Open Journal of Nephrology
Vol. 2 No. 4 (2012) , Article ID: 26354 , 13 pages DOI:10.4236/ojneph.2012.24020
A Prospective Study on Clinical Profile of Autosomal Dominant Polycystic Kidney Disease (ADPKD) in Jammu for a Period of 1 Year
1Department of Medicine, Government Medical College, Chandigarh, India
2Department of Medicine, SKIMS Medical College, Srinagar, India
3SKIMS Medical College, Srinagar, India
4Government Medical College, Jammu, India
5Department of Medicine, Government Medical College, Srinagar, India
6Sher-i-Kashmir Institute of Medical Sciences, Kashmir, India
Email: drsajadk@rediffmail.com
Received September 25, 2012; revised October 28, 2012; accepted November 13, 2012
Keywords: Autosomal Dominant Polycystic Kidney Disease; Ronic Kidney Disease; End-Stage Renal Disease
ABSTRACT
The Present Study was conducted in department of Medicine, Govt. Medical College, jammu, Where a total of 41 patients—29 males and 12 females—fulfilled the inclusion criteria of ADPKD, were gathered during the period of 1 year starting from Nov. 2011 to Oct. 2012. All the patients were subjected to a detailed history, clinical examination and laboratory investigations. X-ray chest (PA view), ECG and ultrasound of abdomen for kidneys, liver and spleen were done. Intravenous pyelogram and CT scan of abdomen was done when a definitive diagnosis of (ADPKD) could not be made on abdominal ultrasound. Echocardiography was done to evaluate cardiac murmurs and associated mitral valve prolapse, based on standard criteria. Male to female patients with ADPKD was 2.42:1. Maximum 17 (41.5%) patients of both gender were seen in 30 - 40 years age group, Family history of ADPKD was present in 18 (43.9%) patients; Hypertension, alone or in combination with renal failure, was present in 65.8% patients; Hypertension alone was present in 19 (46.3%) patients; 8 (19.5%) patients with hypertension had renal failure; Low back pain was present in 24 (58.5%) and abdominal pain in 22 (53.7%) patients; 15 (36.6%) patients presented with at least one episode of gross haematuria; Headache was experienced by 18 (43.9%) patients. On clinical examination, 24 (58.5%) were found to have palpable kidney and 10 (24.4%) had palpable liver. Spleen was palpable in 1 (2.4%) patient, Murmur of mitral valve prolapse was found in 2 (4.9%) Patients; 3 (7.3%) patients having left ventricular hypertrophy; mean Hb was 11.2 g/dL. The liver cysts were found in 24.4% of the patients; Out of 10 (24.4%) patients with hepatic cyst involvement, 1 patient each was found to have evidence of portal hypertension and evidence of hepatic cyst infection. In the present study, hypertension was most common presentation of this disease. So, control of hypertension is very important to prevent progression of this disease. Patients who are detected to have ADPKD should be regularly followed-up to prevent further progression by timely intervention. Also, family members of patients should be screened for disease and initiate treatment as early as possible.
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a monogenic, multi-systemic disorder characterized by the development of renal cysts and various extra-renal manifestations. Although the disease begins in-utero, it may take several decades for the signs to surface and is a leading cause for end stage renal failure. It affects all ethnic groups worldwide with an estimated prevalence of approximately 1 in 800 live births [1]. Being an autosomal dominant disorder, the offspring runs a 50% risk of inheriting the mutated gene. However, only about one half of these cases are diagnosed during their lifetime, as the disease usually runs an asymptomatic course [2].
At least two distinct gene defects have been identified in the pathogenesis of ADPKD—PKD1 and PKD2 [3]. PKD1 and PKD2 are expressed in most organs and tissues of human body. The proteins that are encoded by PKD1 and PKD2 seem to function together to regulate morphologic configuration of cells [4]. Type 1 ADPKD caused by mutation in the PKD1 gene accounts for nearly 85% of the cases with its causative gene localized to the short arm of chromosome 16 in the PKD1 locus [5]. The PKD1 gene codes for polycystin-1 (PC1), which plays a vital role in cell-cell and cell-matrix interactions [6].
The function of polycystins have been scrutinized to the greater extent in epithelial tissues of the kidneys, liver and vascular smooth muscle. Mutation in either polycystins leads to phenotype of ADPKD. The hallmarks of this inherited condition are massively enlarged kidneys caused by sustained expansion of innumerable fluid filled cysts ranging in equivalent size from a pea to a grapefruit. Cysts derive from microscopic tubule precursors. They are seen in liver (~80%), and with lesser frequency in pancreas (10%) and arachnoid membranes (~8%). Aneurysms occur approximately in 5% of patients with ADPKD and higher frequency in those with a family history of aneurysm (~20%).
Type 2 ADPKD caused by a mutation in the PKD2 gene accounts for about 10% - 15% of the cases, with the defective gene located on chromosome 4 [7] The rest (around 5%) have a defect unrelated to either locations and may be accounted by spontaneous mutations. The PKD2 gene codes for polycystin-2 (PC2), which is involved in cell calcium signaling [4]. The occurrence of cysts and end-stage renal disease are delayed in PKD2 disease (mean age 74.0 as against 54.3 years in PKD1) [8].
The disease is characterized by a variable renal disease progression and manifestation. The variable phenotypic expression of the disease within families (intra-familial variability) is often linked to the occurrence of a somatic mutation in the intact allele within epithelial tubular cells (2 nd hit hypothesis) [4,9]. However, the inter-familial variability can be accounted by genetic heterogeneity (PKD1 versus PKD2). Genetic modifiers (any gene unlinked to the PKD1 or PKD2 loci whose genotype correlates with the ADPKD phenotype) also play a role as far as clinical manifestation and disease progression in PKD1 is considered [10].
In ADPKD each epithelial cells within a tubule harbours a germ line mutation, yet only a tiny fraction of the tubules develop renal cysts. It is currently held that the cells are protected by the allele inherited from the parent without ADPKD. When this allele is inactivated by a somatic event (mutation or otherwise) within a solitary renal tubule cell, the cell divides repeatedly until a cyst develops, with an aberrant growth program causing endless expansion. The severity of ADPKD is thought to be a direct consequence of the number of times and the frequency with which this cytogenic process occurs within the kidneys over the life of the patient. Hyperplasia occurs in renal cyst epithelial cells. The hyperplastic cells cause an out pocketing of the tubule wall, with formation of saccular cyst that fills with fluid derived from glomerular filtrate that enters from the afferent tubule segment. Progressive expansion eventually causes most of the emerging cysts to separate from the parent tubule, leaving an isolated sac that fills with fluid by transepithelial secretion. This isolated cyst expands relentlessly as a result of continued proliferation of the mural epithelium together with transepithelial secretion of NaCl and water into the lumen [11]. The expanding fluid filled tumor masses elicit secondary and tertiary changes within the renal interstitium evinced by thickening and lamination of tubule basement membranes, infiltration of macrophages and neovascularisation [12]. Fibrosis within the interstitium begins early in the course of disease. Thus, cysts function as autonomous structures and are responsible for progressive kidney enlargement in ADPKD.
Hypertension is a relatively early finding in ADPKD with an average onset at 30 years of age [13]. Proteinuria observed in those with large (mean combined renal volume 1190 ml) rather than small volume (578 ml) renal volumes, is also associated with a greater likelihood of a subsequent loss of renal function. The renal cysts develop in tiny fraction of nephrons (estimated to be less than 1%)
1.1. Different Presentations of ADPKD Patients
1.1.1. Hemorrhage and Hematuria
Polycystic kidneys are unusually susceptible to traumatic injury [14-17]. Pain: Pain with or without hemorrhage is the most frequent symptom (~60%) reported by adult patients with ADPKD and frequently begins in individuals with normal renal function [18].
