the cleavage of F-1,6-BP by adolase and lead to collapsed glycolysis.
Oxamate is another type of glycolytic blocker that prevents conversion of pyruvate to lactate through its inhibition of LDH [152]. LDH functions at the downstream of HK, PFK and PK and only becomes very active in anaerobic environment (see Acidification of the Figure 1). Thus, oxamate blocks glycolysis at a different level compared to 2-deoxyglucose, and is selective to kill anaerobic cells in in vitro cell cultures [150,152] . Because the conversion of pyruvate to lactate only occurs in reduced oxygen conditions, tumor cells with aerobic metabolism are not affected by this drug. From this point of view, oxamate can be considered as a selective inhibitor of glycolysis compared to 2-deoxyglucose, which inhibits both aerobic and anaerobic cells.
Dichloroacetate (DCA) is another anti-cancer agent, which targets glycolysis by inhibiting activity of PDK [24], which can negatively regulate PDH activity in the mitochondrion. As shown in Figure 1 (TCA cycle), PDH converts pyruvate to acetyl Co-A and, therefore, controls the flow of pyruvate to the citric acid cycle to generate ATP in the mitochondria [153] . It has been shown that DCA could reverse the glycolytic phenotype in a number of tumor cells through increasing the flux of pyruvate into the mitochondria and promoting glucose oxidation over glycolysis. This reverses the suppressed mitochondrial apoptosis in cancer and results in suppression of tumour growth in vitro and in vivo [24,30,154] . Therefore, it is a promising anti-breast cancer strategy to reverse glycolytic phenotype via inhibition of PDK with DCA.
For those cancers that have normal mitochondria glucose metabolism [137] , glycolysis inhibition may not be efficient enough to arrest tumor cell growth while it may be more appropriate in this case to employ OXPHOS– inhibiting drugs such as rotenone, rhodamine (rho) and oligomycin [138]. A novel strategy for the treatment of tumor cells with aerobic metabolism is to use both OXPHOS inhibitors and glycolytic inhibitors. It has been demonstrated that when human breast carcinoma cells (MCF-7) were treated with rho 123 and 2-dg, hundred percent inhibition was observed as colony formation whereas rho 123 alone only achieved 50% inhibition and 2-dg alone did not inhibit colony formation, respectively [155]. Similar results were observed in xenografts of human breast cancer cells in nude mice. Combination of 2-dg and rho 123 abolished the tumor development while only partial or no inhibition was obtained with either drug alone respectively [156]. This result provides evidence that inhibition of both OXPHOS and glycolysis would significantly enhance therapeutic efficacy compared to each alone. In addition, inhibition of OXPHOS would convert tumor cells from aerobic to anaerobic glycolysis and hypersensitize these cells to glycolytic inhibitors [157,158] . Therefore, therapeutic approaches designed to inhibit both glycolysis and OXPHOS are considered a novel therapeutic strategy to selectively kill cancer cells [159,160] .
Cells within the inner core and periphery of a tumor have different accessibilities to oxygen and nutrients. At the inner core, cancer cells are in a poorly oxygenated environment and rely heavily on anaerobic metabolism. The glycolytic inhibitors mentioned above are particularly effective against cancer cells with mitochondrial defects or under hypoxia condition, which are frequently associated with cellular resistance to conventional chemotherapy and radiation therapy [141,161] . Thus, it will be innovative to develop an anti-cancer protocol that employs standard chemotherapy, radiation therapy and inhibition of glucose metabolism to increase the efficacy of cancer treatment.
3.2. Targeting de novo FA Synthesis
Because cancer cells rely greatly on fatty acid for their survival (de novo FA synthesis in the Figure 1), it is plausible that cancer treatment can utilize the suppression of FAS activity. Chemical blockage of fatty acid generation with a FAS inhibitor (cerulenin) exhibited cytotoxicity to a large range of human cancer cell lines ADDIN EN.CITE ADDIN EN.CITE.DATA [154] . Moreover, inhibition of cancer cell proliferation by tea polyphenol(-)-epigallocatechin 3 gallate (EGCG) and theaflavin (TF) was also associated with suppression of FAS expression through inhibiting PI3K-Akt-Sp1 signal pathway [162] . Diosgenin is a steroid sapogenin from plant and was shown to suppress FAS in HER-2-overexpressing breast cancer cells through modulating Akt, mTOR and JNK phosphorylation [163] . Furthermore, it was documented that amentoflavone isolated from S. tamariscina induced breast cancer cell apoptosis through suppressing the biosynthesis of FA [164].
In human breast cancers, increased expression of FAS is observed in infiltrating breast cancer tissue, and was always associated with malignant phenotypes and poor prognosis [165] . Ample evidence is available that supports FAS as an ideal target of anti-breast cancer therapy. For example, in estrogen dependent human breast cancer cells, inhibition of cancer lipogenesis by a FAS inhibitor significantly reduced ER protein and mRNA expression, leading to increased apoptosis [166] . Moreover, pharmacological inhibition of FAS activity in breast cancer cell lines SK-Br3, MCF-7 and MDA-MB-231 using cerulenin enhanced anti-cancer efficacy of Fluorouracil (5-FU) no matter of whether cerulenin and 5-FU were applied at the same time or one prior to the other. This finding indicates that FAS-catalyzed de novo fatty acid biogenesis plays a key role in regulation of breast cancer cells in response to other anti-cancer drugs [167]. Furthermore, up-regulation of FAS rendered human breast cancer cells (MCF-7 and MDA-MB-468 cells) to resist drug-induced apoptosis while reduction of FAS expression was able to efficiently inverse resistance to drug-induced apoptosis [168] . Therefore, FAS over-expression in cancer cells is a new mechanism of drug resistance and may be served as an ideal target for chemo-sensitization in breast cancer chemo-therapy ADDIN EN.CITE ADDIN EN.CITE.DATA [168] .
There are two mechanisms that may explain how FA synthesis regulates malignant breast phenotype: one is through direct activation of pro-apoptotic factors and/or inactivation of anti-apoptotic proteins and the other is through alterations of cellular lipid metabolism. For the induction of apoptosis, it has been documented that inhibition of FAS with small interfering RNA (siRNA) significantly up-regulated pro-apoptotic factors such as Bcl-2/ adenovirus E1B 19 kDa interacting protein-3 (BNIP-3), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and death-associated protein kinase-2 (DAPK- 2) [169] . Moreover, application of BNIP-3 siRNA in this setting significantly counteracted the effect of FAS siRNA induced apoptosis [170] . Furthermore, there was a significant inverse correlation between abundances of FAS and BNIP-3 in clinical samples of human breast cancer [169] . In addition, cerulenin-induced apoptosis in MDA-MB-468 breast cancer cells could be enhanced by down-regulation of anti-apoptotic factors such as XIAP, cIAP-1 and Akt, and up-regulation of apoptotic factor such as Bak in mitochondria [171] . For the alternation of cellular lipid metabolism, silencing ACC alpha (a rate limiting enzyme in FA biosynthesis) or FAS by siRNA technique revealed a significant decrease in palmitic acid synthesis, which led to induction of apoptosis concomitantly with reactive oxygen species (ROS) formation and mitochondrial impairment. Further study indicated that ACC was critical to breast tumor survival because overexpression of ACC alpha mutant that was resistant to ACC siRNA rescued the siRNA induced apoptosis. However, the same strategy did not work in the case of FAS mutant. Interestingly, the addition of palmitate or Vitamin E completely prevented the apoptosis induced by both ACC and FAS siRNA, suggesting an interaction among lipid metabolism, ROS formation and mitochondrial function [172] . Furthermore, besides lipogenesis, breakdown of fatty acids can also regulate fatty acid metabolism through carnitine palmitoyltransferase-1 (CPT- 1). It has been observed that inhibition of CPT-1 with etomoxir resulted in cerulenine-like effects with downregulation of FA synthesis [173] . Therefore, there is an increasing interest in identifying and developing new anti-tumor compounds that modulate FA metabolism.
3.3. GLUTs as a Target of Metabolic Therapy
It has been suggested that malignant transformation involves stimulation of glucose transporters and consequently elevation of glucose metabolism and glycolysis [174] , therefore, understanding GLUTs participation in cancer could be helpful for the diagnosis and treatment of tumor tissue. Because GLUT1 was exclusively found in human breast cancer biopsies and its expression was strongly associated with tumor grade, proliferation as well as invasiveness [111,120,121] , GLUT1 has been proposed to be a new marker for diagnosis and prognosis of human breast cancers, an indication of tumor aggressiveness, and a possible therapeutic target for patients with breast cancer [114,175] . It has been demonstrated that drug mediated inhibition of glucose transport increased the effectiveness of several anti-tumor drugs and effectively induced growth arrest and apoptosis in lung, breast cancer and leukemia cell lines [175,176] . For example, combination of GLUT1 inhibitor with chemotherapeutic drug daunorubicin preferentially sensitized anti-cancer activity of daunorubicin and overcame drug resistance of cancer cells in hypoxia environment [176] . Since GLUT5 is specifically expressed in human breast cancer tissue rather than its normal counterpart, GLUT5 may also service as a novel diagnostic marker and therapeutic target for human breast cancer [177] .
