Advances in Biological Chemistry
Vol.3 No.1(2013), Article ID:28111,11 pages DOI:10.4236/abc.2013.31012
Cysteine-associated distribution of aromatic residues in disulfide-stabilized extracellular protein families
![]()
Department of Science and Mathematics, Alvernia University, Reading, USA
Email: stephen.campion@alvernia.edu
Received 20 December 2012; revised 21 January 2013; accepted 30 January 2013
Keywords: EGF; Sushi; Laminin; Immunoglobulin
ABSTRACT
Cysteine-dependent protein sequences were downloaded from annotated database resources to generate comprehensive EGF, Sushi, Laminin and Immunoglobulin (IgC) motif-specific sequence files. Each dataset was vertically registered and the cumulative distribution of amino acid functional group chemistry determined relative to the respective complement of cysteine residues providing critical disulfide stabilization of these four well-known modular motif families. The cysteine-aligned amino acid distribution data revealed limited ionic, polar, hydrophobic or other side chain preferences, unique to each protein scaffold. In contrast, all four cysteine-dependent protein families exhibited strong positional preference for the aromatic residues phenylalanine (Phe) and tyrosine (Tyr), relative to analogous cysteine landmarks. More than eighty percent of the members in each protein family were found to possesses the same conserved -Cys- (Xxx)3-4-(Phe/Tyr)- arrangement, placing an aromatic amino acid at analogous EGF-C5+4, Sushi-C2+4, Laminin-C7+4 and IgC-C1+5. Over seventy percent of EGF, Sushi and IgC sequences exhibited a second obvious Cys-associated aromatic site -(Phe/Tyr)-XxxCysat EGF-C4-2, Sushi-C2-2 and IgC-C2-2. The cysteine-associated placement of aromatic amino acid chemistry in four major disulfide-dependent protein families likely represents conservation of a molecular determinant of global importance in the structurefunction of this large and diverse subset of extracellular proteins.
1. INTRODUCTION
One of the major impacts of advanced protein bioinformatics has been to demonstrate the wide-spread deployment of a few selected “modular” protein structures across the range of modern genomes [1,2]. The incorporation of one or more epidermal growth factor (EGF), Immunoglobulin (IgC), complement-like Sushi and/or Laminin motif structures into numerous circulating enzymes, growth factors and biochemical modulators, mixed-module and poly-modular membrane-bound receptors and extracellular matrix proteins is well documented in the protein database [3-6]. The prevalence of EGF, Immunoglobulin, Sushi and Laminin homologues in characterized genomes, from slime mold to humans, clearly demonstrates evolutionary selection of some ancestral utility. However, whether these heavily employed and often repetitive protein modules simply represent convenient peptide backbones upon which to incorporate diverse usage-defined determinants of molecular interaction, or whether they contribute some inherent motifspecific or global functionality to the larger protein super-structures in which they are incorporated remains largely unknown.
The reliable annotation of individual EGF, Sushi, Laminin and IgC domain sequences and subsequent assembly into separate motif-specific sequence files, based on extended taxonomical relationships identified by respected sequence alignment tools [7,9], makes secondary analysis of these now well-established families of related protein orthologs and paralogs possible. The Immunoglobulin, Sushi, EGF and Laminin sequence families exhibit distinct consensus patterns of two, four, six or eight cysteine residues, respectively. These familial cysteine landmarks, which constitute the basis for four distinct disulfide-stabilized native protein structures, are easily recognized and aligned despite often dramatic variability in the length, amino acid composition and sequence of the inter-cysteine strands that comprise each member of these large and diverse protein families. The exhaustive compilation and vertical registration of cysteine-aligned EGF, Sushi, Laminin and IgC domain sequence files have made it possible to visualize this sequence diversity, as well as to identify both motif-specific and shared elements of structure-function occurring within the respective cysteine-defined consensus structures.
2. EXPERIMENTAL PROCEDURES
2.1. Compiling Sequence Families
The continuously updated taxonomical listing of available EGF, Sushi, Laminin and Immunoglobulin constant domain (IgC) protein sequences was accessed through the National Center for Biotechnology Information’s network of linked bioinformatics resources at
www.ncbi.nlm.nih.gov [10,11]. This study evaluated more than fifteen hundred individual EGF and calciumbinding EGF (cbEGF) motif modules, including the EGF sequence families cd00053, pfam00008, smart00181, cd00054, and smart00179 registered in NCBI’s Conserved Domains database [12]. Similarly, nearly six hundred immunoglobulin constant (IgC) domains from cd00098, as well as five hundred Sushi modules, including all of the archived cd00033, pfam00084, and smart00032 sequence families were examined. Finally, almost four hundred Laminin protein sequences including cd00055, pfam00053, and smart00180 families were systematically retrieved from the protein database.
