 J. Biomedical Science and Engineering, 2010, 3, 59-64 doi:10.4236/jbise.2010.31009 Published Online January 2010 (http://www.SciRP.org/journal/jbise/ JBiSE ). Published Online January 2010 in SciRes. http://www.scirp.org/journal/jbise Identifying predictive markers of chemosensitivity of breast cancer with random forests Wei Hu Department of Computer Science, Houghton College, Houghton, NY, USA. Email: wei.hu@houghton.edu Received 15 September 2009; revised 10 October 2009; accepted 20 October 2009. ABSTRACT Several gene signatures have been identified to build predictors of chemosensitivity for breast cancer. It is crucial to understand how each gene in a signature contributes to the prediction, i.e., to make the predic- tion model interpretable instead of using it as a black box. We utilized Random Forests (RFs) to build two interpretable predictors of pathologic complete re- sponse (pCR) based on two gene signatures. One sig- nature consisted of the top 31 probe sets (27 genes) differentially expressed between pCR and residual disease (RD) chosen from a previous study, and the other consisted of the genes involved in Notch sin- gling pathway (113 genes). Both predictors had a higher accuracy (82% v 76% & 79% v 76%), a higher specificity (91% v 71% & 98% v 71%), and a higher positive predictive value (PPV) (68% v 52% & 73% v 52%)) than the predictor in the previous study. Furthermore, Random Forests were employed to calculate the importance of each gene in the two sig- natures. Findings of our functional annotation sug- gested that the important genes identified by the fea- ture selection scheme of Random Forests are of bio- logical significance. Keywords: Random Forests; Breast Cancer; Chemosen- sitivity; Gene Signature; Notch Signaling Pathway; Pathologic Complete response; Predictor 1. INTRODUCTION Breast cancer is a clinically heterogeneous disease that demonstrates a wide variation in its clinical courses and response to chemotherapy. This complexity is a reflec- tion of the molecular oncogenic aberration in DNA re- pair, cell cycle control, cell survival, and signal trans- duction in breast tumors. Microarray analysis has identi- fied breast cancer subtypes with distinct gene expression profiles and clinical behavior [1,2,3]. There are several major molecular classes of breast cancers indentified by different research groups. Some studies [2,3] suggested five major classes of breast cancer: normal breast-like, luminal-A, luminal-B, basal-like, and human epidermal growth factor receptor 2 (HER2)-positive cancers. An- other study [4] proposed three major classes: ER+/ HER2-, ER-/HER2-, and HER2+. The heterogeneity of breast cancer characterized by these subtypes brings great challenge to its research. In a significant proportion of breast cancer patients, chemotherapy does not result in response, but can induce significant side effects and financial costs. The ability to identify predictors of re- sponse or resistance to cancer drugs will provide better treatment to the individual patient. Several studies