3.4. Summary and Potential in the Application of Herbal Research
The proposal of the “Warburg effect” and recent observations of 18F-labeled FDG uptake in tumor through PET opened a new era for cancer treatment by modulating glucose metabolism in tumor cells. Since glucose metabolism appears differently in tumor and normal cells, such as aerobic glycolysis in normal cells and anaerobic glycolysis in tumor cells, inhibition of anaerobic glycolysis in cancer cells provides a promising therapeutic approach to arrest cancer proliferation. The therapeutic approach can be achieved by the following strategies: the first is to inhibit glucose transport into the cells by targeting GLUT1 or GLUT5 gene expression. This can be fulfilled by employing short interfering RNA technology or drugs that inhibit GLUT function. The second is to inhibit glucose metabolism in cancer cells by employing drugs that target anaerobic glycolysis directly. The third is to inhibit both aerobic and anaerobic glycolysis in cancer cells. In this case, cancer cells that still utilize aerobic glycolysis will turn to utilize anaerobic glycolysis, and anaerobic glycolysis inhibitors will then significantly inhibit cancer cell proliferation. The fourth is to inhibit de novo fatty acid synthesis by inhibition of FAS activity. This can be achieved by direct inhibition of either enzymes involved in FA synthesis such as FAS and ACC or in combination of those with anaerobic glycolysis.
One of the strategies has been in the phase I and phase II clinical trials at present time [178], which employs the anti-diabetic drug metformin for the treatment of human breast carcinoma [179]. In addition, this strategy of inhibiting cancer through nutrient metabolism may provide opportunities to further understand anti-cancer mechanism of natural herbs. Accordingly, herbs that have hypoglycemic and/or hypolipidemic properties may be of use in cancer treatment. For example, berberine (BBR) is an isoquinoline alkaloid isolated from many botanical materials such as Cortex Phellodendri and Rhizoma Coptidis. Though the anti-tumor mechanism of BBR is not fully revealed, it has been demonstrated that BBR could induce cell cycle arrest in human breast cancer cells [180]. Moreover, it has been reported that BBR has comparable hypoglycemic and potent hypolipidemic activities in patients with type 2 diabetes [181]. Therefore, it is plausible that BBR regulation of glucose-lipid metabolism may contribute to its tumor suppression. Another example is that several herbs such as Panax Ginseng, Ganoderma Lucidum, Astragalus and Lycium Barbarum have been used in the most populated fighting cancer formulation [182-185] . and their polysaccharides extract did have the strong capability in improving metabolic abnormalities in in vivo studies [186-189] . Furthermore, a saponin extract from Gynostemma Pentaphyllum (GP) - gypenoside has been demonstrated to inhibit tumors through inducing mitochondria-dependent apoptosis and cell cycle arrest at G0/G1 [190] . It has been demonstrated that GP is able to modulate blood glucose levels and reduce hyperlipidemia in the Zucker fatty rat [191]. In addition, the polyphenolic compound such as curcumins from Curcuma Longa, has been utilized for cancer therapy through different mechanisms such as cell cycle arrest, apoptosis, mitochondrial dysfunction and protein kinase dysfunctions [192,193] . Curcumins are also good anti-oxidants, which ameliorate high-glucose induced mitochondrial oxidative stress [194]. Therefore, understanding of herbal regulation of glucose metabolism in cancer cells will significantly contribute our knowledge how herbs can be effective in the treatment of cancer, and which will also provide fundamental evidence for clinical application of traditional herbs in the field of cancer.
Transportation of glucose/fructose into cells is mediated through glucose transporters - GLUTs. Once glucose enters the cell, it is phosphorylated by HK to produce G-6-P, which is further metabolized to pyruvate by glycolytic enzymes such as PFK, aldolase, PGM, enolase and PK. A part of carbon derived from glycolysis may flux into PPP, participating in the generation of ribose-5- phophate to support nucleic acid biosynthesis. Standing at the crossroad between oxidative and glycolytic pathways, pyruvate can be converted to either lactate by LDH-A in cytosol or to acetyl-CoA by PDH in mitochondria. Accumulation of lactate would lead to cellular acidification. In the presence of citrate synthase in mitochondrial TCA cycle, the formed acetyl-CoA would be condensed with OAA to generate citrate, which is then transported to cytoplasm and broken down back to acetyl-CoA and OAA by ACL. Meanwhile, acetyl-CoA is carboxylated to malonyl-CoA by ACC. FAS functions to condense acetyl-CoA with malonyl-CoA to complete de novo FA synthesis, which relies on the provision of NADPH by PPP. The increased presence of malonylCoA could inhibit the oxidation of FA through suppressing CPT-1, which is a suppressor of FA biosynthesis.
Warburg phenomenon originally hypothesized an impairment of mitochondrial OXPHOS in tumor cells. In order to transiently adapt to increased demands of nutrients and oxygen, tumor cells boost glycolysis rather than OXPHOS as their main cellular source of ATP through increasing activities of glucose transporters and glycolytic enzymes. Augmented cellular acidification by lactate formation would facilitate tumor growth through acidmediated extracellular matrix degradation, which then favors tumor invasion and metastasis. The persistency of this adaptive mechanism suggests that alternative pathways are necessary to support the constant supply of building-materials for cell growth. Glycolytic and TCA cycle intermediates can be siphoned off by enzymes that use them as biosynthetic precursors. Promoted PPP and FA synthesis pathways are two strategies that tumor cells use for de novo biosynthesis.
Bold arrows marked in this figure indicate preferential pathways in tumor cells; slashed arrow stands for metabolic pathway in non-malignant cells.
4. Acknowledgements
This work is financially supported by FDCT (029/2007/ A2), Macao SAR, China.
REFERENCES
- O. Warburg, “On the Origin of Cancer Cells,” Science, Vol. 123, No. 3191, 1956, pp. 309-314. doi:10.1126/science.123.3191.309
- H. Simonnet, et al., “Low Mitochondrial Respiratory Chain Content Correlates with Tumor Aggressiveness in Renal Cell Carcinoma,” Carcinogenesis, Vol. 23, No. 5, 2002, pp. 759-768. doi:10.1093/carcin/23.5.759
- V. Ganapathy, M. Thangaraju and P. D. Prasad, “Nutrient Transporters in Cancer: Relevance to Warburg Hypothesis and Beyond,” Pharmacology & Therapeutics, Vol. 121, No. 1, 2009, pp. 29-40. doi:10.1016/j.pharmthera.2008.09.005
- M. Board, S. Humm and E. A. Newsholme, “Maximum Activities of Key Enzymes of Glycolysis, Glutaminolysis, Pentose Phosphate Pathway and Tricarboxylic Acid Cycle in Normal, Neoplastic and Suppressed Cells,” Biochemical Journal, Vol. 265, No. 2, 1990, pp. 503-509.
- A. Hennipman, et al., “Glycolytic Enzymes in Breast Cancer, Benign Breast Disease and Normal Breast Tissue,” Tumor Biology, Vol. 8, No. 5, 1987, pp. 251-263. doi:10.1159/000217529
- S. Mazurek, H. Grimm, C. B. Boschek, P. Vaupel and E. Eigenbrodt, “Pyruvate Kinase Type M2: A Crossroad in the Tumor Metabolome,” British Journal of Nutrition, Vol. 87, Suppl. 1, 2002, pp. S23-S29. doi:10.1079/BJN2001454
- J. A. Cooper, N. A. Reiss, R. J. Schwartz and T. Hunter, “Three Glycolytic Enzymes are Phosphorylated at Tyrosine in Cells Transformed by Rous Sarcoma Virus,” Nature, Vol. 302, No. 5905, 1983, pp. 218-223. doi:10.1038/302218a0
- M. I. Koukourakis, A. Giatromanolaki, C. Simopoulos, A. Polychronidis and E. Sivridis, “Lactate Dehydrogenase 5 (LDH5) Relates to up-Regulated Hypoxia Inducible Factor Pathway and Metastasis in Colorectal Cancer,” Clinical and Experimental Metastasis, Vol. 22, No. 1, 2005, pp. 25-30. doi:10.1007/s10585-005-2343-7
- J. W. Kim and C. V. Dang, “Cancer’s Molecular Sweet Tooth and the Warburg Effect,” Cancer Research, Vol. 66, No. 18, 2006, pp. 8927-8930. doi:10.1158/0008-5472.CAN-06-1501
- S. P. Mathupala, C. Heese and P. L. Pedersen, “Glucose Catabolism in Cancer Cells. The Type 2 Hexokinase Promoter Contains Functionally Active Response Elements for the Tumor Suppressor P53,” Journal of Biological Chemistry, Vol. 272, No. 36, 1997, pp. 22776-22780. doi:10.1074/jbc.272.36.22776
- J. D. Gordan and M. C. Simon, “Hypoxia-Inducible Factors: Central Regulators of the Tumor Phenotype,” Current Opinion in Genetics & Development, Vol. 17, No. 1, 2007, pp. 71-77. doi:10.1016/j.gde.2006.12.006
- G. L. Semenza, “Hypoxia-Inducible Factor 1 (Hif-1) Pathway,” Science’s STKE, Vol. 2007, No. 407, 2007, p. cm8. doi:10.1126/stke.4072007cm8
- J. W. Kim and C. V. Dang, “Multifaceted Roles of Glycolytic Enzymes,” Trends in Biochemical Sciences, Vol. 30, No. 3, 2005, pp. 142-150. doi:10.1016/j.tibs.2005.01.005
- K. H. Ibsen, et al., “Expression of Multimolecular Forms of Pyruvate Kinase in Normal, Benign, and Malignant Human Breast Tissue,” Cancer Research, Vol. 42, No. 3, 1982, pp. 888-892.