2.2. Mapping of Amino-Acid Distribution
With nearly absolute conservation of their motif-defining disulfide-bond structures, the alignment of analogous cysteines within respective Laminin, EGF, Sushi, and IgC protein families was relatively uncomplicated. Guided by the NCBI’s Multiple Alignment Viewer, the extensive sequence files generated for each protein family were visually aligned by simple vertical registration of analogous cysteine (Cys) residues, with variable-length intercysteine strands each divided mechanically at the midpoint, with no attempt to align other homologous amino acid chemistry (see Figure 1). The resulting vertically
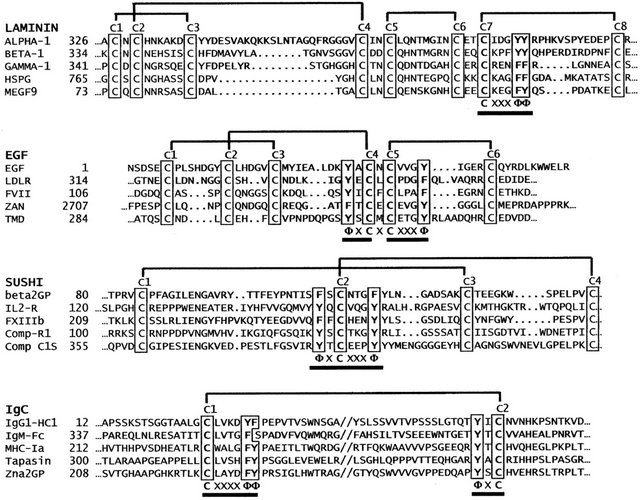
Figure 1. The alignment of selected motif modules from each of the four major protein families illustrates the vertical registration of analogous cysteine (C) residues highlighted, employed in this study. Inter-cysteine strands were subdivided at the mid-point, with any odd number residues placed into the amino-terminal segment of the strand. Selected Laminin-like protein sequences include domain-2 from human Laminins alpha-1 [GI:281185471], beta-1 [GI:317373377], gamma-1 [GI:317373377], human heparan-sulfate proteoglycan core protein [GI:29470], and human brain protein MEGF9 [GI:3449310]. The EGF-like protein sequences shown include human epidermal growth factor [GI:208524], human LDL receptor EGF domain-1 [GI: 126073], human coagulation factor VII EGF domain-1 [GI:182801], human Zonadhesin [GI:325511408], and EGF domain-2 of human thrombomodulin [GI:339657]. Complement like Sushi sequences shown include sushi domain-2 of human beta-2-glycoprotein [GI:28812], sushi domain-2 of human Interleukin-2 receptor [GI:124317], sushi domain-4 of human coagulation factor XIII [GI:179417], sushi domain-2 of human complement receptor-1 [GI:306680], and domain-2 of human complement protease C1s [GI:115205]. Immunoglobulin domains include human IgG heavy-chain domain-1 [GI:121039], and IgM-Fc region [GI: 12054080], human major histocompatibility complex Ia [GI:13509243], human Tapasin [GI:12643361] and human zinc-alpha-2-glycoprotein [GI:292495049]. The regions where cysteine-associated aromatic groups occur are underlined [f = Phe or Tyr].
“registered” sequence arrays represent the actual linear arrangement of amino acids in the vicinity of each landmark cysteine, with none of the gaps introduced by methods employing traditional homology scoring and local alignment algorithms. In order to ensure that every EGF, Sushi, Laminin and IgC sequence represented a unique entry in the database, specific effort was made to exclude the many identical sequence entries bearing different identifiers. As long as each entry satisfied NCBI’s statistical homology-based scoring (E-value) standards to be included in one of the respective protein families, no effort to filter or otherwise adjudicate the inclusion or exclusion of specific individual or groups of protein orthologs or paralogs was applied.