1.1.2. Cosmetic Deformation of Abdomen
The kidneys in some patients enlarge to such an extent that belt and dress sizes must be increased substantially. Enlarged kidneys increase the risk that seat belts may cause injury [19].
1.1.3. Hypertension
Hypertension is an early and frequent finding of ADPKD, occurring in approximately 60% of patients before renal function has become impaired [20-23]. Hypertension is associated with a faster progression to end-stage renal disease (ESRD) and is most importantly potentially treatable variable in ADPKD [14].
1.1.4. Urinary Tract Infections
Urinary tract infection is a common presenting symptom in ADPKD patients, with some having recurrent episodes and have been found to increase the rate of deterioration of renal function [24-27].
1.2. Extrarenal Manifestations of ADPKD
1.2.1. Hepatic Cysts
Hepatic cysts are most common extrarenal manifestation of ADPKD, with a highly variable prevalence ranging from 10 to 88% [28-30]. Both in males and females patients liver function is well preserved and liver enzymes and bilirubin usually remain normal [31]. The potential for complications of hepatic cystic involvement in ADPKD is significant [32].
1.2.2. Intracranial Aneurysm
Rupture of intracranial aneurysm (ICA) is one of the dreadest complication of ADPKD. The reported frequency ranges from 0% to 41% [33,34].
1.2.3. Cardiac Disease
Since initial report of [35], cardiovascular abnormalities, including mitral and aortic valvular prolapse and regurgitation and annuloaortic ectasia have been considered important extrarenal manifestation of ADPKD.
1.2.4. Diverticular Disease
Both an increased prevalence and an increased risk of complications from complicated diverticular disease are features of ADPKD. [36] It has been found that the prevalence of diverticulosis to be 83% in ADPKD patients on hemodialysis versus 32% of controls with chronic renal failure and 38% of age and gender-matched controls.
1.3. Diagnosis and Screening
The diagnosis of ADPKD relies primarily on imaging modalities that include ultrasonography, Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) [37-40]. However, genetic testing is made use of in certain settings when a definitive diagnosis is required. The age of the patient, presence or absence of a family history of the disease, family history of other genetic disorders, clinical manifestations of the disease, as well as the number and types of renal cysts are significant details that help in the diagnosis of ADPKD [17].
A Study was conducted which provide evidence for increased cardiac pre-load, as assessed by plasma atrial natriuretic factor and cardiac index [41].
Bourquia et al. (1989) [42] identified 21 cases of autosomal dominant polycystic kidney disease (ADPKD) with end-stage chronic renal failure (CRF) requiring dialysis and transplantation over a 5-year period.
Al-Muhanna et al. (1995) [43] analysed 30 cases of autosomal dominant polycystic kidney disease (ADPKD), at King Fahd Hospital, Al-Khobar over a period of 8 years, with respect to clinical features, laboratory investtigations, radiological findings, complications and outcome.
Bajwa et al. (2001) [44] outlined a detailed stepwise approach for acute and chronic pain strategies for the patient with ADPKD.
Graf et al. (2002) [45] studied ADPKD patients with a positive family history for a cerebral event, including cerebral stroke and intracranial bleeding or known intracranial saccular aneurysms (ICA) to facilitate the defiition of subgroups who were at risk for ICA and to determine the prevalence of ICA in these subgroups.
Schrier et al. (2002) [46] investigated the cardiac and renal effects of rigorous (<120/800 mmHg) versus standard (135 - 140/80 - 90 mmHg) blood pressure control on left ventricular mass index (LVMI) and kidney function in 75 hypertensive ADPKD patients with left ventricular hypertrophy.
Bajwa et al. (2004) [47] analyzed 171 completed questionnaires distributed to polycystic kidney disease (PKD) patients whose renal function ranged from normal to endstage renal disease (ESRD).
Wolyniec et al. (2008) [48] evaluated diagnostic procedures of autosomal dominant polycystic kidney disease (ADPKD).
Amico et al. (2009) [49] presented a case study of a 47- year old woman with family history of autosomal dominant polycystic kidney disease (ADPKD), who underwent living-donor kidney transplantation in 2000 Bae et al. (2009) [50] evaluated interand intra reader reliability and the effect of gadolinium enhancement on kidney volume measurements obtained from preand postgadolinium T1 MR images in patients with autosomal dominant polycystic kidney disease (ADPKD).
Barua et al. (2009) [51] explored whether a family history of renal disease severity predicts the mutated gene in ADPKD.
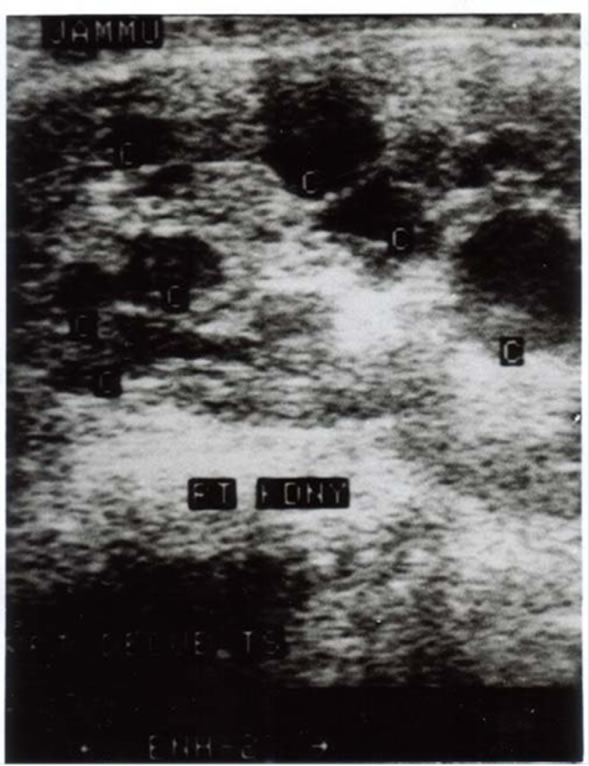
Image 1. Right kidney showing multiple cyst with lost CMD.
Ecder and Schrier (2009) [52] reported that cardiovascular problems are a major cause of morbidity and mortality in patients with autosomal-dominant polycystic kidney disease (ADPKD).
Hadimeri et al. (2009) [53] studied echocardiographic findings in kidney transplant patients with autosomal dominant polycystic kidney disease (ADPKD).
Hajj et al. (2009) [54] reviewed retrospectively all the nephrectomies performed between 1982 and 2003 in patients with autosomal dominant polycystic kidney disease (ADPKD) and chronic renal failure and studied the prevalence and the characteristics of renal cell carcinoma (RCC) in patients with ADPKD.
Idrizi et al. (2009) [55] evaluated the frequency of the manifestations like renal stones, urinary tract infections (UTI) and gross hematuria (GH) in the patients with of autosomal dominant polycystic kidney disease ADPKD and their impact on renal function.
Ramunni et al. (2009) [56] verified the existence of a microvascular reactivity in th early stages of ADPKD in their study. Sallee et al. (2009) [57] conducted a retrospective study on patients who had ADPKD with likely or definite renal and/or hepatic cyst infection.
Sawicki et al. (2009) [58] assessed the clinical usefulness of duplex Doppler sonography in ADPKD patients by measuring the resistive indices (RIs) and pulsatility indices (PIs) of renal and interlobar arteries and investigating the correlation between these parameters and relevant clinical data.
Alam and Perrone (2010) [59] reported the overall outcomes of individuals with ADPKD after ESRD as compared with non-ADPKD patients.
Bae and Grantham (2010) [60] In a review article discussed the overwhelming evidence in support of the view that imaging is an invaluable tool to monitor the onset and progression of autosomal dominant polycystic kidney disease (ADPKD) and is well-suited to gauge the response of this disease to targeted therapy before renal function begins to decline.