- L. G. Baggetto, “Deviant Energetic Metabolism of Glycolytic Cancer Cells,” Biochimie, Vol. 74, No. 11, 1992, pp. 959-974. doi:10.1016/0300-9084(92)90016-8
- P. S. Coleman and B. B. Lavietes, “Membrane Cholesterol, Tumorigenesis, and the Biochemical Phenotype of Neoplasia,” CRC Critical Reviews in Biochemistry, Vol. 11, No. 4, 1981, pp. 341-393.
- W. L. McKeehan, “Glycolysis, Glutaminolysis and Cell Proliferation,” Cell Biology International Reports, Vol. 6, No. 7, 1982, pp. 635-650. doi:10.1016/0309-1651(82)90125-4
- Z. Kovacevic and J. D. McGivan, “Mitochondrial Metabolism of Glutamine and Glutamate and Its Physiological Significance,” Physiological Reviews, Vol. 63, No. 2, 1983, pp. 547-605.
- H. Shim, et al., “C-Myc Transactivation of Ldh-A: Implications for Tumor Metabolism and Growth,” Proceedings of the National Academy of Sciences of the USA, Vol. 94, No. 13, 1997, pp. 6658-6663. doi:10.1073/pnas.94.13.6658
- C. V. Dang and G. L. Semenza, “Oncogenic Alterations of Metabolism,” Trends in Biochemical Sciences, Vol. 24, No. 2, 1999, pp. 68-72. doi:10.1016/S0968-0004(98)01344-9
- G. L. Semenza, P. H. Roth, H. M. Fang and G. L. Wang, “Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia-Inducible Factor 1,” Journal of Biological Chemistry, Vol. 269, No. 38, 1994, pp. 23757-23763.
- J. W. Kim, I. Tchernyshyov, G. L. Semenza and C. V. Dang, “Hif-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia,” Cell Metabolism, Vol. 3, No. 3, 2006, pp. 177-185. doi:10.1016/j.cmet.2006.02.002
- I. Papandreou, R. A. Cairns, L. Fontana, A. L. Lim and N. C. Denko, “Hif-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption,” Cell Metabolism, Vol. 3, No. 3, 2006, pp. 187-197. doi:10.1016/j.cmet.2006.01.012
- E. D. Michelakis, L. Webster and J. R. Mackey, “Dichloroacetate (Dca) as a Potential Metabolic-Targeting Therapy for Cancer,” British Journal of Cancer, Vol. 99, No. 7, 2008, pp. 989-994. doi:10.1038/sj.bjc.6604554
- J. Chesney, “6-Phosphofructo-2-Kinase/Fructose-2, 6-Bisphosphatase and Tumor Cell Glycolysis,” Current Opinion in Clinical Nutrition & Metabolic Care, Vol. 9, No. 5, 2006, pp. 535-539. doi:10.1097/01.mco.0000241661.15514.fb
- G. L. Semenza, et al., “Hypoxia Response Elements in the Aldolase a, Enolase 1, and Lactate Dehydrogenase a Gene Promoters Contain Essential Binding Sites for Hypoxia-Inducible Factor 1,” Journal of Biological Chemistry, Vol. 271, No. 51, 1996, pp. 32529-32537. doi:10.1074/jbc.271.51.32529
- V. R. Fantin, J. St-Pierre and P. Leder, “Attenuation of Ldh-a Expression Uncovers a Link between Glycolysis, Mitochondrial Physiology, and Tumor Maintenance,” Cancer Cell, Vol. 9, No. 6, 2006, pp. 425-434. doi:10.1016/j.ccr.2006.04.023
- M. Wu, et al., “Multiparameter Metabolic Analysis Reveals a Close Link between Attenuated Mitochondrial Bioenergetic Function and Enhanced Glycolysis Dependency in Human Tumor Cells,” American Journal of Physiology-Cell Physiology, Vol. 292, No. 1, 2007, pp. C125- C136. doi:10.1152/ajpcell.00247.2006
- J. G. Pastorino, J. B. Hoek and N. Shulga, “Activation of Glycogen Synthase Kinase 3beta Disrupts the Binding of Hexokinase 2 to Mitochondria by Phosphorylating Voltage-Dependent Anion Channel and Potentiates Chemotherapy-Induced Cytotoxicity,” Cancer Research, Vol. 65, No. 22, 2005, pp. 10545-10554. doi:10.1158/0008-5472.CAN-05-1925
- J. G. Pan and T. W. Mak, “Metabolic Targeting as an Anticancer Strategy: Dawn of a New Era?” Science’s STKE, Vol. 2007, No. 381, 2007, pp. pe14.
- R. L. Elstrom, et al., “Akt Stimulates Aerobic Glycolysis in Cancer Cells,” Cancer Resarch, Vol. 64, No. 11, 2004, pp. 3892-3899. doi:10.1158/0008-5472.CAN-03-2904
- K. Gottlob, et al., “Inhibition of Early Apoptotic Events by Akt/Pkb Is Dependent on the First Committed Step of Glycolysis and Mitochondrial Hexokinase,” Genes & Development, Vol. 15, No. 11, 2001, pp. 1406-1418. doi:10.1101/gad.889901
- K. Bensaad, et al., “Tigar, a P53-Inducible Regulator of Glycolysis and Apoptosis,” Cell, Vol. 126, No. 1, 2006, pp. 107-120. doi:10.1016/j.cell.2006.05.036
- S. Matoba, et al., “P53 Regulates Mitochondrial Respiration,” Science, Vol. 312, No. 5780, 2006, pp. 1650-1653. doi:10.1126/science.1126863
- M. C. Brahimi-Horn, J. Chiche and J. Pouyssegur, “Hypoxia Signalling Controls Metabolic Demand,” Current Opinion in Cell Biology, Vol. 19, No. 2, 2007, pp. 223-229. doi:10.1016/j.ceb.2007.02.003
- F. Dayan, N. M. Mazure, M. C. Brahimi-Horn and J. Pouyssegur, “A Dialogue between the Hypoxia-Inducible Factor and the Tumor Microenvironment,” Cancer Microenvironment, Vol. 1, No. 1, 2008, pp. 53-68.
- S. Zhou, et al., “Frequency and Phenotypic Implications of Mitochondrial DNA Mutations in Human Squamous Cell Cancers of the Head and Neck,” Proceedings of the National Academy of Sciences of the USA, Vol. 104, No. 18, 2007, pp. 7540-7545. doi:10.1073/pnas.0610818104
- S. Dasgupta, M. O. Hoque, S. Upadhyay and D. Sidransky, “Mitochondrial Cytochrome B Gene Mutation Promotes Tumor Growth in Bladder Cancer,” Cancer Research, Vol. 68, No. 3, 2008, pp. 700-706. doi:10.1158/0008-5472.CAN-07-5532
- W. C. Copeland, J. T. Wachsman, F. M. Johnson and J. S. Penta, “Mitochondrial DNA Alterations in Cancer,” Cancer Investigation, Vol. 20, No. 4, 2002, pp. 557-569. doi:10.1081/CNV-120002155
- B. E. Baysal, “Role of Mitochondrial Mutations in Cancer,” Endocrine Pathology, Vol. 17, No. 3, 2006, pp. 203-212. doi:10.1385/EP:17:3:203
- J. S. Carew and P. Huang, “Mitochondrial Defects in Cancer,” Molecular Cancer, Vol. 1, 2002, p. 9. doi:10.1186/1476-4598-1-9
- K. Plak, A. M. Czarnecka, T. Krawczyk, P. Golik and E. Bartnik, “Breast Cancer as a Mitochondrial Disorder (Review),” Oncology Reports, Vol. 21, No. 4, 2009, pp. 845-851.
- T. Pfeiffer, S. Schuster and S. Bonhoeffer, “Cooperation and Competition in the Evolution of Atp-Producing Pathways,” Science, Vol. 292, No. 5516, 2001, pp. 504- 507. doi:10.1126/science.1058079
- N. Bellance, P. Lestienne and R. Rossignol, “Mitochondria: From Bioenergetics to the Metabolic Regulation of Carcinogenesis,” Frontiers in Bioscience, Vol. 14, 2009, pp. 4015-4034.