The Cys-aligned sequence text files were then imported into standard spreadsheet format and subsequently converted to aligned “sequence tables” for automated tabulation of amino acid distribution. The cumulative occurrence of each of the twenty possible amino acids was assessed at each position in every Laminin, EGF, Sushi and IgC sequence in their respective datasets. A total of more than one-hundred-and-fifty-thousand individual amino acids were evaluated, and the cumulative positional occurrence of each residue plotted relative to the nearest Cys landmark. This relative positional mapping permitted examination of these enormously diverse sequence datasets despite often dramatic variation in the length of analogous inter-cysteine strands, both within and across protein families.
3. RESULTS
As a consequence of compiling and evaluating the distribution of individual amino acids in motif-specific datasets, general amino acid composition data for each protein family was generated (Table 1). The most obvious difference between the Laminin, EGF, Sushi and IgC families, which employ sequentially fewer disulfides, was an expected sharp decline in the percent of cysteine. An accompanying increase in aliphatic content, from fifteen percent in Laminin (eight cysteines) up to twentysix percent in IgC (two cysteines), likely reflects an increased dependence on the hydrophobic-based stabilizing forces employed in protein structures with fewer disulfides.
The distribution of amino acids in disulfide-dependent protein families was systematically evaluated and the cumulative occurrence of homologous amino acid chemistry determined relative to essential familial cysteine landmarks. Data for the different amino acid functional classes, acidic residues, basic residues, alcohol-based and amide/imidazole-containing polar amino acids, residues with aliphatic and aromatic sidechains, and the structural residues glycine and proline (not shown) were determined separately for each protein family.
Table 1. Amino acid composition of motif-specific datasets.
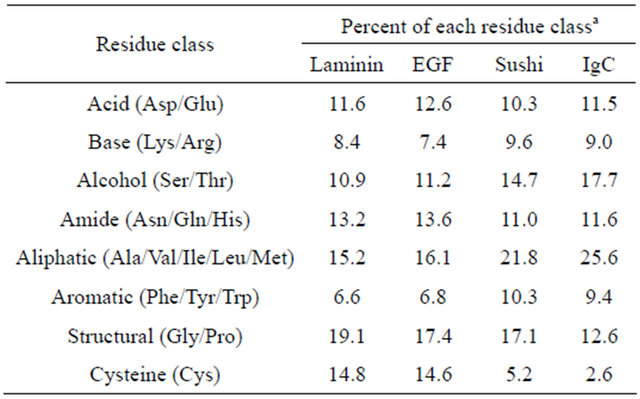
aThe total number of residues identified in each class for each protein family were calculated as a percent of 19,054 total Laminin residues; 62,185 total EGF residues; 27,017 total Sushi residues; and 58,821 total IgC residues, respectively.
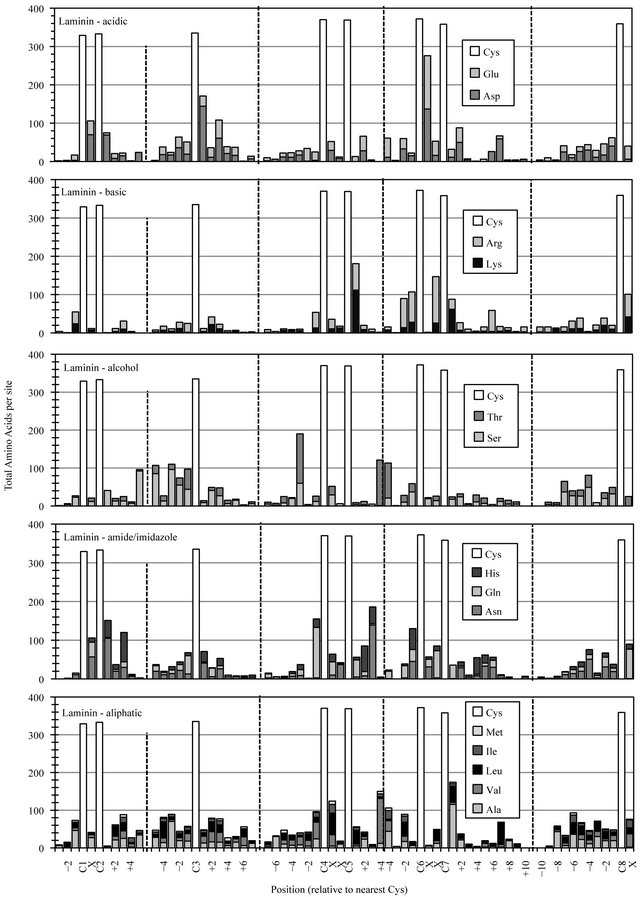
Full-length Laminin motif modules employ eight consistently located cysteines that represent landmarks around which the distribution/accumulation of all other residues was determined (Figure 2(a)). The most notable conservation of amino acid functionality in the Laminin motif dataset was the incorporation of a preferred site for acid residues at Laminin-C6+1, with over two-thirds of Laminin domains employing an aspartic acid (Asp) or glutamic acid (Glu) residue at this location. The accumulation of some other amino acid functional classes, occurring at preferred sites in one-third to one-half of compiled Laminin sequences likely reflect a lesser degree of diversity in a dataset dominated by the three polypeptides of the mature Laminin cruciform superstructure.