have suggested that the gene-exp- ression profiles of chemo sensitive tumors are different from those of chemo resistant ones [5]. Gene expression profiling with a measurement of thousands of mRNA transcripts in a single experiment is widely used in hu- man cancer research. Due to the high dimensionality of microarray data, a feature selection step to find a subset of discriminative genes, referred to as a signature, is often necessary for building robust predictors [6,7]. Ayers et al. [8] developed a multigene predictor of pCR to sequential weekly paclitaxel and FAC (T/FAC) neoadjuvant chemotherapy for breast cancer patients. The study involved 42 patients: 24 patients were used in the training set and 18 patients in the validation set. pCR was obtained in 13 patients (31%). A gene set of 74 markers (P < 0.09) was built using data from the training set and tested on the validation set. Overall, a 78% pre- dictive accuracy was achieved, with a 100% positive predictive value for pCR, a 73% negative predictive value, a sensitivity of 43%, and a specificity of 100%. Later, a follow-up study [9] included 133 patients with stage I-III breast cancer, with a pCR rate of 26% (n=34). A 30-probe set Diagonal Linear Discriminant Analysis (DLDA-30) classifier was selected for independent vali- dation. It showed a significantly higher sensitivity (92% v 61%) than a clinical predictor including age, grade, and estrogen receptor status. This 30-probe set pharma- cogenomic predictor correctly identified all but one of the patients who achieved pCR (12 of 13 patients) and all but one of those who were predicted to have residual disease had residual cancer (27 of 28 patients).  W. Hu / J. Biomedical Science and Engineering 3 (2010) 59-64 SciRes Copyright © 2010 JBiSE 60 Chemosensitivity is better predicted by multigene signatures than by a single molecular discrimination because biological phenomena occur through the con- certed expression of multiple genes [10,11,12]. However, within a signature of genes, the important question of how each individual gene contributes to the prediction has not been studied. We attempted in this work to iden- tify predictors and gene signatures that have better pre- diction performance than the DLDA-30 and to quantify the importance of each gene in a signature in the predic- tion of pCR. In [9], an exhaustive search of a good predictor of pCR was conducted. Different machine learning tech- niques were tested including support vector machines with linear, radial, and polynomial kernels (SVM), Di- agnal Linear Discriminant Analysis (DLDA), and K- nearest neighbor (KNN) using Euclidean distance. One interesting discovery was that SVM provided the worst performance of pCR prediction among all these different techniques in this particular data set. Random Forest has demonstrated its comparable performances to SVM in many bioinformatics applications. In the current study, we sought to explore the utility of Random Forests were utilized to construct two predictors based on two signa- tures, the top 31 probe sets and the Notch signature, and take advantage of the feature selection capability of Ran- dom Forests to measure the importance of each gene in these signatures. 