- J. S. Modica-Napolitano and K. K. Singh, “Mitochondrial Dysfunction in Cancer,” Mitochondrion, Vol. 4, No. 5-6, 2004, pp. 755-762. doi:10.1016/j.mito.2004.07.027
- R. G. Jones and C. B. Thompson, “Tumor Suppressors and Cell Metabolism: A Recipe for Cancer Growth,” Genes & Development, Vol. 23, No. 5, 2009, pp. 537-548. doi:10.1101/gad.1756509
- D. E. Bauer, G. Hatzivassiliou, F. Zhao, C. Andreadis, and C. B. Thompson, “Atp Citrate Lyase is an Important Component of Cell Growth and Transformation,” Oncogene, Vol. 24, No. 41, 2005, pp. 6314-6322. doi:10.1038/sj.onc.1208773
- X. Tong, F. Zhao and C. B. Thompson, “The Molecular Determinants of De Novo Nucleotide Biosynthesis in Cancer Cells,” Current Opinion in Genetics & Development, Vol. 19, No. 1, 2009, pp. 32-37. doi:10.1016/j.gde.2009.01.002
- T. Wood, “Physiological Functions of the Pentose Phosphate Pathway,” Cell Biochemistry and Function, Vol. 4, No. 4, 1986, pp. 241-247. doi:10.1002/cbf.290040403
- E. W. McDermott, E. T. Barron, P. P. Smyth and N. J. O’Higgins, “Premorphological Metabolic Changes in Human Breast Carcinogenesis,” British Journal of Surgery, Vol. 77, No. 10, 1990, pp. 1179-1182. doi:10.1002/bjs.1800771029
- R. Dutu, M. Nedelea, G. Veluda and V. Burculet, “Cytoenzymologic Investigations on Carcinomas of the Cervix Uteri,” Acta Cytologica, Vol. 24, No. 2, 1980, pp. 160-166.
- K. Ikezaki, K. L. Black, S. G. Conklin, and D. P. Becker, “Histochemical Evaluation of Energy Metabolism in Rat Glioma,” Neurological Research, Vol. 14, No. 4, 1992, pp. 289-293.
- R. J. De Berardinis, et al., “Beyond Aerobic Glycolysis: Transformed Cells can Engage in Glutamine Metabolism that Exceeds the Requirement for Protein and Nucleotide Synthesis,” Proceedings of the National Academy of Sciences of the USA, Vol. 104, No. 49, 2007, pp. 19345- 19350. doi:10.1073/pnas.0709747104
- F. P. Kuhajda, et al., “Fatty Acid Synthesis: A Potential Selective Target for Antineoplastic Therapy,” Proceedings of the National Academy of Sciences of the USA, Vol. 91, No. 14, 1994, pp. 6379-6383. doi:10.1073/pnas.91.14.6379
- C. Pompeia, et al., “Effect of Fatty Acids on Leukocyte Function,” Brazilian Journal of Medical and Biological Research, Vol. 33, No. 11, 2000, pp. 1255-1268. doi:10.1590/S0100-879X2000001100001
- N. F. Boyd, M. Cousins, G. Lockwood and D. Tritchler, “Dietary Fat and Breast Cancer Risk: The Feasibility of a Clinical Trial of Breast Cancer Prevention,” Lipids, Vol. 27, No. 10, 1992, pp. 821-826. doi:10.1007/BF02535857
- H. A. Risch, M. Jain, L. D. Marrett and G. R. Howe, “Dietary Fat Intake and Risk of Epithelial Ovarian Cancer,” Journal of the National Cancer Institute, Vol. 86, No. 18, 1994, pp. 1409-1415. doi:10.1093/jnci/86.18.1409
- H. A. Risch, L. D. Marrett, M. Jain and G. R. Howe, “Differences in Risk Factors for Epithelial Ovarian Cancer by Histologic Type. Results of a Case-Control Study,” American Journal of Epidemiology, Vol. 144, No. 4, 1996, pp. 363-372.
- K. K. Carroll, “Dietary Fat and Breast Cancer,” Lipids, Vol. 27, No. 10, 1992, pp. 793-797. doi:10.1007/BF02535852
- B. S. Reddy, “Dietary Fat and Colon Cancer: Animal Model Studies,” Lipids, Vol. 27, No. 10, 1992, pp. 807-813. doi:10.1007/BF02535855
- B. J. Thompson and S. Smith, “Biosynthesis of Fatty Acids by Lactating Human Breast Epithelial Cells: An Evaluation of the Contribution to the Overall Composition of Human Milk Fat,” Pediatric Research, Vol. 19, No. 1, 1985, pp. 139-143. doi:10.1203/00006450-198501000-00036
- H. S. Sul and D. Wang, “Nutritional and Hormonal Regulation of Enzymes in Fat Synthesis: Studies of Fatty Acid Synthase and Mitochondrial Glycerol-3-Phosphate Acyltransferase Gene Transcription,” Annual Review of Nutrition, Vol. 18, 1998, pp. 331-351. doi:10.1146/annurev.nutr.18.1.331
- G. Medes, A. Thomas and S. Weinhouse, “Metabolism of Neoplastic Tissue. IV. A Study of Lipid Synthesis in Neoplastic Tissue Slices in Vitro,” Cancer Research, Vol. 13, No. 1, 1953, pp. 27-29.
- L. Z. Milgraum, L. A. Witters, G. R. Pasternack and F. P. Kuhajda, “Enzymes of the Fatty Acid Synthesis Pathway Are Highly Expressed in Situ Breast Carcinoma,” Clinical Cancer Research, Vol. 3, No. 11, 1997, pp. 2115-2120.
- P. L. Alo, et al., “Expression of Fatty Acid Synthase (Fas) as a Predictor of Recurrence in Stage 1 Breast Carcinoma Patients,” Cancer, Vol. 77, No. 3, 1996, pp. 474-482. doi:10.1002/(SICI)1097-0142(19960201)77:3<474::AID-CNCR8>3.0.CO;2-K
- A. Rashid, et al., “Elevated Expression of Fatty Acid Synthase and Fatty Acid Synthetic Activity in Colorectal Neoplasia,” American Journal of Pathology, Vol. 150, No. 1, 1997, pp. 201-208.
- P. L. Alo, et al., “Fatty Acid Synthase (Fas) Predictive Strength in Poorly Differentiated Early Breast Carcinomas,” Tumori, Vol. 85, No. 1, 1999, pp. 35-40.
- J. I. Epstein, M. Carmichael and A. W. Partin, “Oa-519 (Fatty Acid Synthase) as an Independent Predictor of Pathologic State in Adenocarcinoma of the Prostate,” Urology, Vol. 45, No. 1, 1995, pp. 81-86. doi:10.1016/S0090-4295(95)96904-7
- T. S. Gansler, W. Hardman, D. A. Hunt, S. Schaffel and R. A. Hennigar, “Increased Expression of Fatty Acid Synthase (Oa-519) in Ovarian Neoplasms Predicts Shorter Survival,” Human Pathology, Vol. 28, No. 6, 1997, pp. 686-692. doi:10.1016/S0046-8177(97)90177-5
- E. S. Pizer, S. F. Lax, F. P. Kuhajda, G. R. Pasternack and R. J. Kurman, “Fatty Acid Synthase Expression in Endometrial Carcinoma: Correlation with Cell Proliferation and Hormone Receptors,” Cancer, Vol. 83, No. 3, 1998, pp. 528-537. doi:10.1002/(SICI)1097-0142(19980801)83:3<528::AID-CNCR22>3.0.CO;2-X
- M. S. Shurbaji, J. H. Kalbfleisch and T. S. Thurmond, “Immunohistochemical Detection of a Fatty Acid Synthase (Oa-519) as a Predictor of Progression of Prostate Cancer,” Human Pathology, Vol. 27, No. 9, 1996, pp. 917-921. doi:10.1016/S0046-8177(96)90218-X
- F. P. Kuhajda, “Fatty-Acid Synthase and Human Cancer: New Perspectives on Its Role in Tumor Biology,” Nutrition, Vol. 16, No. 3, 2000, pp. 202-208. doi:10.1016/S0899-9007(99)00266-X
- J. A. Menendez and R. Lupu, “Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis,” Nature Reviews Cancer, Vol. 7, No. 10, 2007, pp. 763-777. doi:10.1038/nrc2222
- M. Ookhtens, R. Kannan, I. Lyon and N. Baker, “Liver and Adipose Tissue Contributions to Newly Formed Fatty Acids in an Ascites Tumor,” American Journal of Physiology, Vol. 247, No. 1, Pt 2, 1984, pp. R146-R153.
- E. Kalkhoven, L. Kwakkenbos-Isbrucker, S. W. de Laat, P. T. van der Saag and B. van der Burg, “Synthetic Progestins Induce Proliferation of Breast Tumor Cell Lines via the Progesterone or Estrogen Receptor,” Molecular and Cellular Endocrinology, Vol. 102, No. 1-2, 1994, pp. 45-52. doi:10.1016/0303-7207(94)90096-5
- C. Kumar-Sinha, K. W. Ignatoski, M. E. Lippman, S. P. Ethier and A. M. Chinnaiyan, “Transcriptome Analysis of Her2 Reveals a Molecular Connection to Fatty Acid Synthesis,” Cancer Research, Vol. 63, No. 1, 2003, pp. 132-139.