The EGF superfamily represents perhaps the largest and most diverse collection of related protein structures in all of nature. A few sites of preferred amino acid chemistry can be observed for most functional group classes (Figure 2(b)). Most notably was the conserved placement of an amide or imidazole group at EGF-C2+2. The accumulation of asparigine (Asn), glutamine (Gln) or histidine (His) at this site occurred in nearly two-thirds of EGF-like domains. Moderate elevation in acid residue content was detected in the regions preceding the initial EGF-C1 and following the final EGF-C6, reflecting an anionic functional group preference in the predicted calcium-binding regions located between successive cbEGF modules in a subset of calcium-binding proteins like Fibrilin [13].
The Sushi domain data demonstrated a slightly higher and more uniform distribution of all amino acid classes (Figure 2(c)). Only a few sites of elevated amino acid preference were observed, most notably, the increased conservation of aliphatic Ile, Leu and Val at Sushi-C2-4, Sushi-C2+6 and Sushi-C3-2 potentially correlate with the presence of fewer cysteines. Over half of all Sushi se-
 (a)
(a)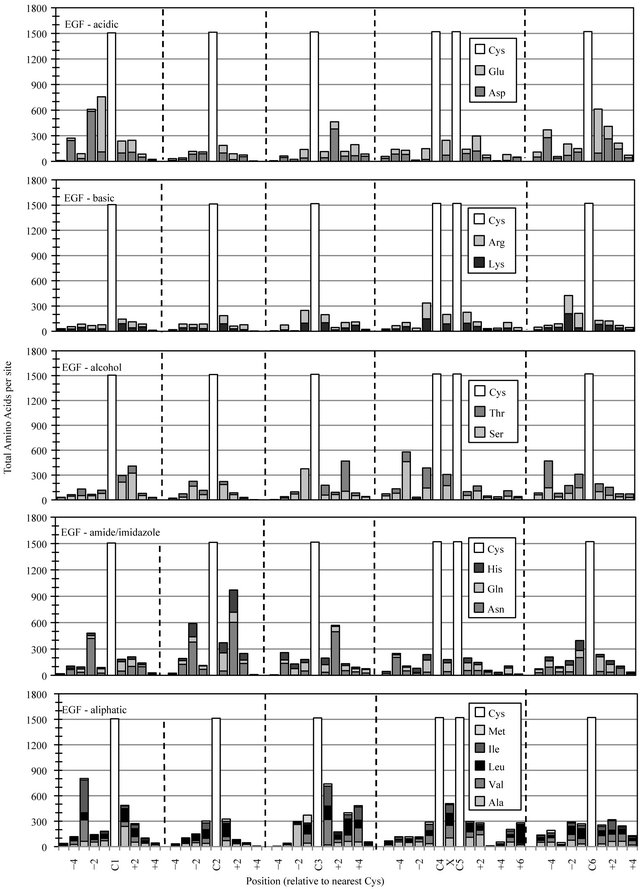 (b)
(b) (c)
(c)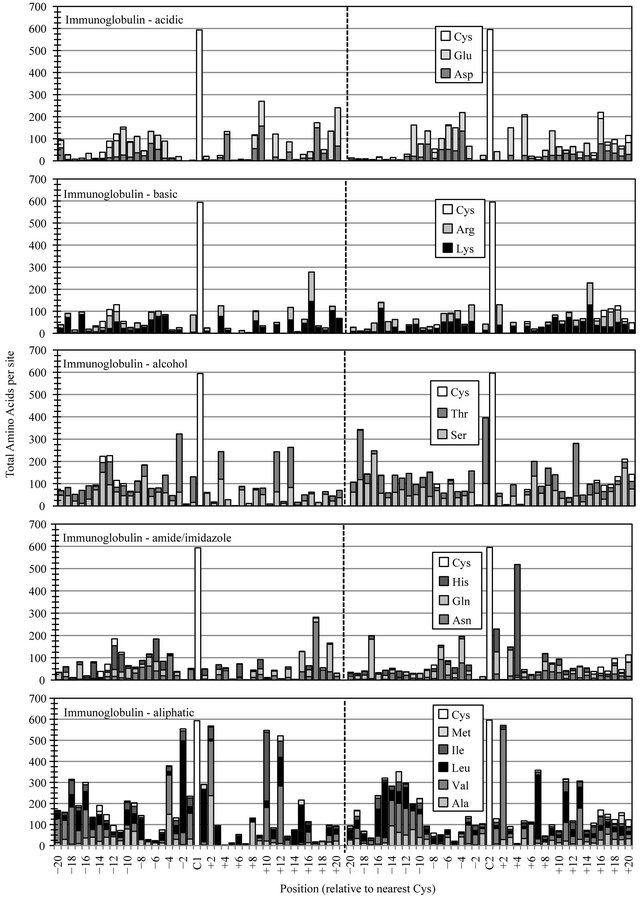 (d)
(d)
Figure 2. Cysteine-based alignment of motif-specific sequence data. Motif critical Cys residues were vertically registered and the distribution of amino acid chemistry determined relative to (a) the eight conserved Cys found in Laminin sequences, (b) the six conserved Cys observed in EGF sequence (c) the four Cys of Sushi and (d) the two Cys conserved in Immunglobulin constant domains. Residue classes, as noted from top panel to bottom panel, were acidic residues Asp and Glu, basic residues Lys and Arg, polar alcohol residues Ser and Thr, polar amide/imidazole residues Asn Gln and His, and aliphatic residues Ala, Val, Leu, Ile and Met.
quences incorporate an amide/imidazole site at SushiC1+8, a similar degree of conservation as observed for this functional class in the EGF family.
Immunoglobulin constant (IgC) domains have the longest peptide chain-length of the four families and a single disulfide (Figure 2(d)). The data presented in this figure was limited to regions nearest the two conserved cysteines (Cys+/−20), and so some internal sequence data is not displayed here. Like each of the other three protein families, the IgC domain showed several sites and classes of elevated amino acid preference in these regions. Continuing the trend, relative to cysteine content, IgC data showed a much higher level of preferred aliphatic chemistry, with five sites showing marked preference for Ile, Leu or Val in seventy to ninety percent of members. Of several sites with elevated preference for polar amino acid chemistry, the most striking conservation was the Ig-C2+4 placement of the His imidazole group in eighty-four percent of IgC domains.