Barua and Pei (2010) [61] reviewed the clinical utilities and limitations of the imagingand molecular-based diagnostic tests, and outlined their approach for the evaluation of individuals suspected to have autosomal-dominant polycystic kidney disease (ADPKD).
Chang et al. (2010) [62] compared intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH) in patients with autosomal-dominant polycystic kidney disease (ADPKD).
Chapman et al. (2010) [63] carried out a study on hypertension in autosomal dominant polycystic kidney disease (ADPKD).
Harris and Rossetti (2010) [64] reported that like Mendelian diseases, the presentation and progression of autosomal dominant polycystic kidney disease (ADPKD) vary widely in the population.
Meijer et al. (2010) [65] evaluated the prevalence of renal abnormalities in 103 prevalent ADPKD patients (Ravine criteria) at different ages.
Mufti and Nalagatla (2010) [66] reviewed nephrolithiasis in autosomal dominant polycystic kidney disease (ADPKD).
Pirson (2010) [67] studied extrarenal manifestations of autosomal dominant polycystic kidney disease (AKPKD).
Qian (2010) [68] reported that isolated polycystic liver disease (PCLD) is an autosomal dominant disease with genetic and clinical heterogeneity.
Rabbani et al. (2010) [69] reported a case of a renal transplant recipient who suffered an isolated Mycobacterium tuberculosis infection of a native polycystic kidney.
Zheng et al. (2010) [70] treated two uremic autosomal dominant polycystic kidney disease (ADPKD) patients who developed refractory ascites after 1 year on hemodialysis with continuous ambulatory peritoneal dialysis (CAPD).
Since no study related to ADPKD has been undertaken in Jammu region, this study was conducted to study the various manifestations related with this heritable disease.
2. Materials & Methods
The present prospective study on clinical profile of autosomal dominant polycystic kidney disease (ADPKD) was conducted in Postgraduate Department of Medicine, Government Medical College, Jammu for a period of one year w.e.f. November, 2011 to October, 2012. A total of 41 patients fulfilled criteria of ADPKD.
2.1. Methods
All patients were subjected to a detailed history, clinical examination and laboratory investigations which were recorded in the predesigned proforma for this study. The following points were considered while taking history:
• Onset of symptoms
• Underlying systemic disease
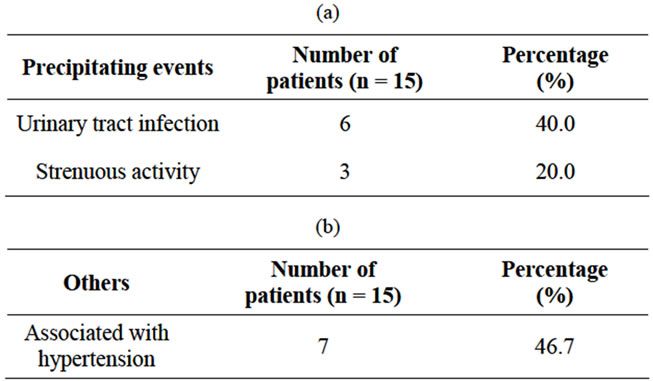
• Haematuria (Table 1, Figure 1)
• Abdominal pain (Table 2, Figure 2)
• Abdominal mass
• Headache (Table 3, Figure 3)
• Non-specific symptoms like weakness, fever, nausea/ vomiting, malaise
• Shortness of breath/dyspnoea
2.2. Clinical Examination
Clinical examination was focused on general physical examination, blood pressure, altered level of conscious ness, pallor, abdominal mass, cardiovascular system to exclude valvular lesions and left ventricular hypertrophy. Exa-

mination of all other systems, including CVS, CNS and respiratory system was done and recorded in the given proforma.
All patients were subjected to investigations which included haemolobin, TLC, DLC, PBF, ESR, blood sugar, renal function tests—serum urea, serum creatinine, serum electrolytes (Na+, K+), liver function tests—serum bilirubin, SGOT, SGPT, serum alkaline phosphatase, serum proteins, serum albumin, complete urine examination with special attention to proteinuria, sediment, specific gravity, 24-hour urinary protein, X-ray chest (PA view), ECG and ultrasound of abdomen for kidneys, liver and spleen (Table 4). Intravenous pyelogram and CT scan of abdomen was done in selected cases when a definitive diagnosis of autosomal dominant polycystic kidney disease
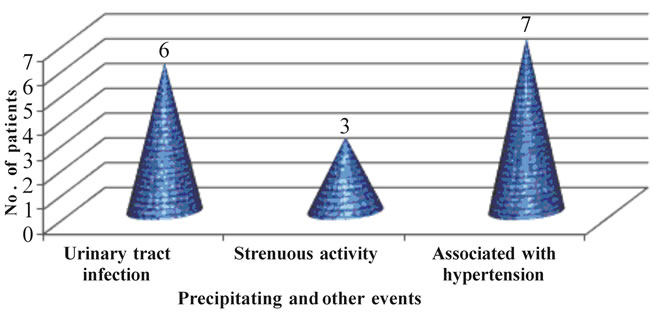
Figure 1. Profile of haematuria in ADPKD patients

Figure 2. Profile of abdominal pain frequency in ADPKD patients.
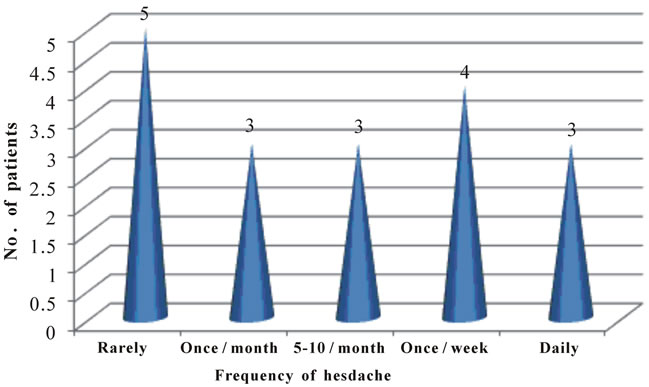
Figure 3. Profile of headache frequency in ADPKD patients.
(ADPKD) could not be made on abdominal ultrasound. The requirement for diagnosis of associated hepatic was the presence of at least one cyst in the liver. Echocardiography was done to evaluate cardiac murmurs and associated mitral valve prolapse, based on standard criteria.
• 2.3. Inclusion Criteria
• More male ADPKD patients (29; 70.73%) were observed than female ADPKD patients (12; 29.27%) during the study period.
• Male to female ratio was 2.42:1.
• Most male patients 12 (41.4%) were observed in 30 - 40 years age group, followed by 11 (37.9%) in 41 -

Table 4. Laboratory investigations results in ADPKD patients.
50 years age group.
• Similarly, most female patients 5 (41.7%) were observed in 30 - 40 years age group, followed by 4 (33.3%) in 41 - 50 years age group.
• Least number of male patients 2 (6.9%) were observed in 61 - 70 years age group.
• Similarly, only 1 (8.3%) female patient was observed in 61 - 70 years age group.
• Most common form of clinical presentation was hypertension in 27 (65.85%) patients.
• Low back pain was seen in 24 (58.54%) and abdominal pain in 22 (53.66%) patients.
• 18 (43.90%) patients presented with headache and 15 (36.59%) presented with hematuria.
• 13 (31.70%) patients presented with renal failure (defined as elevated serum creatinine level of > 1.8 mg/dL for more than 8 weeks).
• Of these 13, 8 (19.51%) had hypertension and 5 (12.19%) were normotensive.