- Y. Chang, J. Wang, X. Lu, D. P. Thewke and R. J. Mason, “Kgf Induces Lipogenic Genes through a Pi3k and Jnk/Srebp-1 Pathway in H292 Cells,” Journal of Lipid Research, Vol. 46, No. 12, 2005, pp. 2624-2635. doi:10.1194/jlr.M500154-JLR200
- J. V. Swinnen, et al., “Stimulation of Tumor-Associated Fatty Acid Synthase Expression by Growth Factor Activation of the Sterol Regulatory Element-Binding Protein Pathway,” Oncogene, Vol. 19, No. 45, 2000, pp. 5173-5181. doi:10.1038/sj.onc.1203889
- H. Pelicano, et al., “Mitochondrial Respiration Defects in Cancer Cells Cause Activation of Akt Survival Pathway through a Redox-Mediated Mechanism,” Journal of Cell Biology, Vol. 175, No. 6, 2006, pp. 913-923. doi:10.1083/jcb.200512100
- S. Bandyopadhyay, et al., “Fas Expression Inversely Correlates with Pten Level in Prostate Cancer and a Pi 3-Kinase Inhibitor Synergizes with Fas Sirna to Induce Apoptosis,” Oncogene, Vol. 24, No. 34, 2005, pp. 5389-5395. doi:10.1038/sj.onc.1208555
- A. M. D’Erchia, A. Tullo, K. Lefkimmiatis, C. Saccone and E. Sbisa, “The Fatty Acid Synthase Gene is a Conserved P53 Family Target from Worm to Human,” Cell Cycle, Vol. 5, No. 7, 2006, pp. 750-758. doi:10.4161/cc.5.7.2622
- T. Porstmann, et al., “Pkb/Akt Induces Transcription of Enzymes Involved in Cholesterol and Fatty Acid Biosynthesis Via Activation of Srebp,” Oncogene, Vol. 24, No. 43, 2005, pp. 6465-6481.
- E. Furuta, et al., “Fatty Acid Synthase Gene is up-Regulated by Hypoxia Via Activation of Akt and Sterol Regulatory Element Binding Protein-1,” Cancer Research, Vol. 68, No. 4, 2008, pp. 1003-1011. doi:10.1158/0008-5472.CAN-07-2489
- M. A. Hediger and D. B. Rhoads, “Molecular Physiology of Sodium-Glucose Cotransporters,” Physiological Reviews, Vol. 74, No. 4, 1994, pp. 993-1026.
- A. Carruthers, “Facilitated Diffusion of Glucose,” Physiological Reviews, Vol. 70, No. 4, 1990, pp. 1135-1176.
- M. L. Macheda, S. Rogers and J. D. Best, “Molecular and Cellular Regulation of Glucose Transporter (Glut) Proteins in Cancer,” Journal of Cellular Physiology, Vol. 202, No. 3, 2005, pp. 654-662. doi:10.1002/jcp.20166
- C. Postic, et al., “Development and Regulation of Glucose Transporter and Hexokinase Expression in Rat,” American Journal of Physiology, Vol. 266, No. 4, Pt 1, 1994, pp. E548-E559.
- T. Santalucia, et al., “Developmental Regulation of Glut- 1 (Erythroid/Hep G2) and Glut-4 (Muscle/Fat) Glucose Transporter Expression in Rat Heart, Skeletal Muscle, and Brown Adipose Tissue,” Endocrinology, Vol. 130, No. 2, 1992, pp. 837-846. doi:10.1210/en.130.2.837
- M. Mueckler, et al., “Sequence and Structure of a Human Glucose Transporter,” Science, Vol. 229, No. 4717, 1985, pp. 941-945. doi:10.1126/science.3839598
- M. J. Birnbaum, H. C. Haspel and O. M. Rosen, “Cloning and Characterization of a Cdna Encoding the Rat Brain Glucose-Transporter Protein,” Proceedings of the National Academy of Sciences of the USA, Vol. 83, No. 16, 1986, pp. 5784-5788. doi:10.1073/pnas.83.16.5784
- H. Fukumoto, S. Seino, H. Imura, Y. Seino and G. I. Bell, “Characterization and Expression of Human Hepg2/ Erythrocyte Glucose-Transporter Gene,” Diabetes, Vol. 37, No. 5, 1988, pp. 657-661. doi:10.2337/diabetes.37.5.657
- B. Thorens, H. K. Sarkar, H. R. Kaback and H. F. Lodish, “Cloning and Functional Expression in Bacteria of a Novel Glucose Transporter Present in Liver, Intestine, Kidney, and Beta-Pancreatic Islet Cells,” Cell, Vol. 55, No. 2, 1988, pp. 281-290. doi:10.1016/0092-8674(88)90051-7
- T. Kayano, et al., “Evidence for a Family of Human Glucose Transporter-Like Proteins. Sequence and Gene Localization of a Protein Expressed in Fetal Skeletal Muscle and Other Tissues,” Journal of Biological Chemistry, Vol. 263, No. 30, 1988, pp. 15245-15248.
- M. J. Birnbaum, “Identification of a Novel Gene Encoding an Insulin-Responsive Glucose Transporter Protein,” Cell, Vol. 57, No. 2, 1989, pp. 305-315. doi:10.1016/0092-8674(89)90968-9
- T. Kayano, et al., “Human Facilitative Glucose Transporters. Isolation, Functional Characterization, and Gene Localization of Cdnas Encoding an Isoform (Glut5) Expressed in Small Intestine, Kidney, Muscle, and Adipose Tissue and an Unusual Glucose Transporter PseudogeneLike Sequence (Glut6),” Journal of Biological Chemistry, Vol. 265, No. 22, 1990, pp. 13276-13282.
- C. F. Burant, J. Takeda, E. Brot-Laroche, G. I. Bell and N. O. Davidson, “Fructose Transporter in Human Spermatozoa and Small Intestine Is Glut5,” Journal of Biological Chemistry, Vol. 267, No. 21, 1992, pp. 14523- 14526.
- M. E. Phelps, “Pet: The Merging of Biology and Imaging into Molecular Imaging,” Journal of Nuclear Medicine, Vol. 41, No. 4, 2000, pp. 661-681.
- T. A. Smith, “Mammalian Hexokinases and Their Abnormal Expression in Cancer,” British Journal of Biomedical Science, Vol. 57, No. 2, 2000, pp. 170-178.
- E. K. Pauwels, E. J. Sturm, E. Bombardieri, F. J. Cleton and M. P. Stokkel, “Positron-Emission Tomography with [18f] Fluorodeoxyglucose. Part 1. Biochemical Uptake Mechanism and Its Implication for Clinical Studies,” Journal of Cancer Research and Clinical Oncology, Vol. 126, No. 10, 2000, pp. 549-559. doi:10.1007/PL00008465
- M. J. Birnbaum, H. C. Haspel and O. M. Rosen, “Transformation of Rat Fibroblasts by Fsv Rapidly Increases Glucose Transporter Gene Transcription,” Science, Vol. 235, No. 4795, 1987, pp. 1495-1498. doi:10.1126/science.3029870
- J. S. Flier, M. M. Mueckler, P. Usher and H. F. Lodish, “Elevated Levels of Glucose Transport and Transporter Messenger Rna Are Induced by Ras or Src Oncogenes,” Science, Vol. 235, No. 4795, 1987, pp. 1492-1495. doi:10.1126/science.3103217
- T. Murakami, et al., “Identification of Two Enhancer Elements in the Gene Encoding the Type 1 Glucose Transporter from the Mouse Which are Responsive to Serum, Growth Factor, and Oncogenes,” Journal of Biological Chemistry, Vol. 267, No. 13, 1992, pp. 9300- 9306.
- T. C. Yen, et al., “18f-Fdg Uptake in Squamous Cell Carcinoma of the Cervix is Correlated with Glucose Transporter 1 Expression,” Journal of Nuclear Medicine, Vol. 45, No. 1, 2004, pp. 22-29.
- R. Bos, et al., “Biologic Correlates of (18) Fluorodeoxyglucose Uptake in Human Breast Cancer Measured by Positron Emission Tomography,” Journal of Clinical Oncology, Vol. 20, No. 2, 2002, pp. 379-387. doi:10.1200/JCO.20.2.379
- K. Higashi, et al., “Correlation of Glut-1 Glucose Transporter Expression With,” European Journal of Nuclear Medicine, Vol. 27, No. 12, 2000, pp. 1778-1785. doi:10.1007/s002590000367
- T. Kurokawa, et al., “Expression of Glut-1 Glucose Transfer, Cellular Proliferation Activity and Grade of Tumor Correlate with [F-18]-Fluorodeoxyglucose Uptake by Positron Emission Tomography in Epithelial Tumors of the Ovary,” International Journal of Cancer, Vol. 109, No. 6, 2004, pp. 926-932. doi:10.1002/ijc.20057
- R. P. Beaney, “Positron Emission Tomography in the Study of Human Tumors,” Seminars in Nuclear Medicine, Vol. 14, No. 4, 1984, pp. 324-341. doi:10.1016/S0001-2998(84)80006-9
- G. Di Chiro, et al., “Glucose Utilization of Cerebral Gliomas Measured by [18f] Fluorodeoxyglucose and Positron Emission Tomography,” Neurology, Vol. 32, No. 12, 1982, pp. 1323-1329.