Analysis of the data compiled for aromatic residues showed a strong preference for site-specific accumulation/distribution of phenylalanine (Phe) and tyrosine (Tyr) residues in all four protein families (Figure 3). The restricted deployment of aromatic functionality was reflected across each protein scaffold, with the exception of two sites where Phe and Tyr together occurred in seventy to ninety percent of members from all four protein families. One of these sites showed the identical cysteine-associated construct -C-X-X-X-(F/Y)- at EGF-C5+ 4, Sushi-C2+4 and Laminin-C7+4, with an offset in IgC domains -C-X-X-X-X-(F/Y)-, occurring at IgC-C1+5. The EGF, Sushi and IgC sequence families each exhibited a second high frequency aromatic construct, -(F/Y)-X-C-, restricted to analogous cysteine-associated EGF-C4-2, Sushi-C2-2, and IgC-C2-2. About half of both the Laminin and IgC domain sequences exhibited the double aromatic construct -C-(X)3-4-(F/Y)-(F/Y)-.
4. DISCUSSION
Over time, the usual mechanisms of genetic variation, acting on protein structure-function at the molecular level, have resulted in the duplication, mobilization and adaptation of ancestral EGF, Sushi, Laminin and Immunoglobulin genes to establish the recognized families now widely employed across modern eukaryotic genomes [1,2,14,15]. More than three thousand annotated EGF, Sushi, Laminin and Immunoglobulin sequences, each representing a unique protein ortholog, paralog or other polymorphism were systematically extracted from the protein database. The composition of each of the resulting motif-specific sequence datasets was not manipulated or purposefully constructed in any intentional way, to avoid introducing any preconceived bias into the data, but this otherwise blind approach to assemble the most comprehensive datasets possible does certainly reflect selective trends of past and present sequence discovery efforts. The inclusion of a few highly repetitive or heavily represented parologs might be observable, but minimally affect accumulation data of the size and diversity employed here. To fully appreciate the sequence diversity of relevant protein families, readers are directed to representative alignments of original source data at www.ncbi.nlm.nih.gov/cdd [12].