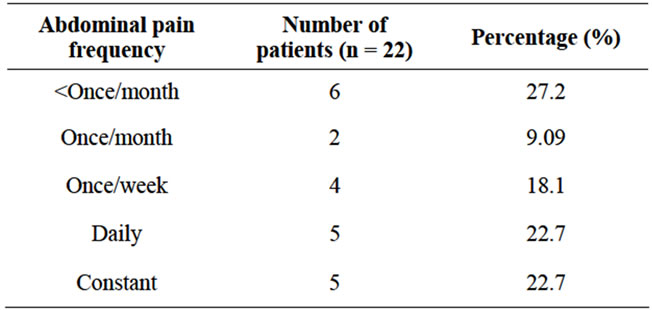
• Urinary tract infection was observed in 6 (40%) patients.
• Strenuous activity was observed in 3 (20%) patients.
• Association with hypertension was observed in 7 (46.7%) patients.
• Combined precipitating events were observed in some patients.
• 6 (27.2%) patients experienced abdominal pain once a month.
• 5 (22.7%) patients each experienced abdominal pain daily or the pain was constant.
• 4 (18.1%) patients experienced abdominal pain once a week.
• 24 (58.5%) patients had palpable kidney.
• 10 (24.4%) patients had palpable liver.
• 2 (4.9%) had murmur of mitral valve prolapse and 1 (2.4%) had palpable spleen.
• 5 (27.7%) patients had headache rarely.
• 4 (22.2%) patients had headache once a week.
• 3 (13.6%) patients had headache every day.
• 24 (58.5%) patients had palpable kidney.
• 10 (24.4%) patients had palpable liver.
• 2 (4.9%) had murmur of mitral valve prolapse and 1 (2.4%) had palpable spleen.
• 2 (4.9%) patients had mitral valve prolapse and 3 (7.3%) had left ventricular hypertrophy.
• No case of tricuspid valve prolapse, tricuspid regurgitation, aortic dilatation or aortic regurgitation were found.
• 2 (4.9%) patients had mitral valve prolapse and 3 (7.3%) had left ventricular hypertrophy.
• No case of tricuspid valve prolapse, tricuspid regurgitation, aortic dilatation or aortic regurgitation were found.
• 10 (24.4%) patients had hepatic cysts.
• 1 (2.4%) patient had evidence of hypertension (ascites, splenomegaly and Grade I ×3 varices) on UGI endoscopy
• 1 (2.4%) had evidence of hepatic cyst infection (fever, right upper quadrant pain and debris in cyst) on USG abdomen.
• The above table depicts mean ± standard deviation of laboratory parameters of 41 patients included in the study.
3. Discussion
Autosomal dominant polycystic kidney disease (ADPKD) is the most common form of polycystic kidney disease in which cysts are distributed throughout the cortex and medulla. The age of presentation in the present study was 30 - 70 years (Table 5, Figure 4). Glassberg et al. (1981) [71] in a study found that most cases of ADPKD were identified between 30 and 50 years of age.In the present study, the proportion of male to female patients with ADPKD was 2.42:1 (Table 6, Figure 5). Rabbani et al. 2008) [72] found a ratio of 2.5:1 in a study conducted in Pakistan on 56 ADPKD patients.In the present study, family history of ADPKD was present in 18 (43.9%) (Patients. Rabbani et al. (2008) [72] found 44.6% patients of ADPKD with family history of ADPKD (Table 7). In the present study, hypertension alone or in combination with renal failure was present in65.8% patients (Table 8, Figure 6). Out of 41 patients, hypertension alone was present in 19 patients (46.3%). 13 presented with renal failure, defined as presently elevated
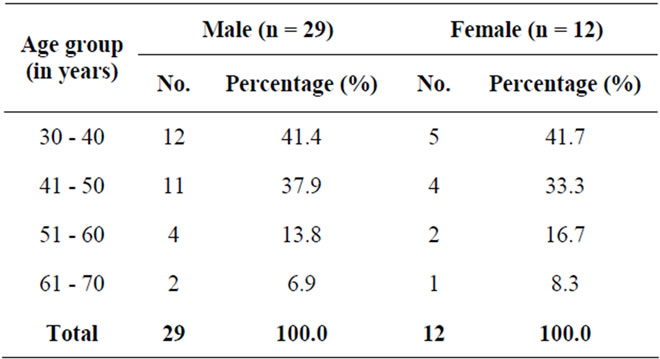


Table 7. Ultrasonographic criteria for diagnosing ADPKD as mentioned below was followed.

serum creatinine of greater than 1.8 mg/dL for more than 8 weeks. Of these 13 patients, 8 had hypertension, whereas 5 were normotensive. Due to early screening, hypertension has become the most common form of presentation
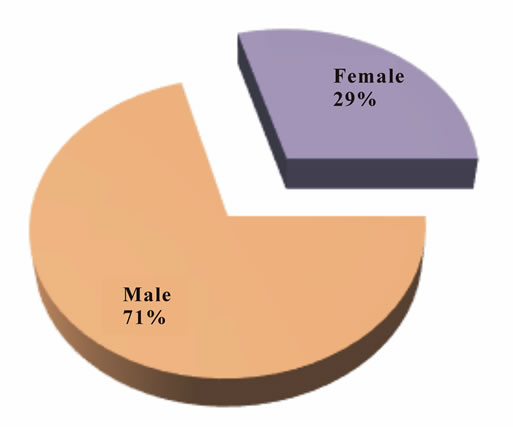
Figure 4. Genderwise distribution of ADPKD patients.

Figure 5. Profile of abdominal pain frequency in ADPKD patients.

Figure 6. Clinical presentation in ADPKD patients.
in patients with ADPKD (Gabow et al., 1990b) [73]. In 1949, Rall and Odel [74] found a 73% incidence of hypertension, Dalgaard (1957) [75], found 41% incidence, Hansson et al. (1974) [76] observed 75% incidence. Milutinovic et al. (1984) [77] reported a 33% incidence, Gabow et al. (1984) [78] found that the prevalence of hypertension was 62% in ADPKD patients. In the present study, low back pain was present in 24 (58.5%) of the patients, abdominal pain was reported by 22 (53.6%) patients (Table 2, Figure 2). Causes of pain are multiple and can include consequences related to cysts—cyst enlargement, cyst rupture and cyst infection. Severe back, flank and abdominal pain are known con sequences of cyst enlargement in patients with polycystic kidney disease (PKD). Massive kidney and liver cysts commonly produce mechanical low back pain from exaggerated pelvic tilt and subsequent lumbar lordosis (Bajwa et al., 2001) [44]. In the present study, headaches were experienced by 18 (43.9%) patients (Table 3, Figure 3). Headaches might be due to uncontrolled hypertension in these patients. Renal stones were present in 6 (14.6%) patients in our study which is in contrast to 18% reported by Dalgaard (1957) [75] and 20% reported by Segal et al. (1977) [79]. In the present study on clinical examination (Table 9, Figure 7), 24 (58.5%) patients were found to have palpable kidney and 10 (24.4%) were found to have palpable liver. Spleen was palpable in 1 (2.4%) patient, who was found to have portal hypertension leading to ascites and oesophageal varices. The murmur of mitral valve prolapse was found in 2 (4.9%) patients which was confirmed on echocardiography (Table 10, Figure 8).
Cerebral aneurysm is well known complication of ADPKD. The reported frequency ranges from 0% - 41%. This wide variation reflects the use of different diagnostic techniques, selection biases in some studies due to evaluation only of symptomatic patients or patients with a family history of ICA, differences in racial or ethnic background and possibly the association of ICA with particularly mutations of the PKD1 or PKD2 genes that might be shared kindreds (Perrone, 1997) [80]. In the present study, no patient was found to have ICA. The reason might be that only patients with headaches were evaluated for ICA.
