 J. Biomedical Science and Engineering, 2010, 3, 1-12 doi:10.4236/jbise.2010.31001 Published Online December 2009 (http://www.SciRP.org/journal/jbise/ JBiSE ). Published Online December 2010 in SciRes. http://www.scirp.org/journal/jbise The interaction between the 2009 H1N1 influenza A hemagglutinin and neuraminidase: mutations, co-mutations, and the NA stalk motifs Wei Hu Department of Computer Science, Houghton College, Houghton, NY, USA. Email: wei.hu@houghton.edu Received 29 October 2009; revised 6 November 2009; accepted 9 November 2009. ABSTRACT As the world is closely watching the current 2009 H1N1 pandemic unfold, there is a great interest and need in understanding its origin, genetic structures, virulence, and pathogenicity. The two surface proteins, hemagglutinin (HA) and neuraminidase (NA), of the influenza virus have been the focus of most flu re- search due to their crucial biological functions. In our previous study on 2009 H1N1, three aspects of NA were investigated: the mutations and co-mutations, the stalk motifs, and the phylogenetic analysis. In this study, we turned our attention to HA and the interac- tion between HA and NA. The 118 mutations of 2009 H1N1 HA were found and mapped to the 3D homology model of H1, and the mutations on the five epitope regions on H1 were identified. This information is es- sential for developing new drugs and vaccine. The dis- tinct response patterns of HA to the changes of NA stalk motifs were discovered, illustrating the functional dependence between HA and NA. With help from our previous results, two co-mutation networks were un- covered, one in HA and one in NA, where each muta- tion in one network co-mutates with the mutations in the other network across the two proteins HA and NA. These two networks residing in HA and NA separately may provide a functional linkage between the muta- tions that can impact the drug binding sites in NA and those that can affect the host immune response or vac- cine efficacy in HA. Our findings demonstrated the value of conducting timely analysis on the 2009 H1N1 virus and of the integrated approach to studying both surface proteins HA and NA together to reveal their interdependence, which could not be accomplished by studying them individually. Keywords: Co-Mutations; Entropy; Epitope; H1N1; Hemagglutinin; Influenza; Mutation; Mutual Information; Neuraminidase; Phylogenetic Analysis; Stalk Motif; Swine Flu 1. INTRODUCTION Influenza viruses caused several pandemics in history such as the Spanish flu (H1N1, 1918), the Asian flu (H2N2, 1958), and the Hong Kong flu (H3N2, 1968), where the H1N1 virus has the longest recorded history of human infection. In March and April 2009, a new A (H1N1) influenza virus first emerged in Mexico and the United States. Antigenically the new virus is similar to North American swine A (H1N1) viruses but distinct from seasonal human A (H1N1). This virus consisting of gene segments in swine or humans has acquired the ca- pacity to spread quickly by human-to-human transmis- sion across the globe and therefore has attracted interna- tional attention. On June 11, 2009, the World Health Organization (WHO) declared the H1N1 virus a pan- demic. The influenza A viral genome is composed of 8 genes encoding 11 proteins, including two surface glycopro- teins, hemagglutinin (HA) and neuraminidase (NA). The main influenza antigens targeted by the human immune system are these two proteins, and influenza A subtypes are classified by the antigenic distinctions of the HA and NA proteins. There are 16 subtypes of HA and 9 sub- types of NA. HA mediates virus binding to sialic acid receptor on a host cell surface to initiate infection, and NA cleaves the binding to promote release of viral progeny. Otherwise, the viral progeny particles will re- main aggregated at the cell surface. HA is also the main target of the host immune system. Once in a host cell, the HA protein comes under selective pressure for change to evade the host immune response. HA and NA have been of great interest in flu research due to their pivotal role in viral infection and replication. The HA is a cylindrically shaped homotrimer mole- cule composed of three identical HA polypeptides, which are cleaved by protease into two subunits HA1 and HA2 during virus maturation. The globular region of the molecule is based mainly on the HA1 residues, and  2 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE the stem contains some residues of HA1 and all of HA2. The enzymatic domain of NA is held away from the vi- rus surface by a long, thin stalk of variable length. The replication efficiency of viruses in eggs and mice corre- lates the NA stalk length [1]. NA with a short stalk was found to be inefficient in virion progeny since an active site located too close to the viral envelop could not ac- cess its substrate correctly [2]. The percentage of viruses with a short NA