- R. E. Airley and A. Mobasheri, “Hypoxic Regulation of Glucose Transport, Anaerobic Metabolism and Angiogenesis in Cancer: Novel Pathways and Targets for Anticancer Therapeutics,” Chemotherapy, Vol. 53, No. 4, 2007, pp. 233-256. doi:10.1159/000104457
- C. Rudlowski, et al., “Glut1 Messenger Rna and Protein Induction Relates to the Malignant Transformation of Cervical Cancer,” American Journal of Clinical Pathology, Vol. 120, No. 5, 2003, pp. 691-698. doi:10.1309/4KYNQM5862JW2GD7
- M. Younes, R. W. Brown, D. R. Mody, L. Fernandez and R. Laucirica, “Glut1 Expression in Human Breast Carcinoma: Correlation with Known Prognostic Markers,” Anticancer Research, Vol. 15, No. 6B, 1995, pp. 2895- 2898.
- G. Cantuaria, et al., “Glut-1 Expression in Ovarian Carcinoma: Association with Survival and Response to Chemotherapy,” Cancer, Vol. 92, No. 5, 2001, pp. 1144-1150. doi:10.1002/1097-0142(20010901)92:5<1144::AID-CNCR1432>3.0.CO;2-T
- S. C. Baer, L. Casaubon and M. Younes, “Expression of the Human Erythrocyte Glucose Transporter Glut1 in Cutaneous Neoplasia,” Journal of the American Academy of Dermatology, Vol. 37, No. 4, 1997, pp. 575-577. doi:10.1016/S0190-9622(97)70174-9
- S. S. Kang, et al., “Clinical Significance of Glucose Transporter 1 (Glut1) Expression in Human Breast Carcinoma,” Japanese Journal of Cancer Research, Vol. 93, No. 10, 2002, pp. 1123-1128.
- M. Younes, L. V. Lechago, J. R. Somoano, M. Mosharaf, and J. Lechago, “Immunohistochemical Detection of Glut3 in Human Tumors and Normal Tissues,” Anticancer Research, Vol. 17, No. 4A, 1997, pp. 2747-2750.
- S. P. Zamora-Leon, et al., “Expression of the Fructose Transporter Glut5 in Human Breast Cancer,” Proceedings of the National Academy of Sciences of the USA, Vol. 93, No. 5, 1996, pp. 1847-1852. doi:10.1073/pnas.93.5.1847
- C. Binder, L. Binder, D. Marx, A. Schauer, and W. Hiddemann, “Deregulated Simultaneous Expression of Multiple Glucose Transporter Isoforms in Malignant Cells and Tissues,” Anticancer Research, Vol. 17, No. 6D, 1997, pp. 4299-4304.
- R. S. Brown and R. L. Wahl, “Overexpression of Glut-1 Glucose Transporter in Human Breast Cancer. An Immunohistochemical Study,” Cancer, Vol. 72, No. 10, 1993, pp. 2979-2985. doi:10.1002/1097-0142(19931115)72:10<2979::AID-CNCR2820721020>3.0.CO;2-X
- A. Godoy, et al., “Differential Subcellular Distribution of Glucose Transporters Glut1-6 and Glut9 in Human Cancer: Ultrastructural Localization of Glut1 and Glut5 in Breast Tumor Tissues,” Journal of Cellular Physiology, Vol. 207, No. 3, 2006, pp. 614-627. doi:10.1002/jcp.20606
- P. L. Alo, et al., “Immunohistochemical Expression of Human Erythrocyte Glucose Transporter and Fatty Acid Synthase in Infiltrating Breast Carcinomas and Adjacent Typical/Atypical Hyperplastic or Normal Breast Tissue,” American Journal of Clinical Pathology, Vol. 116, No. 1, 2001, pp. 129-134. doi:10.1309/5Y2L-CDCK-YB55-KDK6
- M. Grover-McKay, S. A. Walsh, E. A. Seftor, P. A. Thomas and M. J. Hendrix, “Role for Glucose Transporter 1 Protein in Human Breast Cancer,” Pathology & Oncology Research, Vol. 4, No. 2, 1998, pp. 115-120. doi:10.1007/BF02904704
- A. M. Meneses, et al., “Regulation of Glut3 and Glucose Uptake by the Camp Signalling Pathway in the Breast Cancer Cell Line Zr-75,” Journal of Cellular Physiology, Vol. 214, No. 1, 2008, pp. 110-116. doi:10.1002/jcp.21166
- S. Rogers, S. E. Docherty, J. L. Slavin, M. A. Henderson and J. D. Best, “Differential Expression of Glut12 in Breast Cancer and Normal Breast Tissue,” Cancer Letters, Vol. 193, No. 2, 2003, pp. 225-233. doi:10.1016/S0304-3835(03)00010-7
- S. Rogers, et al., “Identification of a Novel Glucose Transporter-Like Protein-Glut-12,” American Journal of Physiology-Endocrinology and Metabolism, Vol. 282, No. 3, 2002, pp. E733-E738.
- G. L. Semenza, “Targeting Hif-1 for Cancer Therapy,” Nature Reviews Cancer, Vol. 3, No. 10, 2003, pp. 721-732. doi:10.1038/nrc1187
- R. A. Gatenby, et al., “Cellular Adaptations to Hypoxia and Acidosis During Somatic Evolution of Breast Cancer,” British Journal of Cancer, Vol. 97, No. 5, 2007, pp. 646-653. doi:10.1038/sj.bjc.6603922
- J. Li, et al., “Knockdown of Hypoxia-Inducible Factor-1alpha in Breast Carcinoma Mcf-7 Cells Results in Reduced Tumor Growth and Increased Sensitivity to Methotrexate,” Biochemical and Biophysical Research Communications, Vol. 342, No. 4, 2006, pp. 1341-1351. doi:10.1016/j.bbrc.2006.02.094
- P. Burgman, J. A. Odonoghue, J. L. Humm and C. C. Ling, “Hypoxia-Induced Increase in Fdg Uptake in Mcf7 Cells,” Journal of Nuclear Medicine, Vol. 42, No. 1, 2001, pp. 170-175.
- S. L. Pankratz, E. Y. Tan, Y. Fine, A. M. Mercurio and L. M. Shaw, “Insulin Receptor Substrate-2 Regulates Aerobic Glycolysis in Mouse Mammary Tumor Cells Via Glucose Transporter 1,” Journal of Biological Chemistry, Vol. 284, No. 4, 2009, pp. 2031-2037. doi:10.1074/jbc.M804776200
- A. Barthel, et al., “Regulation of Glut1 Gene Transcription by the Serine/Threonine Kinase Akt1,” Journal of Biological Chemistry, Vol. 274, No. 29, 1999, pp. 20281-20286. doi:10.1074/jbc.274.29.20281
- R. C. Osthus, et al., “Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by C-Myc,” Journal of Biological Chemistry, Vol. 275, No. 29, 2000, pp. 21797-21800. doi:10.1074/jbc.C000023200
- J. C. Rathmell, et al., “Akt-Directed Glucose Metabolism Can Prevent Bax Conformation Change and Promote Growth Factor-Independent Survival,” Molecular and Cellular Biology, Vol. 23, No. 20, 2003, pp. 7315-7328. doi:10.1128/MCB.23.20.7315-7328.2003
- D. Rivenzon-Segal, S. Boldin-Adamsky, D. Seger, R. Seger and H. Degani, “Glycolysis and Glucose Transporter 1 as Markers of Response to Hormonal Therapy in Breast Cancer,” International Journal of Cancer, Vol. 107, No. 2, 2003, pp. 177-182. doi:10.1002/ijc.11387
- M. Neeman and H. Degani, “Metabolic Studies of Estrogen and Tamoxifen-Treated Human Breast Cancer Cells by Nuclear Magnetic Resonance Spectroscopy,” Cancer Research, Vol. 49, No. 3, 1989, pp. 589-594.
- P. Laudanski, et al., “Expression of Glucose Transporter Glut-1 and Estrogen Receptors Er-Alpha and Er-Beta in Human Breast Cancer,” Neoplasma, Vol. 51, No. 3, 2004, pp. 164-168.
- G. Wilding, M. E. Lippman and E. P. Gelmann, “Effects of Steroid Hormones and Peptide Growth Factors on Protooncogene C-Fos Expression in Human Breast Cancer Cells,” Cancer Research, Vol. 48, No. 4, 1988, pp. 802-805.
- M. Guppy, P. Leedman, X. Zu and V. Russell, “Contribution by Different Fuels and Metabolic Pathways to the Total Atp Turnover of Proliferating Mcf-7 Breast Cancer Cells,” Biochemical Journal, Vol. 364, Pt. 1, 2002, pp. 309-315.