The method of simple vertical registration has been similarly used in previous tabulation of antibody framework residues in examining large sets of Immunoglobulin variable (IgV) domains [16,17]. The study presented here looked exclusively at amino acid functional group chemistry with respect to actual linear sequence relationships only, unlike algorithm-based alignment methods complicated by the many natural insertions and deletions responsible for variable length inter-cysteine strands. The mechanical splitting of inter-cysteine sequences of different length makes data recorded at sites farther from landmark cysteines generally less significant, especially for the highly variable-length strands of the EGF family. The less variable strands in Sushi, Laminin and IgC families exhibited sequence homology even some distance from relevant cysteines. This method for examining the evolutionary success of four major motif modules has made it possible to illustrate the sequence diversity that has accumulated, as well as to demonstrate the extreme conservation of selected cysteine-associated sites of preferred amino acid chemistry shared within and across modern protein families.
Selective pressure asserted at the molecular level has provided for conservation of those elements of protein structure-function ultimately beneficial to survival, allowing diversity to exist elsewhere in protein primary structure, as long as the biochemical “fitness” of the native protein has not been too severely impaired. It would seem reasonable that in the evolutionary progression of primordial proteomes, the forces of diversification that introduce new function must be balanced by conservation of those elements of protein structure-function necessary for any modified protein to achieve and maintain a viable protein structure and permit the altered molecular entity to be successfully maintained or integrated into existing macromolecular or cellular structures, biochemical processes or physiological pathways [18,19].
While this study identified some preferred amino acid chemistry at a limited number of sites in each of the families examined, the data clearly demonstrate that all four of these disulfide-dependent modular motif structures represent readily diversifiable peptide scaffolds onto which a significant number of differently arranged amino acid chemical functionalities can be deployed to
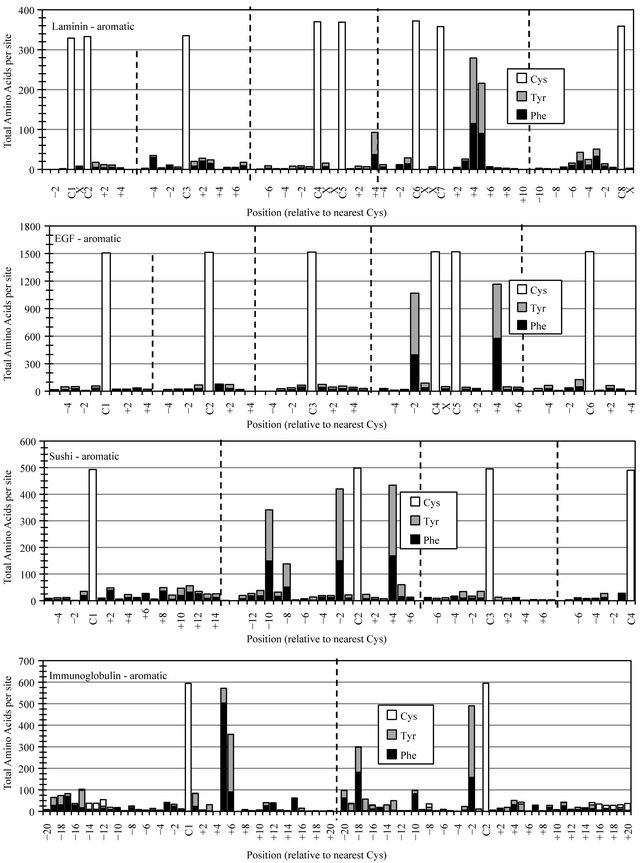
Figure 3. Cysteine-associated distribution of aromatic residues. The cumulative occurrence of phenylalanine(Phe) and tyrosine (Tyr) were plotted relative to landmark cysteines (Cys) found in motif-specific sequence files as noted from top panel to bottom panel, Laminin, EGF, Sushi and Immunoglobulin.
meet the biological purposes of a range of protein superstructures. The events responsible for introducing and maintaining the shared aromatic determinants -C-(X)3-4- (F/Y)- and -(F/Y)-X-Cmust, however, represent an early evolutionary parallel in a diversification/selection process leading to their accumulation in the multitude of protein orthologs and paralogs now comprising these four major protein families. Being conserved in four different modular motif structures, the observed positional restriction of aromatic chemistry must provide beneficial biochemical function beyond the level of individual motif structure and likely has global ramifications for protein structure-function, potentially representing some shared determinant of common regulation, cellular trafficking, or quality control in the folding and assembly of diverse multimeric/multidomain proteins.