2. MATERIALS AND METHODS 2.1. Patient Cohorts and Clinical Information One breast cancer patient cohort was obtained from a previous publication [9] (n=133). Needle-biopsy sam- ples were collected from 133 patients with stage I, II, or III breast cancer who received preoperative weekly pa- clitaxel and a combination of fluorouracil, doxorubicin, and cyclophosphamide (T/FAC). These 133 patients were divided into two subsets, one training set of size 81 and one validation set of size 52. These data contain clinical information including patient age, gender, race, histological classification, stage, nuclear grade, ER (es- trogen receptor), PR (progesterone receptor), and HER2 (human epidermal growth factor 2) status, pathologic complete response, and residual disease. These data also contain each patient’s genome-scale gene expression profiles generated using Affymetrix U133A chip (Santa Clara, CA). pCR was defined as no residual invasive cancer in the breast or lymph nodes. pCR is presently accepted as a reasonable early indicator for long-term survival. 2.2. Top 31 Probe Set Signature To build a predictor of pCR, the genes that are highly expressed in either the pCR cases or the RD cases need to be identified. To achieve this goal, t-tests for unequal variances for all the probe sets on Affymetrix U133A chip were carried out. The 31 probe sets (27 genes) with the smallest t-test P values (FDR=0.05%) were selected in [9], which was used as our first signature. 2.3. Notch Signature Notch genes encode highly conserved cell surface re- ceptors. The Notch signaling pathway consists of Notch receptors, ligands, negative and positive modifiers, and transcription factors. It plays a key role in the normal development of many tissues and cell types, through diverse effects on cell regulation, proliferation, and dif- ferentiation. Aberrant Notch signaling has been observed in several human cancers including acute T-cell lym- phoblastic leukemia and cervical cancer. Recent evi- dences implied that it might be associated with breast cancer [13,14]. Selecting a gene signature based on differentially ex- pressed genes between two conditions, such as pCR and RD in our study, is a common strategy nowadays. Here we endeavored to take a quite different approach, i.e., to identify a signature of genes involved in a particular pathway that has a key impact on human cancers. The Oligo GEArray Human Notch Signaling Pathway Microarray [15] was designed for profiling expression of 113 genes involved in Notch signaling. Our second sig- nature was these 113 genes as shown in Table 1. We were particularly interested in uncovering what genes in these two signatures are important for the pre- diction of pCR and what biological or medical signifi- cance they might have. 2.4. False Discovery Rate The standard P value was designed for testing individual hypotheses. When applied in a multiple testing problem such as selecting informative genes in microarray data, it may result in many false positives. While there are a number of methods to overcome the problems due to mul- tiple testing, False Discovery Rate (FDR) approach [16,17] was used to help select the top 31 probe sets in [9]. 