One of the associated manifestation of ADPKD is occurrence of cardiovascular lesions in a significant num-

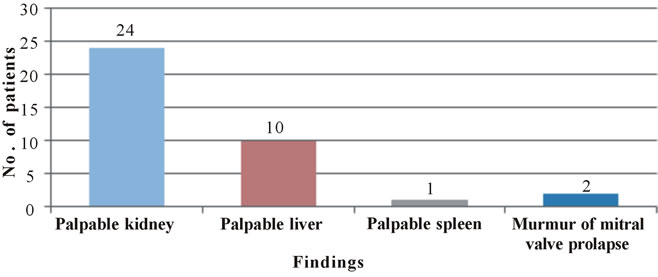
Figure 7. Clinical examination of ADPKD patients.

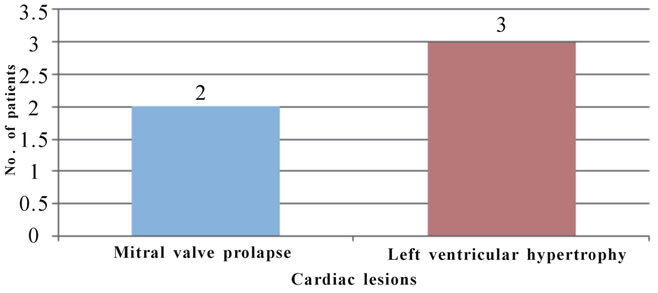
Figure 8. Cardiac diseases in ADPKD patients.
ber of patients (Leier et al., 1984) [35]. In the present study, 2 (4.9%) patients were found to have mitral valve prolapse and 3 (7.3%) patients were found to have left ventricular hypertrophy.
In the present study, no patient was found to have any diverticular disease. Rabbani et al. (2008) [72] also reported that none of their patients had splenic or pancreatic cyst and diverticulosis. No splenic or pancreatic cyst was detected in the present study either.
As patients with ADPKD have an increased renal mass, erythropoietin levels are increased, making anaemia unusual even when end-stage renal disease is present (Gabow et al., 1993) [33]. The mean haemoglobin in the present series was 11.2 g/dL. Blood coagulation studies and, leukocyte and platelet counts were normal.
Whether insulin resistance and elevation of serum insulin, triglycerides, low density lipoprotein and cholesterol occur in ADPKD-associated hyperension has not been ascertained. Nevertheless, reduced high density lipoprotein (HDL) and elevated LDL levels have been identified as risk factors for renal disease progression in ADPKD (Klahr et al., 1995) [81]. The level of HDL and LDL in our patients were satisfactory.
The early stages of ADPKD usually are not reflected in the urinalysis. Patients with early ADPKD may have a diminished ability to maximal concentrate the urine (Martinez-Maldonado et al., 1977) [82]. Nocturia may be the only symptom of this defect. Massive proteinuria is a rare finding (Montoyo et al., 1992) [83], and none of our patients had proteinuria in the nephrotic range. The reason of proteinuria in ADPKD is still unclear, however, possible explanation would be damage to capillary endothelium and glomerulosclerosis due to hypertension (Reckelhoff and Granger, 1999) [84]. Haematuria is usually due to rupture of a cyst into the pelvis of the kidney.
Dedi et al. (2001) [85] observed that ~68% of the patients had haematuria on urinalysis.
The liver is diffusely cystic in 20% - 50% of patients with ADPKD (Chauveau et al., 2000) [86] (Table 11, Figure 9). The frequency of liver cysts in our series was 24.4%. Out of 10 patients with hepatic cyst involvement, 1 patient was found to have evidence of portal hypertension and one was found to have evidence of hepatic cyst infection.
4. Summary
The present study was conducted in the Postgraduate Department of Medicine, Government Medical College, Jammu, where a total of 41 patients—29 males and 12 females—fulfilled the criteria of ADPKD. The results of the study are summarised as follow:
Ø Proportion of male to female patients with ADPKD was 2.42:1.
Ø Maximum 17 (41.5%) patients of both gender were seen in 30 - 40 years age group.
Ø Family history of ADPKD was present in 18 (43.9%) patients.
Ø Hypertension, alone or in combination with renal failure, was present in 65.8% patients.
Ø Hypertension alone was present in 19 (46.3%) patients.
Ø 8 (19.5%) patients with hypertension had renal failure.
Ø Low back pain was present in 24 (58.5%) and abdominal pain in 22 (53.7%) patients.
Ø 15 (36.6%) patients presented with atleast one episode of gross haematuria.
Ø Headache was experienced by 18 (43.9%) patients.
Ø On clinical examination, 24 (58.5%) were found to have palpable kidney and 10 (24.4%) had palpable liver.
Ø Spleen was palpable in 1 (2.4%) patient, who was found to have portal hypertension leading to ascites and oesophageal varices.
Ø Murmur of mitral valve prolapse was found in 2 (4.9%) patients which was confirmed on echocardiography.
Ø 3 (7.3%) patients were found to having left ventricular hypertrophy confirmed on echocardiography.
Ø No patient was found to have ICA and valvular abnormality.
Ø The mean haemoglobin in the present series was 11.2 g/dL.
Ø Blood coagulation studies and, leukocyte and platelet counts were normal.
Ø The level of HDL and LDL in our patients were satisfactory.
Ø The liver cysts were found in 24.4% of the patients.
Ø Out of 10 (24.4%) patients with hepatic cyst in volve-
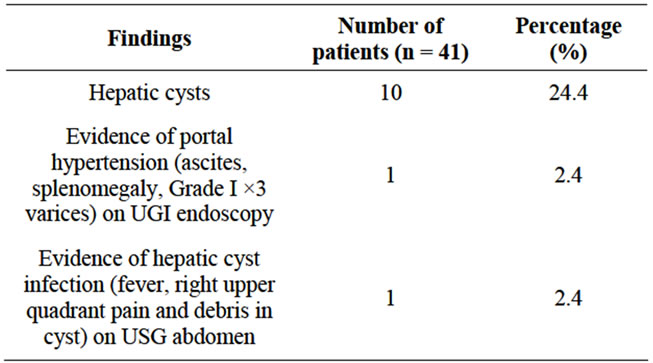

Figure 9. Hepatic involvement in ADPKD patients.
ment, 1 patient each was found to have evidence of portal hypertension and evidence of hepatic cyst infection.
5. Conclusion
As ADPKD is one of the common inherited renal diseases in our part of the country, more research is needed to study the pattern of this disease. In the present study, hypertension was most common presentation of this disease. So, control of hypertension is very important to prevent progression of this disease. Patients who are detected to have ADPKD should be regularly followed-up to prevent further progression by timely intervention. Also, family members of patients should be screened for disease and initiate treatment as early as possible.
REFERENCES
- P. D. Wilson, “Polycystic Kidney Disease,” New England Journal of Medicine, Vol. 350, No. 2, 2004, pp. 151-164. doi:10.1056/NEJMra022161
- F. Davies, G. A. Coles, P. S. Harper, et al., “Polycystic Kidney Disease Re-Evaluated: A Population-Based Study,” QJM: An International Journal of Medicine, Vol. 79, No. 3, 1991, pp. 477-485.
- P. S. Parfrey, J. C. Bear, J. Morgan, et al., “The Diagnosis and Prognosis of Autosomal Dominant Polycystic Kidney Disease,” New England Journal of Medicine, Vol. 323, No. 16, 1990, pp. 1085-1090. doi:10.1056/NEJM199010183231601
- A. C. Ong and P. C. Harris, “Molecular Pathogenesis of ADPKD: The Polycystin Complex Gets Complex,” Kidney International, Vol. 67, No. 4, 2005, pp. 1234-1247. doi:10.1111/j.1523-1755.2005.00201.x
- V. E. Torres and P. C. Harris, “Autosomal Dominant Polycystic Kidney Disease,” Nefrologia, Vol. 23, Suppl. 1, 2003, pp. 14-22.