Although amino acid sequence conservation is generally thought of in context of mature native protein structure, the possibility that the cysteine-associated distribution of aromatic residues detected in linear protein sequence data might be conserved to intentionally function in nascent protein or other premature non-native protein structures must also be considered. The presence of an appropriate export leader sequence or other extracellular reference was identified for each of the proteins evaluated here, confirming that all of these cysteine-rich proteins are targeted for export via the endoplasmic reticulum (ER), where they would all be subjected to the same protein folding environment and quality control inspection. Analogous positioning of aromatic residues near essential cysteines in multiple extracellular protein families may be relevant to protein folding, quality control or trafficking, in light of evidence suggesting an aromaticbased recognition of ER folding substrates by Protein Disulfide Isomerase [20,21].
We postulate that the Cys-associated placement of aromatic functionality, observed in four major disulfidestabilized protein families, represents the conservation of a common element for either facilitating and/or signaling changes in protein conformation as part of the protein folding/quality control process in the ER. In particular, placement of a conserved aromatic site near essential cysteines might allow one or more of the growing list of ER-resident chaperones, like Disulfide Isomerase, to more easily recognize unfolded or misfolded domains where critical disulfides are either reduced or scrambled, by virtue of interaction with aromatic groups whose positions have been evolutionarily restricted to facilitate detection in non-native folding intermediates. Subsequent achievement of native disulfide configuration and the consequential establishment of a mature protein conformation where exposed aromatic groups become sufficiently concealed from any Phe/Tyr-specific ER binding proteins could contribute to signaling readiness for export.
In theory, the incorporation of a single, simple, generic determinant marking non-native protein structure would constitute the most efficient system for inspecting the enormous variety of disulfide configurations and protein conformations presented by the diverse population of disulfide-dependent protein structures that transit the ER. This capability could be especially important in monitoring the folding/assembly of repetitive or multi-subunit superstructures where the presence of an easily recognized, position-restricted determinant that demarcates and potentially facilitates the segregation of individual modules within these larger proteins during folding and assembly would seem advantageous. If or how such an aromatic “folding sensor” might cooperate with established glyco-based protein inspection systems should be considered.
REFERENCES
- Henikoff, S., Greene, E.A., Pietrokovski, S., Bork, P., Attwood, T.K. and Hood, L. (1997) Gene families: The taxonomy of protein paralogs and chimeras. Science, 278, 609-614. doi:10.1126/science.278.5338.609
- Chothia, C., Gough, J., Vogel, C. and Teichmann, S.A. (2003) Evolution of the protein repertoire. Science, 300, 1701-1703. doi:10.1126/science.1085371
- Appella, E., Weber, I.T. and Blasi, F. (1988) Structure and function of epidermal growth factor-like regions in proteins. FEBS Letters, 231, 1-4. doi:10.1016/0014-5793(88)80690-2
- Bork, P., Downing, A.K., Kieffer, B. and Campbell, I.D. (1996) Structure and distribution of modules in extracellular proteins. Quarterly Reviews of Biophysics, 29, 119- 167. doi:10.1017/S0033583500005783
- Kirkitadze, M.D. and Barlow, P.N. (2001) Structure and flexibility of the multiple domain proteins that regulate complement activation. Immunological Reviews, 180, 146-161. doi:10.1034/j.1600-065X.2001.1800113.x
- Hegyi, H. and Bork, P. (1997) On the classification and evolution of protein modules. Journal of Protein Chemistry, 16, 545-551. doi:10.1023/A:1026382032119
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. Journal of Molecular Biology, 215, 403-410.