2.5. Random Forests Random Forest, proposed by Leo Breiman in 1999 [18], is an ensemble classifier based on many decision trees. Each tree is built on a bootstrap sample from the original training set. The variables used for splitting the tree nodes are a random subset of the whole variable set. The classification decision of a new instance is made by ma- jority voting over all trees. About one-third of the in- stances are left of the bootstrap sample and not used in the construction of the tree. These instances in the train- ing set are called “out-of-bag” instances and are used to evaluate the performance of the classifier. 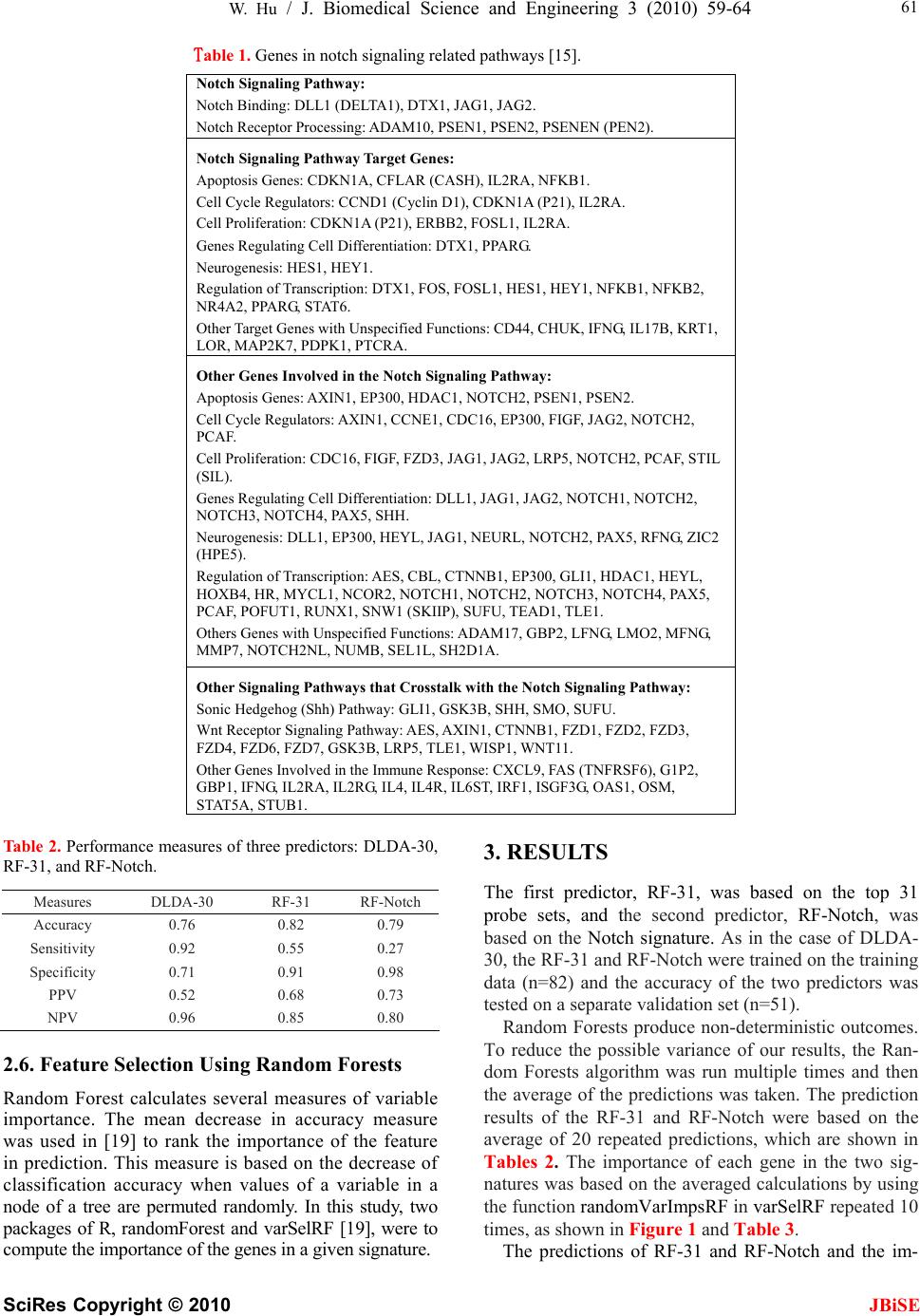 W. Hu / J. Biomedical Science and Engineering 3 (2010) 59-64 SciRes Copyright © 2010 JBiSE 61 Table 1. Genes in notch signaling related pathways [15]. Notch Signaling Pathway: Notch Binding: DLL1 (DELTA1), DTX1, JAG1, JAG2. Notch Receptor Processing: ADAM10, PSEN1, PSEN2, PSENEN (PEN2). Notch Signaling Pathway Target Genes: Apoptosis Genes: CDKN1A, CFLAR (CASH), IL2RA, NFKB1. Cell Cycle Regulators: CCND1 (Cyclin D1), CDKN1A (P21), IL2RA. Cell Proliferation: CDKN1A (P21), ERBB2, FOSL1, IL2RA. Genes Regulating Cell Differentiation: DTX1, PPARG. Neurogenesis: HES1, HEY1. Regulation of Transcription: DTX1, FOS, FOSL1, HES1, HEY1, NFKB1, NFKB2, NR4A2, PPARG, STAT6. Other Target Genes with Unspecified Functions: CD44, CHUK, IFNG, IL17B, KRT1, LOR, MAP2K7, PDPK1, PTCRA. Other Genes Involved in the Notch Signaling Pathway: Apoptosis Genes: AXIN1, EP300, HDAC1, NOTCH2, PSEN1, PSEN2. Cell Cycle Regulators: AXIN1, CCNE1, CDC16, EP300, FIGF, JAG2, NOTCH2, PCAF. Cell Proliferation: CDC16, FIGF, FZD3, JAG1, JAG2, LRP5, NOTCH2, PCAF, STIL (SIL). Genes Regulating Cell Differentiation: DLL1, JAG1, JAG2, NOTCH1, NOTCH2, NOTCH3, NOTCH4, PAX5, SHH. Neurogenesis: DLL1, EP300, HEYL, JAG1, NEURL, NOTCH2, PAX5, RFNG, ZIC2 (HPE5). Regulation of Transcription: AES, CBL, CTNNB1, EP300, GLI1, HDAC1, HEYL, HOXB4, HR, MYCL1, NCOR2, NOTCH1, NOTCH2, NOTCH3, NOTCH4, PAX5, PCAF, POFUT1, RUNX1, SNW1 (SKIIP), SUFU, TEAD1, TLE1. Others Genes with Unspecified Functions: ADAM17, GBP2, LFNG, LMO2, MFNG, MMP7, NOTCH2NL, NUMB, SEL1L, SH2D1A. Other Signaling Pathways that Crosstalk with the Notch Signaling Pathway: Sonic Hedgehog (Shh) Pathway: GLI1, GSK3B, SHH, SMO, SUFU. Wnt Receptor Signaling Pathway: AES, AXIN1, CTNNB1, FZD1, FZD2, FZD3, FZD4, FZD6, FZD7, GSK3B, LRP5, TLE1, WISP1, WNT11. Other Genes Involved in the Immune Response: CXCL9, FAS (TNFRSF6), G1P2, GBP1, IFNG, IL2RA, IL2RG, IL4, IL4R, IL6ST, IRF1, ISGF3G, OAS1, OSM, STAT5A, STUB1. Table 2. Performance measures of three predictors: DLDA-30, RF-31, and RF-Notch. Measures DLDA-30 RF-31 RF-Notch Accuracy 0.76 0.82 0.79 