- O. Ibraghimov-Beskrovnaya, W. R. Dackowski, L. Foggensteiner, et al., “Polycystin: In Vitro Synthesis, in Vivo Tissue Expression, and Subcellular Localization Identifies a Large Membrane Associated Protein,” Proceedings of the National Academy of Sciences, Vol. 94, No. 12, 1997, pp. 6397-6402. doi:10.1073/pnas.94.12.6397
- W. J. Kimberling, S. Kumar, P. A. Gabow, et al., “Autosomal Dominant Polycystic Kidney Disease: Localization of the Second Gene to Chromosome 4q13-q23,” Genomics, Vol. 18, No. 3, 1993, pp. 467-472. doi:10.1016/S0888-7543(11)80001-7
- N. Hateboer, M. A. v Dijk, N. Bogdanova, et al., “Comparison of Phenotypes of Polycystic Kidney Disease Types 1 and 2. European PKD1-PKD2 Study Group,” Lancet, Vol. 353, No. 9147, 1999, pp. 103-107. doi:10.1016/S0140-6736(98)03495-3
- P. Igarashi and S. Somlo, “Genetics and Pathogenesis of Polycystic Kidney Disease,” Journal of the American Society of Nephrology, Vol. 13, No. 9, 2002, pp. 2384-2398. doi:10.1097/01.ASN.0000028643.17901.42
- D. D. Woo, S. Y. Miao, J. C. Pelayo, et al., “Taxol Inhibits Progression of Congenital Polycystic Kidney Disease,” Nature, Vol. 368, 1994, pp. 750-753. doi:10.1038/368750a0
- J. Grantham, B. J. Cowley and V. E. Torres, “Progression of Autosomal Dominant Polycystic Kidney Disease (ADPKD) to Renal Failure,” In: D. Seldin and G. Giebisch, Eds., The Kidney: Physiology and Pathophysiology, Lippincott Williams and Wilkins, Philadelphia, 2000, pp. 2513-2536.
- Z. D. heng, M. Wolfe, B. D. Cowley Jr, et al., “Urinary Excretion of Monocyte Chemoattractant Protein-1 in Autosomal Dominant Polycystic Kidney Disease,” Journal of the American Society of Nephrology, Vol. 14, No. 10, 2003, pp. 2588-2595.
- A. B. Chapman, A. Johnson, P. A. Gabow, et al., “The Renin-Angiotensin-Aldosterone System and Autosomal Dominant Polycystic Kidney Disease,” New England Journal of Medicine, Vol. 323, No. 16, 1990, pp. 1091-1096. doi:10.1056/NEJM199010183231602
- P. A. Gabow, I. Duley and A. M. Johnson, “Clinical Profiles of Gross Hematuria in Autosomal Dominant Polycystic Kidney Disease,” American Journal of Kidney Diseases, Vol. 20, No. 1992, pp. 140-143.
- E. Levine and J. J. Grantham, “Perinephric Hemorrhage in Autosomal Dominant Polycystic Kidney Disease: CT and MR Findings,” Journal of Computer Assisted Tomography, Vol. 11, No. 1, 1987, pp. 108-111. doi:10.1097/00004728-198701000-00021
- Y. Ubara, et al., “Transcatheter Renal Arterial Embolization Therapy on a Patient with Polycystic Kidney Disease on Hemodialysis,” American Journal of Kidney Diseases, Vol. 34, No. 5, 1999, pp. 926-931.
- A. Vijay, A. Vijay and P. Pankaj, “Autosomal Dominant Polycystic Kidney Disease: A Comprehensive Review,” International Urology and Nephrology, Vol. 2, No. 1, 2010, pp. 172-192.
- G. M. Fick-Brosnahan, Z. V. Tran, A. M. Johnson, et al., “Progression of Autosomal-Dominant Polycystic Kidney Disease in Children,” Kidney International, Vol. 59, No. 5, 2001, pp. 1654-1662. doi:10.1046/j.1523-1755.2001.0590051654.x
- W. J. Amend, “Polycystic Kidney Disease and Seatbelts,” Annals of Internal Medicine, Vol. 79, No. 2, 1973, pp. 287-292.
- A. B. Chapman and R. W. Schrier, “Pathogenesis of Hypertension in Autosomal Dominant Polycystic Kidney Disease,” Seminars in Nephrology, Vol. 11, No. 6, 1991, pp. 653-660.
- G. M. Fick, A. M. Johnson and W. S. Hammond, “Causes of Death in Autosomal Dominant Polycystic Kidney Disease,” Journal of the American Society of Nephrology, Vol. 5, No. 12, 1995, pp. 2048-2056.
- M. Loghman-Adham, C. E. Soto, T. Inagami, et al., “The Intrarenal Renin-Angiotensin System in Autosomal Dominant Polycystic Kidney Disease,” American Journal of Physiology—Renal Physiology, Vol. 287, No. 4, 2004, pp. 775-788. doi:10.1152/ajprenal.00370.2003
- T. Nakamura, T. Sugaya, Y. Kawagoe, et al., “Candesartan Reduces Urinary Fatty Acid-Binding Protein Excretion in Patients with Autosomal Dominant Polycystic Kidney Disease,” American Journal of the Medical Sciences, Vol. 330, No. 4, 2005, pp. 161-165. doi:10.1097/00000441-200510000-00002
- E. R. Ahmed, M. A. Tashkandi, S. Nahrir, et al., “Retrospective Analysis of Factors Affecting the Progression of Chronic Renal Failure in Adult Polycystic Kidney Disease,” Saudi Journal of Kidney Diseases and Transplantation, Vol. 17, No. 4, 2006, pp. 511-515.
- G. Gambaro, A. Fabris, D. Puliatta, et al., “Lithiasis in Cystic Kidney Disease and Malformations of the Urinary Tract,” Urological Research, Vol. 34, No. 2, 2006, pp. 102-107. doi:10.1007/s00240-005-0019-z
- V. B. Delaney, S. Adler, F. J. Bruns, et al., “Autosomal Dominant Polycystic Kidney Disease: Presentation, Complications, and Prognosis,” American Journal of Kidney Diseases, Vol. 5, No. 2, 1985, pp. 104-111.
- O. Koslowe, R. Frank, B. Gauthier, et al., “Urinary Tract Infections, VUR and Autosomal Dominant Polycystic Kidney Disease,” Pediatric Nephrology, Vol. 18, No. 8, 2003, pp. 823-825. doi:10.1007/s00467-003-1211-4
- H. Thomsen and J. Taysen, “Frequency of Hepatic Cysts in Adult Polycystic Kidney Disease,” Acta Medica Scandinavica, Vol. 224, No. 4, 1988, pp. 381-384. doi:10.1111/j.0954-6820.1988.tb19598.x
- J. P. Grunfeld, G. Albouze, P. Jungers, et al., “Liver Changes and Complications in Adult Polycystic Kidney Disease,” In: J. F. Bach, J. Crosnier, J. L. Funck-Brentano et al., Eds., Advances in Nephrology, Year Book Medical Publishers, Chicago, 1985, pp. 1-20.
- N. Gretz, M. Zeier, S. Geberth, et al., “Is Gender a Determinant for Evolution of Renal Failure? A Study in Autosomal Dominant Polycystic Kidney Disease,” American Journal of Kidney Diseases, Vol. 14, No. 3, 1989, pp. 178-183.
- A. Telenti, V. Torres, J. J. Gross, et al., “Hepatic Cyst Infection in Autosomal Dominant Polycystic Kidney Disease,” Mayo Clinic Proceedings, Vol. 65, No. 7, 1990, pp. 933-942. doi:10.1016/S0025-6196(12)65154-4
- F. Que, D. M. Nagorney, J. B. Gross Jr., et al., “Liver Resection and Cyst Fenestration in the Treatment of Severe Polycystic Liver Disease,” Gastroenterology, Vol. 108, No. 2, 1995, pp. 487-494. doi:10.1016/0016-5085(95)90078-0
- P. A. Gabow, “Autosomal Dominant Polycystic Kidney Disease,” American Journal of Kidney Diseases, Vol. 22, No. 4, 1993, pp. 511-512.