- Altschul, S.F, Madden, T.L., Schäffer, A.A, Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Research, 25, 3389-3402. doi:10.1093/nar/25.17.3389
- Yu, Y.K., Gertz, E.M., Agarwala, R., Schäffer, A.A. and Altschul, S.F. (2006) Retrieval accuracy, statistical significance and compositional similarity in protein sequence database searches. Nucleic Acids Research, 34, 5966-5973. doi:10.1093/nar/gkl731
- Tatusova, T. (2010) Genomic databases and resources at the National Center for Biotechnology Information. Methods in Molecular Biology, 609, 17-44. doi:10.1007/978-1-60327-241-4_2
- Sayers, E.W., Barrett, T., Benson, D.A., Bolton, E., Bryant, S.H., Canese, K., Chetvernin, V., Church, D.M., Dicuccio, M., Federhen, S., Feolo, M., Geer, L.Y., Helmberg, W., Kapustin, Y., Landsman, D., Lipman, D.J., Lu, Z., Madden, T.L., Madej, T., Maglott, D.R., Marchler-Bauer, A., Miller, V., Mizrachi, I., Ostell, J., Panchenko, A., Pruitt, K.D., Schuler, G.D., Sequeira, E., Sherry, S.T., Shumway, M., Sirotkin, K., Slotta, D., Souvorov, A., Starchenko, G., Tatusova, T.A., Wagner, L., Wang, Y., John, W., Yaschenko, E. and Ye, J. (2010) Database resources of the National Center for Biotechnology Information. Nucleic Acids Research, 38, D5-D16. doi:10.1093/nar/gkp967
- Marchler-Bauer, A., Anderson, J.B., Chitsaz, F., Derbyshire, M.K., DeWeese-Scott, C., Fong, J.H., Geer, L.Y., Geer, R.C., Gonzales, N.R., Gwadz, M., He, S., Hurwitz, D.I., Jackson, J.D., Ke, Z., Lanczycki, C.J., Liebert, C.A., Liu, C., Lu, F., Lu, S., Marchler, G.H., Mullokandov, M., Song, J.S., Tasneem, A., Thanki, N., Yamashita, R.A., Zhang, D., Zhang, N. and Bryant, S.H. (2009) CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Research, 37, D205-D210. doi:10.1093/nar/gkn845
- Downing, A.K., Knott, V., Werner, J.M., Cardy, C.M., Campbell, I.A. and Handford, P.A. (1996) Solution structure of a pair of Calcium-binding epidermal growth factor-like domains: Implications for the Marfan Syndrome and other genetic disorders. Cell, 85, 597-605. doi:10.1016/S0092-8674(00)81259-3
- Buljan, M. and Bateman, A. (2009) The evolution of protein families. Biochemical Society Transactions, 37, 751-755. doi:10.1042/BST0370751
- Worth, C.L., Gong, S. and Blundell, T.L. (2009) Structural and functional constraints in the evolution of protein families. Nature Reviews Molecular Cell Biology, 10, 709-720.
- Padlan, E.A. (1994) Anatomy of the antibody molecule. Molecular Immunology, 31, 169-217. doi10.1016/0161-5890(94)90001-9
- Chothia, C., Gelfand, I. and Kister, A. (1998) Structural determinants in the sequences of immunoglobulin variable domain. Journal of Molecular Biology, 278, 457-479. doi:10.1006/jmbi.1998.1653
- Andreeya, A. and Murzin, A.G. (2006) Evolution of protein fold in the presence of functional constraints. Current Opinion in Structural Biology, 16, 399-408. doi:10.1016/j.sbi.2006.04.003
- Gong, S., Worth, C.L., Bickerton, G.R., Lee, S., Tanramluk, D. and Blundell, T.L. (2009) Structural and functional restraints in the evolution of protein families and superfamilies. Biochemical Society Transactions, 37, 727-733. doi:10.1042/BST0370727
- Ruddock, L.W., Freedman, R.B. and Klappa, P. (2000) Specificity in substrate binding by protein folding catalysts: Tyrosine and tryptophan residues are the recognition motifs for the binding of peptides to the pancreasspecific protein disulfide isomerase PDIp. Protein Science, 9, 758-764. doi:10.1110/ps.9.4.758
- Klappa, P., Freedman, R.B., Langenbuch, M., Lan, M.S., Robinson, G.K. and Ruddock, L.W. (2001) The pancreasspecific protein disulphide-isomerase PDIp interacts with a hydroxyaryl group in ligands. Biochemical Journal, 15, 553-559. doi:10.1042/0264-6021:3540553