Sensitivity 0.92 0.55 0.27 Specificity 0.71 0.91 0.98 PPV 0.52 0.68 0.73 NPV 0.96 0.85 0.80 2.6. Feature Selection Using Random Forests Random Forest calculates several measures of variable importance. The mean decrease in accuracy measure was used in [19] to rank the importance of the feature in prediction. This measure is based on the decrease of classification accuracy when values of a variable in a node of a tree are permuted randomly. In this study, two packages of R, randomForest and varSelRF [19], were to compute the importance of the genes in a given signature. 3. RESULTS The first predictor, RF-31, was based on the top 31 probe sets, and the second predictor, RF-Notch, was based on the Notch signature. As in the case of DLDA- 30, the RF-31 and RF-Notch were trained on the training data (n=82) and the accuracy of the two predictors was tested on a separate validation set (n=51). Random Forests produce non-deterministic outcomes. To reduce the possible variance of our results, the Ran- dom Forests algorithm was run multiple times and then the average of the predictions was taken. The prediction results of the RF-31 and RF-Notch were based on the average of 20 repeated predictions, which are shown in Tables 2. The importance of each gene in the two sig- natures was based on the averaged calculations by using the function randomVarImpsRF in varSelRF repeated 10 times, as shown in Figure 1 and Table 3. The predictions of RF-31 and RF-Notch and the im- 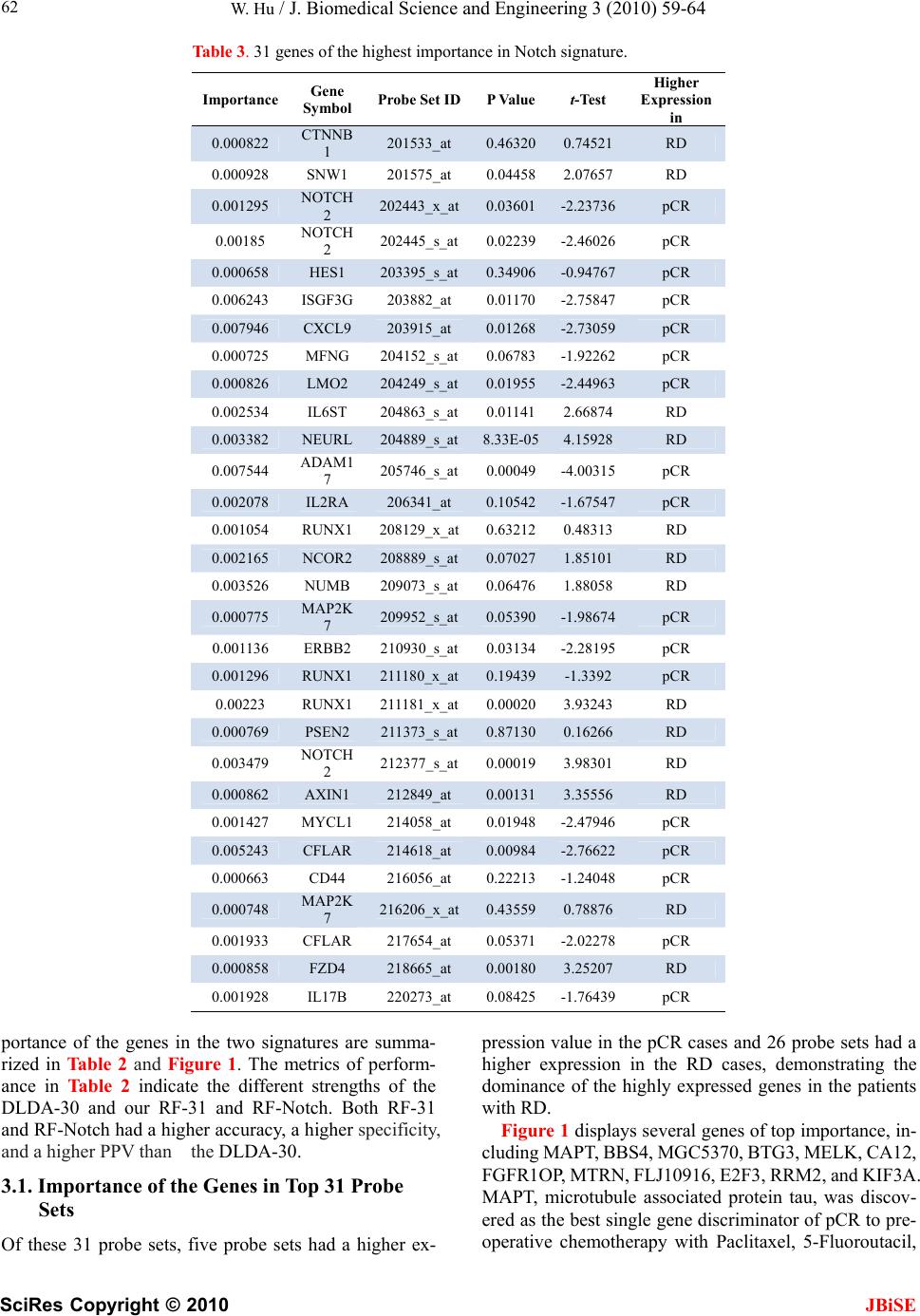 W. Hu / J. Biomedical Science and Engineering 3 (2010) 59-64 SciRes Copyright © 2010 JBiSE 62 Table 3. 31 genes of the highest importance in Notch signature. Importance Gene Symbol Probe Set IDP Valuet-Test Higher Expression in 0.000822 CTNNB 1 201533_at 0.46320 0.74521RD 0.000928 SNW1 201575_at 0.044582.07657RD 0.001295 NOTCH 2 202443_x_at 0.03601 -2.23736pCR 0.00185 NOTCH 2 202445_s_at0.02239-2.46026pCR 0.000658 HES1 203395_s_at 0.34906 -0.94767pCR 0.006243 ISGF3G 203882_at 0.01170-2.75847pCR 0.007946 CXCL9 203915_at 0.01268 -2.73059pCR 0.000725 MFNG 204152_s_at0.06783-1.92262pCR 0.000826 LMO2 204249_s_at 0.01955 -2.44963pCR 0.002534 IL6ST 204863_s_at0.011412.66874RD 0.003382 NEURL 204889_s_at 8.33E-05 4.15928RD 0.007544 ADAM1 7 205746_s_at0.00049-4.00315pCR 0.002078 IL2RA 206341_at 0.10542 -1.67547pCR 0.001054 RUNX1 208129_x_at0.632120.48313RD 0.002165 NCOR2 208889_s_at 0.07027 1.85101RD 0.003526 NUMB 209073_s_at0.064761.88058RD 0.000775 MAP2K 7 209952_s_at 0.05390 -1.98674pCR 0.001136 ERBB2 210930_s_at0.03134-2.28195pCR 0.001296 RUNX1 211180_x_at0.19439-1.3392 pCR 0.00223 RUNX1 211181_x_at0.000203.93243RD 0.000769 PSEN2 211373_s_at 0.87130 0.16266RD 0.003479 NOTCH 2 212377_s_at0.000193.98301RD 0.000862 AXIN1 212849_at 0.00131 3.35556RD 0.001427 MYCL1 214058_at 0.01948-2.47946pCR 0.005243 CFLAR 214618_at 0.00984 -2.76622pCR 0.000663 CD44 216056_at 0.22213-1.24048pCR 0.000748 MAP2K 7 216206_x_at 0.43559 0.78876RD 0.001933 CFLAR 217654_at 0.05371-2.02278pCR 0.000858 FZD4 218665_at 0.00180 3.25207RD 0.001928 IL17B 220273_at 0.08425-1.76439pCR portance of the genes in the two signatures are summa- rized in Table 2 and Figure 1. The metrics of perform- ance in Table 2 indicate the different strengths of the DLDA-30 and our RF-31 and RF-Notch. Both RF-31 and RF-Notch had a higher accuracy, a higher specificity, and a higher PPV than the DLDA-30. 