- W. Schievink, Torres, D. Piepgras, et al., “Saccular Intracranial Aneurysms in Autosomal Dominant Polycystic Kidney Disease,” Journal of the American Society of Nephrology, Vol. 3, No. 1, 1992, pp. 88-95.
- C.V. Leier, P. B. Baker, J. W. Kilman, et al., “Cardiovascular Abnormalities Associated with Adult Polycystic Kidney Disease,” Annals of Internal Medicine, Vol. 100, No. 5, 1984, pp. 683-688.
- R. Schieff, G. Zuckerman, H. Marter, et al., “Diverticular Disease in Patients with Chronic Renal Failure Due to Polycystic Kidney Disease,” Annals of Internal Medicine, Vol. 92, No. 1, 1980, pp. 202-204.
- V. E. Torres, P. C. Harris and Y. Pirson, “Autosomal Dominant Polycystic Kidney Disease,” Lancet, Vol. 369, No. 9569, 2007, pp. 1287-1301. doi:10.1016/S0140-6736(07)60601-1
- W. C. O’Neill, M. L. Robbin, K. T. Bae, et al., “Sonographic Assessment of the Severity and Progression of Autosomal Dominant Polycystic Kidney Disease: The Consortium of Renal Imaging Studies in Polycystic Kidney Disease (CRISP),” American Journal of Kidney Diseases, Vol. 46, No. 2005, pp. 1058-1064. doi:10.1053/j.ajkd.2005.08.026
- H. S. Thomsen, P. Marckmann and V. B. Logager, “Update on Nephrogenic Systemic Fibrosis,” Magnetic Resonance Imaging Clinics of North America, Vol. 16, No. 4, 2008, pp. 551-560. doi:10.1016/j.mric.2008.07.011
- A. D. Kistler, D. Poster, F. Krauer, et al., “Increases in Kidney Volume in Autosomal Dominant Polycystic Kidney Disease Can Be Detected within 6 Months,” Kidney International, Vol. 75, No. 2009, pp. 235-241. doi:10.1038/ki.2008.558
- P. E. Bell, K. F. Hossack, P. A. Gabow, et al., “Hypertension in Autosomal Dominant Polycystic Kidney Disease,” Kidney International, Vol. 34, No. 1988, pp. 683-690. doi:10.1038/ki.1988.233
- A. Bourquia, B. Ramdani, A. J. Jabrane, et al., “Dominant Renal Polycystic Disease,” Revue de Medecine Interne, Vol. 10, No. 4, 1989, pp. 313-318. doi:10.1016/S0248-8663(89)80028-1
- F. A. Al-Muhanna, K. K. Malhotra, I. Saeed, et al., “Autosomal Dominant Polycystic Kidney Disease: Observations from a University Hospital in Saudi Arabia,” Saudi Journal of Kidney Diseases and Transplantation, Vol. 6, 1995, pp. 28-31.
- Z. H. Bajwa, S. Gupta, C. A. Warfield, et al., “Pain Management in Polycystic Kidney Disease,” Kidney International, Vol. 60, No. 5, 2001, pp. 1631-1644. doi:10.1046/j.1523-1755.2001.00985.x
- S. Graf, A. Schischma, K. E. Eberhardt, et al., “Intracranial Aneurysms and Dolichoectasia in Autosomal Dominant Polycystic Kidney Disease,” Nephrology Dialysis Transplantation, Vol. 17, No. 5, 2002, pp. 819-823. doi:10.1093/ndt/17.5.819
- R. Schrier, K. McFann, A. Johnson, et al., “Cardiac and Renal Effects of Standard versus Rigorous Blood Pressure Control in Autosomal-Dominant Polycystic Kidney Disease: Results of a Seven-Year Prospective Randomized Study,” Journal of the American Society of Nephrology, Vol. 13, No. 7, 2002, pp. 1733-1739. doi:10.1097/01.ASN.0000018407.60002.B9
- Z. H. Bajwa, K. A. Sial, A. B. Malik, et al., “Pain Patterns in Patients with Polycystic Kidney Disease,” Kidney International, Vol. 66, No. 4, 2004, pp. 1561-1569. doi:10.1111/j.1523-1755.2004.00921.x
- W. Wolyniec, M. M. Jankowska, E. Krol, et al., “Current Diagnostic Evaluation of Autosomal Dominant Polycystic Kidney Disease,” Polskie Archiwum Medycyny Wewnę- trznej, Vol. 118, No. 12, 2008, pp. 767-773.
- P. Amico, S. Kalbermatter and D. Kiss, “Aliskiren Corrects Recurrent Hyperreninemia and Hyperaldosteronism in Autosomaldominant Polycystic Kidney Disease,” Clinical Nephrology, Vol. 72, No. 3, 2009, pp. 237-239.
- K. T. Bae, C. Tao, F. Zhu, et al., “MRI-Based Kidney Volume Measurements in ADPKD: Reliability and Effect of Gadolinium Enhancement,” Clinical Journal of the American Society of Nephrology, Vol. 4, No. 4, 2009, pp. 719-725. doi:10.2215/CJN.03750708
- M. Barua, O. Cil, A. D. Paterson, et al., “Family History of Renal Disease Severity Predicts the Mutated Gene in ADPKD,” Journal of the American Society of Nephrology, Vol. 20, No. 8, 2009, pp. 1833-1838. doi:10.1681/ASN.2009020162
- T. Ecder and R. W. Schrier, “Cardiovascular Abnormalities in Autosomal-Dominant Polycystic Kidney Disease,” Nature Reviews Nephrology, Vol. 5, No. 4, 2009, pp. 221-228. doi:10.1038/nrneph.2009.13
- H. Hadimeri, K. Caidahl, O. Bech-Hanssen, et al., “Echocardiographic Findings in Kidney Transplant Patients with Autosomal Dominant Polycystic Kidney Disease,” Scandinavian Journal of Urology and Nephrology, Vol. 43, No. 5, 2009, pp. 416-419. doi:10.3109/00365590902972446
- P. Hajj, S. Ferlicot, W. Massoud, et al., “Prevalence of Renal Cell Carcinoma in Patients with Autosomal Dominant Polycystic Kidney Disease and Chronic Renal Failure,” Urology, Vol. 74, No. 3, 2009, pp. 631-634. doi:10.1016/j.urology.2009.02.078
- A. Idrizi, M. Barbullushi, E. Petrela, et al., “The Influence of Renal Manifestations to the Progression of Autosomal Dominant Polycystic Kidney Disease,” Hippokratia, Vol. 13, No. 3, 2009, pp. 161-164.
- A. Ramunni, P. Brescia, D. Quaranta et al., “Cutaneous Microcirculation Is Impaired in Early Autosomal Dominant Polycystic Kidney Disease,” Nephron Clinical Practice, Vol. 113, No. 2, 2009, pp. c71-75. doi:10.1159/000228537
- M. Sallee, C. Rafat, J. R. Zahar, et al., “Cyst Infections in Patients with Autosomal Dominant Polycystic Kidney Disease,” Clinical Journal of the American Society of Nephrology, Vol. 4, No. 7, 2009, pp. 1183-1189. doi:10.2215/CJN.01870309
- M. Sawicki, A. Walecka, J. Rozanski, et al., “Doppler Sonography Measurements of Renal Vascular Resistance in Autosomal-Dominant Polycystic Kidney Disease,” Medical Science Monitor, Vol. 15, No. 8, 2009, pp. 101-104.