- R. Moreno-Sanchez, S. Rodriguez-Enriquez, A. MarinHernandez and E. Saavedra, “Energy Metabolism in Tumor Cells,” Febs Journal, Vol. 274, No. 6, 2007, pp. 1393-1418. doi:10.1111/j.1742-4658.2007.05686.x
- S. Rodriguez-Enriquez, et al., “Control of Cellular Proliferation by Modulation of Oxidative Phosphorylation in Human and Rodent Fast-Growing Tumor Cells,” Toxicology and Applied Pharmacology, Vol. 215, No. 2, 2006, pp. 208-217. doi:10.1016/j.taap.2006.02.005
- R. Diaz-Ruiz, S. Uribe-Carvajal, A. Devin and M. Rigoulet, “Tumor Cell Energy Metabolism and Its Common Features with Yeast Metabolism,” Biochimica et Biophysica Acta, Vol. 1796, No. 2, 2009, pp. 252-265.
- H. Pelicano, D. S. Martin, R. H. Xu and P. Huang, “Glycolysis Inhibition for Anticancer Treatment,” Oncogene, Vol. 25, No. 34, 2006, pp. 4633-4646. doi:10.1038/sj.onc.1209597
- D. Singh, et al., “Optimizing Cancer Radiotherapy with 2-Deoxy-D-Glucose Dose Escalation Studies in Patients with Glioblastoma Multiforme,” Strahlentherapie und Onkologie, Vol. 181, No. 8, 2005, pp. 507-514. doi:10.1007/s00066-005-1320-z
- C. W. Christopher, W. W. Colby and D. Ullrey, “Derepression and Carrier Turnover: Evidence for Two Distinct Mechanisms of Hexose Transport Regulation in Animal Cells,” Journal of Cellular Physiology, Vol. 89, No. 4, 1976, pp. 683-692. doi:10.1002/jcp.1040890427
- R. Keller, “Suppression of Natural Antitumour Defence Mechanisms by Phorbol Esters,” Nature, Vol. 282, No. 5740, 1979, pp. 729-731. doi:10.1038/282729a0
- E. M. Bessell, V. D. Courtenay, A. B. Foster, M. Jones, and J. H. Westwood, “Some in Vivo and in Vitro Antitumour Effects of the Deoxyfluoro-D-Glucopyranoses,” European Journal of Cancer, Vol. 9, No. 7, 1973, pp. 463-470. doi:10.1016/0014-2964(73)90128-X
- J. H. Risse, et al., “18f-Fdg-Pet and Histopathology in 131i-Lipiodol Treatment for Primary Liver Cancer,” Cancer Biotherapy & Radiopharmaceuticals, Vol. 24, No. 4, 2009, pp. 445-452. doi:10.1089/cbr.2008.0560
- M. Kurtoglu, J. C. Maher and T. J. Lampidis, “Differential Toxic Mechanisms of 2-Deoxy-D-Glucose Versus 2-Fluorodeoxy-D-Glucose in Hypoxic and Normoxic Tumor Cells,” Antioxidants and Redox Signaling, Vol. 9, No. 9, 2007, pp. 1383-1390. doi:10.1089/ars.2007.1714
- I. F. Tannock, P. Guttman and A. M. Rauth, “Failure of 2-Deoxy-D-Glucose and 5-Thio-D-Glucose to Kill Hypoxic Cells of Two Murine Tumors,” Cancer Research, Vol. 43, No. 3, 1983, pp. 980-983.
- J. H. Kim, S. H. Kim, E. W. Hahn and C. W. Song, “5-Thio-D-Glucose Selectively Potentiates Hyperthermic Killing of Hypoxic Tumor Cells,” Science, Vol. 200, No. 4338, 1978, pp. 206-207. doi:10.1126/science.635582
- E. M. Bessell, A. B. Foster and J. H. Westwood, “The Use of Deoxyfluoro-D-Glucopyranoses and Related Compounds in a Study of Yeast Hexokinase Specificity,” Biochemical Journal, Vol. 128, No. 2, 1972, pp. 199-204.
- T. J. Lampidis, et al., “Efficacy of 2-Halogen Substituted D-Glucose Analogs in Blocking Glycolysis and Killing ‘Hypoxic Tumor Cells’,” Cancer Chemotherapy and Pharmacology, Vol. 58, No. 6, 2006, pp. 725-734. doi:10.1007/s00280-006-0207-8
- E. L. Coe and R. C. Strunk, “The Effect of Oxamate on Glycolysis in Intact Ascites Tumor Cells. 1. Kinetic Evidence for a Dual Glycolytic System,” Biochimica et Biophysica Acta, Vol. 208, No. 2, 1970, pp. 189-202.
- S. Bonnet, et al., “A Mitochondria-K+ Channel Axis is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth,” Cancer Cell, Vol. 11, No. 1, 2007, pp. 37-51. doi:10.1016/j.ccr.2006.10.020
- J. Y. Wong, G. S. Huggins, M. Debidda, N. C. Munshi, and I. De Vivo, “Dichloroacetate Induces Apoptosis in Endometrial Cancer Cells,” Gynecologic Oncology, Vol. 109, No. 3, 2008, pp. 394-402. doi:10.1016/j.ygyno.2008.01.038
- T. J. Lampidis, S. D. Bernal, I. C. Summerhayes and L. B. Chen, “Selective Toxicity of Rhodamine 123 in Carcinoma Cells in Vitro,” Cancer Research, Vol. 43, No. 2, 1983, pp. 716-720.
- S. D. Bernal, T. J. Lampidis, R. M. McIsaac and L. B. Chen, “Anticarcinoma Activity in Vivo of Rhodamine 123, a Mitochondrial-Specific Dye,” Science, Vol. 222, No. 4620, 1983, pp. 169-172. doi:10.1126/science.6623064
- R. M. Sutherland, “Cell and Environment Interactions in Tumor Microregions: The Multicell Spheroid Model,” Science, Vol. 240, No. 4849, 1988, pp. 177-184. doi:10.1126/science.2451290
- L. Galluzzi, N. Larochette, N. Zamzami and G. Kroemer, “Mitochondria as Therapeutic Targets for Cancer Chemotherapy,” Oncogene, Vol. 25, No. 34, 2006, pp. 4812-4830. doi:10.1038/sj.onc.1209598
- C. E. Griguer, et al., “Pharmacologic Manipulations of Mitochondrial Membrane Potential (Deltapsim) Selectively in Glioma Cells,” Journal of Neuro-Oncology, Vol. 81, No. 1, 2007, pp. 9-20. doi:10.1007/s11060-006-9201-6
- G. J. Arismendi-Morillo and A. V. Castellano-Ramirez, “Ultrastructural Mitochondrial Pathology in Human Astrocytic Tumors: Potentials Implications Pro-therapeutics Strategies,” Journal of Electron Microscopy (Tokyo), Vol. 57, No. 1, 2008, pp. 33-39. doi:10.1093/jmicro/dfm038
- Z. Chen, W. Lu, C. Garcia-Prieto and P. Huang, “The Warburg Effect and Its Cancer Therapeutic Implications,” Journal of Bioenergetics and Biomembranes, Vol. 39, No. 3, 2007, pp. 267-274. doi:10.1007/s10863-007-9086-x
- C. W. Yeh, W. J. Chen, C. T. Chiang, S. Y. Lin-Shiau and J. K. Lin, “Suppression of Fatty Acid Synthase in Mcf-7 Breast Cancer Cells by Tea and Tea Polyphenols: A Possible Mechanism for Their Hypolipidemic Effects,” The Pharmacogenomics Journal, Vol. 3, No. 5, 2003, pp. 267-276.
- C. T. Chiang, T. D. Way, S. J. Tsai and J. K. Lin, “Diosgenin, a Naturally Occurring Steroid, Suppresses Fatty Acid Synthase Expression in Her2-Overexpressing Breast Cancer Cells through Modulating Akt, Mtor and Jnk Phosphorylation,” Febs Letters, Vol. 581, No. 30, 2007, pp. 5735-5742. doi:10.1016/j.febslet.2007.11.021
- J. S. Lee, M. S. Lee, W. K. Oh and J. Y. Sul, “Fatty Acid Synthase Inhibition by Amentoflavone Induces Apoptosis and Antiproliferation in Human Breast Cancer Cells,” Biological & Pharmaceutical Bulletin, Vol. 32, No. 8, 2009, pp. 1427-1432. doi:10.1248/bpb.32.1427
- C. Yan, et al., “A New Targeting Approach for Breast Cancer Gene Therapy Using the Human Fatty Acid Synthase Promoter,” Acta Oncologica, Vol. 46, No. 6, 2007, pp. 773-781. doi:10.1080/02841860601016070
- R. Lupu and J. A. Menendez, “Targeting Fatty Acid Synthase in Breast and Endometrial Cancer: An Alternative to Selective Estrogen Receptor Modulators?” Endocrinology, Vol. 147, No. 9, 2006, pp. 4056-4066. doi:10.1210/en.2006-0486
- A. Vazquez-Martin, S. Ropero, J. Brunet, R. Colomer and J. A. Menendez, “Inhibition of Fatty Acid Synthase (Fasn) Synergistically Enhances the Efficacy of 5-Fluorouracil in Breast Carcinoma Cells,” Oncology Reports, Vol. 18, No. 4, 2007, pp. 973-980.