3.1. Importance of the Genes in Top 31 Probe Sets Of these 31 probe sets, five probe sets had a higher ex- pression value in the pCR cases and 26 probe sets had a higher expression in the RD cases, demonstrating the dominance of the highly expressed genes in the patients with RD. Figure 1 displays several genes of top importance, in- cluding MAPT, BBS4, MGC5370, BTG3, MELK, CA12, FGFR1OP, MTRN, FLJ10916, E2F3, RRM2, and KIF3A. MAPT, microtubule associated protein tau, was discov- ered as the best single gene discriminator of pCR to pre- operative chemotherapy with Paclitaxel, 5-Fluoroutacil, 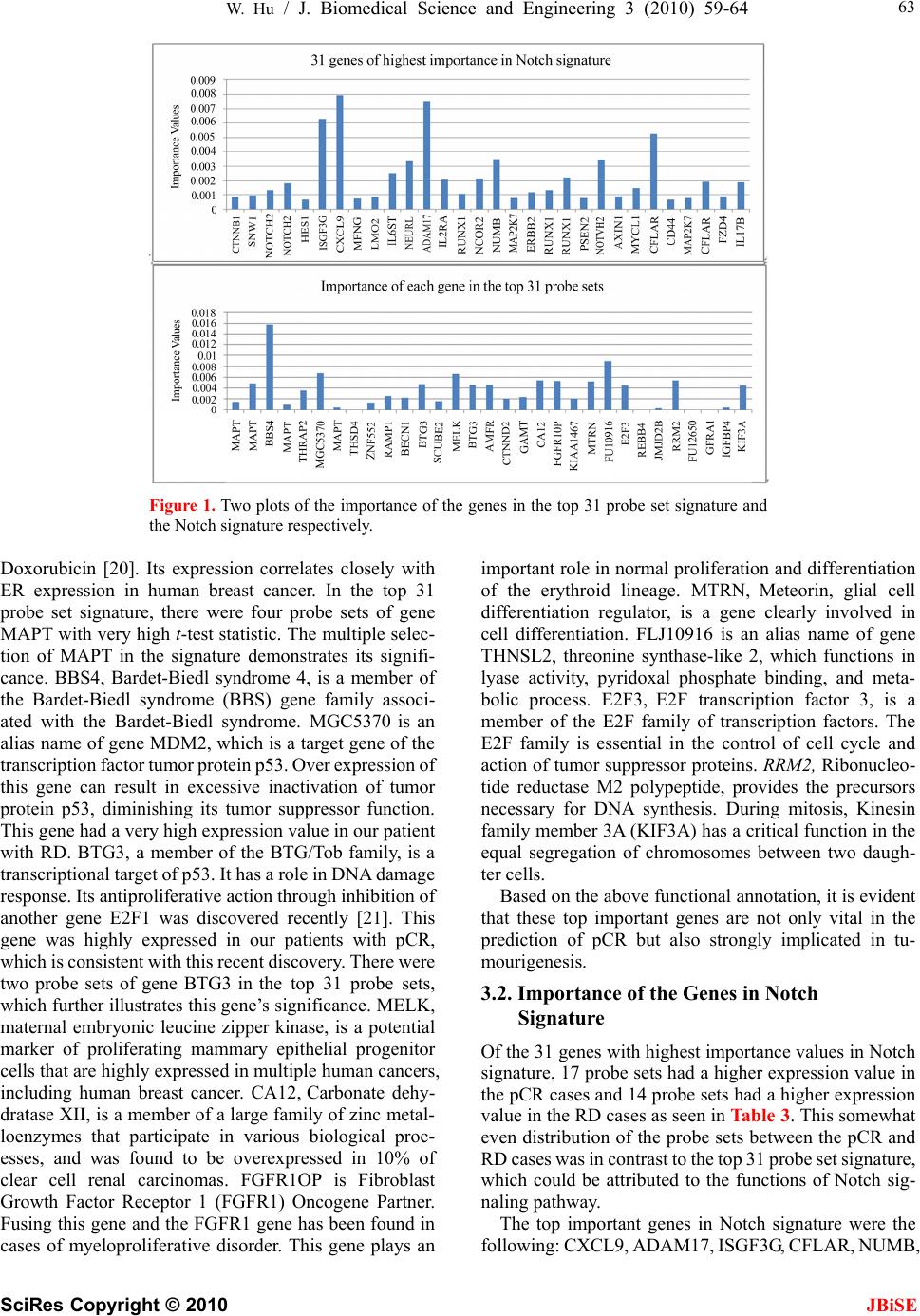 W. Hu / J. Biomedical Science and Engineering 3 (2010) 59-64 SciRes Copyright © 2010 JBiSE 63 Figure 1. Two plots of the importance of the genes in the top 31 probe set signature and the Notch signature respectively. Doxorubicin [20]. Its expression correlates closely with ER expression in human breast cancer. In the top 31 probe set signature, there were four probe sets of gene MAPT with very high t-test statistic. The multiple selec- tion of MAPT in the signature demonstrates its signifi- cance. BBS4, Bardet-Biedl syndrome 4, is a member of the Bardet-Biedl syndrome (BBS) gene family associ- ated with the Bardet-Biedl syndrome. MGC5370 is an alias name of gene MDM2, which is a target gene of the transcription factor tumor protein p53. Over expression of this gene can result in excessive inactivation of tumor protein p53, diminishing its tumor suppressor function. This gene had a very high expression value in our patient with RD. BTG3, a member of the BTG/Tob family, is a transcriptional target of p53. It has a role in DNA damage response. Its antiproliferative action through inhibition of another gene E2F1 was discovered recently [21]. This gene was highly expressed in our patients with pCR, which is consistent with this recent discovery. There were two probe sets of gene BTG3 in the top 31 probe sets, which further illustrates this gene’s significance. MELK, maternal embryonic leucine zipper kinase, is a potential marker of proliferating mammary epithelial progenitor cells that are highly expressed in multiple human cancers, including human breast cancer. CA12, Carbonate dehy- dratase XII, is a member of a large family of zinc metal- loenzymes that participate in various biological proc- esses, and was found to be overexpressed in 10% of clear cell renal carcinomas. FGFR1OP is Fibroblast Growth Factor Receptor 1 (FGFR1) Oncogene Partner. Fusing this gene and the FGFR1 gene has been found in cases of myeloproliferative disorder. This gene plays an important role in normal proliferation and differentiation of the erythroid lineage. MTRN, Meteorin, glial