- A. Alam and R. D. Perrone, “Management of ESRD in Patients with Autosomal Dominant Polycystic Kidney Disease,” Advances in Chronic Kidney Disease, Vol. 17, No. 2, 2010, pp. 164-172. doi:10.1053/j.ackd.2009.12.006
- K. T. Baeand and J. J. Grantham, “Imaging for the Prognosis of Autosomal Dominant Polycystic Kidney Disease,” Nature Reviews Nephrology, Vol. 6, No. 2, 2010, pp. 96-106. doi:10.1038/nrneph.2009.214
- M. Barua and Y. Pei, “Diagnosis of Autosomal-Dominant Polycystic Kidney Disease: An Integrated Approach,” Seminars in Nephrology, Vol. 30, No. 4, 2010, pp. 356-365.
- M. Y. Chang, C. M. Kuok, Y. C. Chen, et al., “Comparison of Intracerebral Hemorrhage and Subarachnoid Hemorrhage in Patients with Autosomal-Dominant Polycystic Kidney Disease,” Nephron Clinical Practice, Vol. 114, No. 2, 2010, pp. c158-c164. doi:10.1159/000256568
- A. B. Chapman, K. Stepniakowski and F. Rahbari-Oskoui, “Hypertension in Autosomal Dominant Polycystic Kidney Disease,” Advances in Chronic Kidney Disease, Vol. 17, No. 2, 2010, pp. 153-163. doi:10.1053/j.ackd.2010.01.001
- P. C. Harris and S. Rossetti, “Determinants of Renal Disease Variability in ADPKD,” Advances in Chronic Kidney Disease, Vol. 17, No. 2, 2010, pp. 131-139. doi:10.1053/j.ackd.2009.12.004
- E. Meijer, M. Rook, H. Tent, et al., “Early Renal Abnormalities in Autosomal Dominant Polycystic Kidney Disease,” Clinical Journal of the American Society of Nephrology, Vol. 5, No. 6, 2010, pp. 1091-1098. doi:10.2215/CJN.00360110
- U. B. Muftiand, S. K. Nalagatla, “Nephrolithiasis in Autosomal Dominant Polycystic Kidney Disease,” Journal of Endourology, Vol. 24, No. 10, 2010, pp. 1557-1561.
- Y. Pirson, “Extrarenal Manifestations of Autosomal Dominant Polycystic Kidney Disease,” Advances in Chronic Kidney Disease, Vol. 17, No. 2, 2010, pp. 173-180. doi:10.1053/j.ackd.2010.01.003
- Q. Qian, “Isolated Polycystic Liver Disease,” Advances in Chronic Kidney Disease, Vol. 17, No. 2, 2010, pp. 181-189. doi:10.1053/j.ackd.2009.12.005
- M. A. Rabbani, B. Ahmed and M. A. Khan, “Mycobacterium Tuberculosis Infection of a Native Polycystic Kidney Following Renal Transplantation,” Transplant Infectious Disease, Vol. 13, No. 1, 2010, pp. 44-46.
- D. Zheng, L. T. Cheng, Q. F. Han, et al., “Refractory Ascites Due to Portal Hypertension in Autosomal Dominant Polycystic Kidney Disease (Adpkd) Patients Successfully Treated with Peritoneal Dialysis,” Peritoneal Dialysis International, Vol. 30, No. 2, 2010, pp. 151-155. doi:10.3747/pdi.2009.00129
- K. I. Glassberg, R. E. Hackett, K. Waterhouse, “Congenital Anomalies of the Kidney, Ureter and Bladder,” In: A. R. Kendall and L. Karafin, Eds., Harry S Coldsmith’s Practice of Surgery: Urology, Harper and Row, Hagerstown, 1981.
- M. A. Rabbani, A. Ahmad, S. S. Ali, et al., “Clinical Presentation and Outcome of Autosomal Dominant Polycystic Kidney Disease in Pakistan: A Single Center Experience,” Japan Pharmaceutical Manufacturers Association, Vol. 58, No. 6, 2008, pp. 305-311.
- P. A. Gabow, A. B. Chapman, A. M. Johnson, et al., “Renal Structure and Hypertension in Autosomal Dominant Polycystic Kidney Disease,” Kidney International, Vol. 38, No. 6, 1990, pp. 1177-1180. doi:10.1038/ki.1990.330
- J. E. Rall and H. M. Odel, “Congenital Polycystic Disease of the Kidney: Review of the Literature, and Data on 207 Cases,” American Journal of the Medical Sciences, Vol. 218, No. 4, 1949, pp. 399-407. doi:10.1097/00000441-194910000-00007
- O. Z. Dalgaard, “Bilateral Polycystic Disease of the Kidneys. A Follow-Up of Two Hundred and Eighty-Four Patients and Their Families,” Acta Medica Scandinavica, Vol. 328, 1957, 1-255. doi:10.3109/00365597409132130
- L. Hansson, L. E. Karlander, W. Lundgren, et al., “Hypertension in Polycystic Kidney Disease,” Scandinavian Journal of Urology and Nephrology, Vol. 8, No. 3, 1974, pp. 203-205.
- J. Milutinovic, P. J. Fialkow, L. Y. Agodoa, et al., “Autosomal Dominant Polycystic Kidney Disease: Symptoms and Clinical Findings,” QJM: An International Journal of Medicine, Vol. 53, No. 1984, pp. 511-522.
- P. A. Gabow, D. W. Ikle and J. H. Holmes, “Polycystic Kidney Disease: Prospective Analysis of Nonazotemic Patients and Family Members,” Annals of Internal Medicine, Vol. 101, No. 101, 1984, pp. 238-247.
- A. J. Segal, R. F. Spataro and Z. L. Barbaric, “Adult Polycystic Kidney Disease: A Review of 100 Cases,” Journal of Urology, Vol. 118, No. 5, 1977, pp. 711-713.
- R. D. Perrone, “Extrarenal Manifestations of ADPKD,” Kidney International, Vol. 51, 1997, pp. 2022-2036. doi:10.1038/ki.1997.276
- S. Klahr, J. Breyer, G. J. Beck, et al. “Dietary Protein Restriction, Blood Pressure Control, and the Progression Polycystic Kidney Disease. Modification of Diet in Renal Disease Study Group,” Journal of the American Society of Nephrology, Vol. 5, 1995, pp. 2037-2047.
- M. Martinez-Maldonado, J. J. Yium, W. N. Suki, et al., “Electrolyte Excretion in Polycystic Kidney Disease: Interrelationship between Sodium, Calcium, Magnesium and Phosphate,” Journal of Laboratory and Clinical Medicine, Vol. 90, No. 6, 1977, pp. 1066-1075.
- C. Montoyo, M. A. Marinez, C. Campo, et al., “Nephrotic syndrome and Focal Glomerculosclerosis in Adult Polycystic Kidney Disease,” Nephron, Vol. 61, No. 1, 1992, pp. 106-110. doi:10.1159/000186845
- J. F. Reckelhoff and J. P. Granger, “Role of Andorgens in Mediating Hypertension and Renal Injury,” Clinical and Experimental Pharmacology and Physiology, Vol. 26, No. 2, 1999, pp. 127-131. doi:10.1046/j.1440-1681.1999.02996.x
- R. Dedi, S. Bhandari, J. H. Turney, et al., “Lesson of the Week: Causes of Hematuria in Adult Polycystic Kidney Disease,” British Medical Journal, Vol. 323, 2001, pp. 386-387. doi:10.1136/bmj.323.7309.386
- D. Chauveau, F. Fakhouri, J. P. Grünfeld, “Liver Involvement in Autosomal-Dominant Polycystic Kidney Disease: Therapeutic Dilemma,” Journal of the American Society of Nephrology, Vol. 11, No. 9, 2000, pp. 1767-1775.