- H. Liu, Y. Liu and J. T. Zhang, “A New Mechanism of Drug Resistance in Breast Cancer Cells: Fatty Acid Synthase Overexpression-Mediated Palmitate Overproduction,” Molecular Cancer Therapeutics, Vol. 7, No. 2, 2008, pp. 263-270. doi:10.1158/1535-7163.MCT-07-0445
- S. Bandyopadhyay, et al., “Mechanism of Apoptosis Induced by the Inhibition of Fatty Acid Synthase in Breast Cancer Cells,” Cancer Research, Vol. 66, No. 11, 2006, pp. 5934-5940. doi:10.1158/0008-5472.CAN-05-3197
- D. Manka, Z. Spicer and D. E. Millhorn, “Bcl-2/Adenovirus E1b 19 Kda Interacting Protein-3 Knockdown Enables Growth of Breast Cancer Metastases in the Lung, Liver, and Bone,” Cancer Research, Vol. 65, No. 24, 2005, pp. 11689-11693. doi:10.1158/0008-5472.CAN-05-3091
- X. Liu, Y. Shi, V. L. Giranda and Y. Luo, “Inhibition of the Phosphatidylinositol 3-Kinase/Akt Pathway Sensitizes Mda-Mb468 Human Breast Cancer Cells to CeruleninInduced Apoptosis,” Molecular Cancer Therapeutics, Vol. 5, No. 3, 2006, pp. 494-501. doi:10.1158/1535-7163.MCT-05-0049
- V. Chajes, M. Cambot, K. Moreau, G. M. Lenoir and V. Joulin, “Acetyl-Coa Carboxylase Alpha is Essential to Breast Cancer Cell Survival,” Cancer Research, Vol. 66, No. 10, 2006, pp. 5287-5294. doi:10.1158/0008-5472.CAN-05-1489
- J. N. Thupari, M. L. Pinn and F. P. Kuhajda, “Fatty Acid Synthase Inhibition in Human Breast Cancer Cells Leads to Malonyl-Coa-Induced Inhibition of Fatty Acid Oxidation and Cytotoxicity,” Biochemical and Biophysical Research Communications, Vol. 285, No. 2, 2001, pp. 217-223. doi:10.1006/bbrc.2001.5146
- D. Rivenzon-Segal, E. Rushkin, S. Polak-Charcon and H. Degani, “Glucose Transporters and Transport Kinetics in Retinoic Acid-Differentiated T47d Human Breast Cancer Cells,” American Journal of Physiology-Endocrinology and Metabolism, Vol. 279, No. 3, 2000, pp. E508-E519.
- S. Rastogi, S. Banerjee, S. Chellappan and G. R. Simon, “Glut-1 Antibodies Induce Growth Arrest and Apoptosis in Human Cancer Cell Lines,” Cancer Letters, Vol. 257, No. 2, 2007, pp. 244-251. doi:10.1016/j.canlet.2007.07.021
- X. Cao, et al., “Glucose Uptake Inhibitor Sensitizes Cancer Cells to Daunorubicin and Overcomes Drug Resistance in Hypoxia,” Cancer Chemotherapy and Pharmacology, Vol. 59, No. 4, 2007, pp. 495-505. doi:10.1007/s00280-006-0291-9
- K. K. Chan, J. Y. Chan, K. K. Chung and K. P. Fung, “Inhibition of Cell Proliferation in Human Breast Tumor Cells by Antisense Oligonucleotides against Facilitative Glucose Transporter 5,” Journal of Cellular Biochemistry, Vol. 93, No. 6, 2004, pp. 1134-1142. doi:10.1002/jcb.20270
- P. J. Goodwin, J. A. Ligibel and V. Stambolic, “Metformin in Breast Cancer: Time for Action,” Journal of Clinical Oncology, Vol. 27, No. 20, 2009, pp. 3271-3273. doi:10.1200/JCO.2009.22.1630
- M. Zakikhani, R. Dowling, I. G. Fantus, N. Sonenberg and M. Pollak, “Metformin is an Amp Kinase-Dependent Growth Inhibitor for Breast Cancer Cells,” Cancer Research, Vol. 66, No. 21, 2006, pp. 10269-10273. doi:10.1158/0008-5472.CAN-06-1500
- J. B. Kim, et al., “The Alkaloid Berberine Inhibits the Growth of Anoikis-Resistant Mcf-7 and Mda-Mb-231 Breast Cancer Cell Lines by Inducing Cell Cycle Arrest,” Phytomedicine, Vol. 17, No. 6, 2009, pp. 436-440.
- J. Yin, H. Xing and J. Ye, “Efficacy of Berberine in Patients with Type 2 Diabetes Mellitus,” Metabolism, Vol. 57, No. 5, 2008, pp. 712-717. doi:10.1016/j.metabol.2008.01.013
- L. M. Harhaji Trajkovic, et al., “Anticancer Properties of Ganoderma Lucidum Methanol Extracts in Vitro and in Vivo,” Nutrition and Cancer, Vol. 61, No. 5, 2009, pp. 696-707. doi:10.1080/01635580902898743
- Q. Luo, et al., “Lycium Barbarum Polysaccharides Induce Apoptosis in Human Prostate Cancer Cells and Inhibits Prostate Cancer Growth in a Xenograft Mouse Model of Human Prostate Cancer,” Journal of Medicinal Food, Vol. 12, No. 4, 2009, pp. 695-703. doi:10.1089/jmf.2008.1232
- P. Wu, J. J. Dugoua, O. Eyawo and E. J. Mills, “Traditional Chinese Medicines in the Treatment of Hepatocellular Cancers: A Systematic Review and Meta-Analysis,” Journal of Experimental & Clinical Cancer Research, Vol. 28, 2009, p. 112. doi:10.1186/1756-9966-28-112
- W. Ni, et al., “Antitumor Activities and Immunomodulatory Effects of Ginseng Neutral Polysaccharides in Combination with 5-Fluorouracil,” Journal of Medicinal Food, Vol. 13, No. 2, 2010, pp. 270-277. doi:10.1089/jmf.2009.1119
- F. Zou, X. Q. Mao, N. Wang, J. Liu and J. P. Ou-Yang, “Astragalus Polysaccharides Alleviates Glucose Toxicity and Restores Glucose Homeostasis in Diabetic States Via Activation of Ampk,” Acta Pharmacologica Sinica, Vol. 30, No. 12, 2009, pp. 1607-1615. doi:10.1038/aps.2009.168
- S. W. Seto, et al., “Novel Hypoglycemic Effects of Ganoderma Lucidum Water-Extract in Obese/Diabetic (+Db/ +Db) Mice,” Phytomedicine, Vol. 16, No. 5, 2009, pp. 426-436. doi:10.1016/j.phymed.2008.10.004
- Q. Luo, Y. Cai, J. Yan, M. Sun and H. Corke, “Hypoglycemic and Hypolipidemic Effects and Antioxidant Activity of Fruit Extracts from Lycium Barbarum,” Life Sciences, Vol. 76, No. 2, 2004, pp. 137-149. doi:10.1016/j.lfs.2004.04.056
- T. B. Ng and H. W. Yeung, “Hypoglycemic Constituents of Panax Ginseng,” General Pharmacology, Vol. 16, No. 6, 1985, pp. 549-552. doi:10.1016/0306-3623(85)90140-5
- J. C. Chen, et al., “Gypenosides Induced G0/G1 Arrest Via Chk2 and Apoptosis through Endoplasmic Reticulum Stress and Mitochondria-Dependent Pathways in Human Tongue Cancer Scc-4 Cells,” Oral Oncology, Vol. 45, No. 3, 2009, pp. 273-283. doi:10.1016/j.oraloncology.2008.05.012
- S. Megalli, N. M. Davies and B. D. Roufogalis, “AntiHyperlipidemic and Hypoglycemic Effects of Gynostemma Pentaphyllum in the Zucker Fatty Rat,” Journal of Pharmacy & Pharmaceutical Sciences, Vol. 9, No. 3, 2006, pp. 281-291.
- D. Lamoral-Theys, et al., “Natural Polyphenols That Display Anticancer Properties through Inhibition of Kinase Activity,” Current Medicinal Chemistry, Vol. 17, No. 9, 812-825. doi:10.2174/092986710790712183
- J. Ravindran, S. Prasad and B. B. Aggarwal, “Curcumin and Cancer Cells: How Many Ways Can Curry Kill Tumor Cells Selectively?” The AAPS Journal, Vol. 11, No. 3, 2009, pp. 495-510. doi:10.1208/s12248-009-9128-x
- T. Osawa and Y. Kato, “Protective Role of Antioxidative Food Factors in Oxidative Stress Caused by Hyperglycemia,” Annals of the New York Academy of Sciences, Vol. 1043, 2005, pp. 440-451. doi:10.1196/annals.1333.050