cell differentiation regulator, is a gene clearly involved in cell differentiation. FLJ10916 is an alias name of gene THNSL2, threonine synthase-like 2, which functions in lyase activity, pyridoxal phosphate binding, and meta- bolic process. E2F3, E2F transcription factor 3, is a member of the E2F family of transcription factors. The E2F family is essential in the control of cell cycle and action of tumor suppressor proteins. RRM2, Ribonucleo- tide reductase M2 polypeptide, provides the precursors necessary for DNA synthesis. During mitosis, Kinesin family member 3A (KIF3A) has a critical function in the equal segregation of chromosomes between two daugh- ter cells. Based on the above functional annotation, it is evident that these top important genes are not only vital in the prediction of pCR but also strongly implicated in tu- mourigenesis. 3.2. Importance of the Genes in Notch Signature Of the 31 genes with highest importance values in Notch signature, 17 probe sets had a higher expression value in the pCR cases and 14 probe sets had a higher expression value in the RD cases as seen in Table 3. This somewhat even distribution of the probe sets between the pCR and RD cases was in contrast to the top 31 probe set signature, which could be attributed to the functions of Notch sig- naling pathway. The top important genes in Notch signature were the following: CXCL9, ADAM17, ISGF3G, CFLAR, NUMB,  W. Hu / J. Biomedical Science and Engineering 3 (2010) 59-64 SciRes Copyright © 2010 JBiSE 64 NOTCH2, NEURL, IL6ST, and RUNX1. NOTCH2 is a multifunctional gene involved in apoptosis, cell prolif- eration, cell differentiation, neurogenesis, and regulation of transcription. There are three probe sets of NOTCH2 and two probe sets of CFLAR in Figure 1, reflecting these genes’ importance. CXCL9, ISGF3G, and IL6ST are all involved in immune response. Since the functions of these genes are illustrated through their pathways in Ta- ble 1, we will not elaborate on them any further here. There were 15 genes in the top 31 probe set signature with importance values above 0.003, and there were seven such genes in Notch signature. This was expected. Because of their high t-test statistics, the top 31 probe sets should be more sensitive to the random permutation employed in the importance calculation than those in the Notch signature. Nonetheless, in Figure 1 the Notch signature genes displayed their significance. 4. CONLUSIONS Random Forests were employed to study the prediction of pathologic complete response in breast cancer, and the results improved the predictions of the DLDA-30. Func- tional annotation demonstrated that the important genes identified by the feature selection scheme of Random Forests are of biological significance. 5. ACKNOWLEDGMENT We thank Houghton College for its financial support. REFERENCES [1] Nguyen, P. L., Taghian, A. G. et al. (2008) Breast cancer subtype approximated by estrogen receptor, progesterone receptor, and HER-2 is associated with local and distant recurrence after breast-conserving therapy, J. Clin Oncol., 26(14), 2373-8. [2] Perou, C. M., Sorlie, T., et al. (2000)Molecular portraits of human breast tumours, Nature, 406(6797), 747-752. [3] Sorlie, T., Perou, C. M. et al. (2001) Gene expression patterns of breast carcinomas distinguish tumor sub- classes with clinical implications, Proc Natl Acad Sci U S A, 98(19), 10869-10874. [4] Kapp, A. V., Jeffrey, S. S. et al. (2006) Discovery and validation of breast cancer subtypes. BMC Genomics, 7, 231. [5] Pusztai, L., (2008) Current status of prognostic profiling in breast cancer, The Oncologist, 13, 350-360. [6] van ’t Veer, L. J., Dai, H., et al. (2002) Gene expression profiling predicts clinical outcome of breast cancer. Na- ture, 415, 530-536. [7] van de Vijver, M. J. et al. (2002) A gene-expression sig- nature as a predictor of survival in breast cancer. N Engl J Med, 347, 1999-2009. [8] Ayers, M., Symmans, W. F., Stec, J. et al. (2004) Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel/FAC chemotherapy in breast cancer. J Clin Oncol, 22, 2284-2293. [9] Hess, K. R., Anderson, K., Symmans, W. F. et al., (2006) Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxoru- bicin, and cyclophosphamide in breast cancer. J Clin Oncol, 24, 4236-4244. [10] Brenton, J. D., Carey L. A., Ahmed, A. A. et al. (2005) Molecular classification and molecular forecasting of breast cancer: ready for clinical application? J Clin On- col, 23, 7350-7360. [11] Wang, Y., Klijn, J. G. et al. (2005) Gene-expression pro- files to predict distant metastasis of lymph-node-negative primary breast cancer, Lancet, 365, 671-679. [12] Ma, X. J., Wang, Z., et al. (2004) A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell, 5(6), 607-16. [13] Brennan, K. and Anthony Brown, M. C. (2003) Is there a role for Notch signalling in human breast cancer? Breast Cancer Res, 5(2), 69-75. [14] Stylianou, S., Clarke, R. B. et al. (2006) Activation of notch signaling in human breast cancer, Cancer Research, 66, 1517-1525. [15] GEArray, O. Human notch signaling pathway microarray. http://www.sabiosciences.com/gene_array_product/HTM L/OHS-059.html [16] Benjamini, Y. and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol, 57, 289-300. [17] Pounds, S. and Morris, S. W. (2003) Estimating the oc- currence of false positive and false negatives in microar- ray studies by approximating and partitioning the em- pirical distribution of p-values. Bioinformatics 19, 1236- 1242. [18] Breiman, L. and Random, F. (2001) Machine Learning, 45 (1), 5-32. [19] Díaz-Uriarte, R. and Alvarez de Andrés, S. (2006) Gene selection and classification of microarray data using random forest. BMC Bioinformatics, 7(3). [20] Rouzier, R., Rajan, R., et al. (2005) Microtubule associ- ated protein tau is a predictive marker and modulator of response to paclitaxel-containing preoperative chemo- therapy in breast cancer. Proc Natl Acad Sci U S A, 102, 8315-8320. [21] Ou, Y. H., Chung, P. H., et al. (2007)The candidate tumor suppressor BTG3 is a transcriptional target of p53 that inhibits E2F1, The EMBO Journal, 26, 3968-3980.
|