stalk has increased steadily in recent years [3]. There is a balanced interplay between HA and NA; one serves as receptor binder and one as receptor de- stroyer, to facilitate efficient virus replication in host cells [4]. Influenza viruses can overcome host restriction and become adapted to a new host by making changes in HA or/and NA [5], i.e., concomitant changes in HA and NA are required for influenza viruses to survive in host cells. The deficiency in NA activity conferred by the shortened protein stalk could be compensated by modu- lating the receptor binding affinity of HA to restore the functional balance between HA and NA [4]. In [6] a spe- cial stalk motif, commonly found in H5N1 in the past, was discovered in the 2009 H1N1 strains for the first time. This finding is significant given the fact that the viruses with this motif tend to have high virulence [3]. One of the goals of this study was to investigate the im- pact of the NA motifs on HA in the 2009 H1N1 strains. As the 2009 H1N1 virus continues to transmit effec- tively from human to human, the occurrence of drug- resistant viruses is expected. One recent study [7] showed that the novel mutations of the 2009 H1N1 virus NA are located at sites that do not interfere with the ac- tive site so the currently used three drugs oseltamivir (Tamiflu®), zanamivir (Relenza®), and peramivir re- main effective. Another study [6] identified two net- works of co-mutations of 2009 H1N1 NA that may af- fect the active site from a greater distance. As the principal antigen on the virus surface, HA is the main viral target for the human immune system, which can neutralize the virus through blocking viral binding to the receptors on host cells. An epitope is a region on the surface of an antigen, such as the HA in this study, capable of eliciting an immune response. An- tigenic variation of HA is one mechanism employed by the flu virus to escape the response of the host immune system. One report found that one single amino acid substitution in 1918 H1N1 HA changes receptor binding specificity [8]. Influenza HA evolution is typically a combination of functional constraint and positive selec- tion in epitope regions. As such, the identification of epitope regions on HA is important for both drug and vaccine development. Epitope mapping using mono- clonal antibodies and the availability of the 3-dimen- sional structure have identified five antigenic sites in the HA of H3 subtype [9,10]. Corresponding antigenic sites have subsequently been mapped to H1 and H2 subtypes [11,12]. With new technology as the one employed in [9,10], a recent refinement of the definition of H1 epi- topes was conducted in [13]. One of the tasks of our study was to map the sequence mutations of the 2009 H1N1 HA relative to the five epitope regions of H1. We were also interested in finding the co-mutations in HA and co-mutations between HA and NA of 2009 H1N1. 2. MATERIALS AND METHODS 2.1. Sequence Data Published HA sequences of 3936 influenza A virus from 2005 to 2009, H1 sequences of 1900 from 1918 to 2008, and H1 sequences of 508 in 2009 were downloaded from the Influenza Virus Resource (http://www.ncbi/nlm.nih- giv/genomes/FLU/FLU.html) of the National Center for Biotechnology Information (NCBI) on Oct. 13, 2009. We were mainly interested in the sequences in 2009, but also needed the sequences in several years before 2009 to provide comparison in the study. All the sequences used in the study were aligned with MAFFT [14]. 2.2. Entropy and Mutual Information In information theory [15], entropy is a measure of dis- order or randomness associated with a random variable. Let be a discrete random variable that has a set of possible values with probabilities where . The entropy H of 123 {,, ,...} n aaa a () ii Px ap 12 pp 3 {,, ,...} n p p is () log ii i xp p The mutual information of two random variables is a quantity that measures the mutual dependence of the two variables or the average amount of information that conveys about , which can be defined as y (, )()()(, ) xyHx Hy Hxy where ()xis the entropy of , and (, )xy is the joint entropy of and. if and only if y(, )Ixy0 and are independent random variables. y In current study, each of the n columns in a multiple sequence alignment of a set of HA sequences of N resi- dues is considered as a discrete random variable i (1 ≤i ≤N) that takes on one of the 20 (n=20) amino acid types with some probability. () i x () i has its minimum value 0 if all the residues at position i are the same, and achieves its maximum if all the 20 amino acid types ap- pear with equal probability at position i, which can be verified by the Lagrange multiplier technique. A position of high entropy means that the amino acids are often varied at this position. While x measures the ge- netic diversity at position i in our current study, measures the correlation between residue sub- (,) j i Ix y 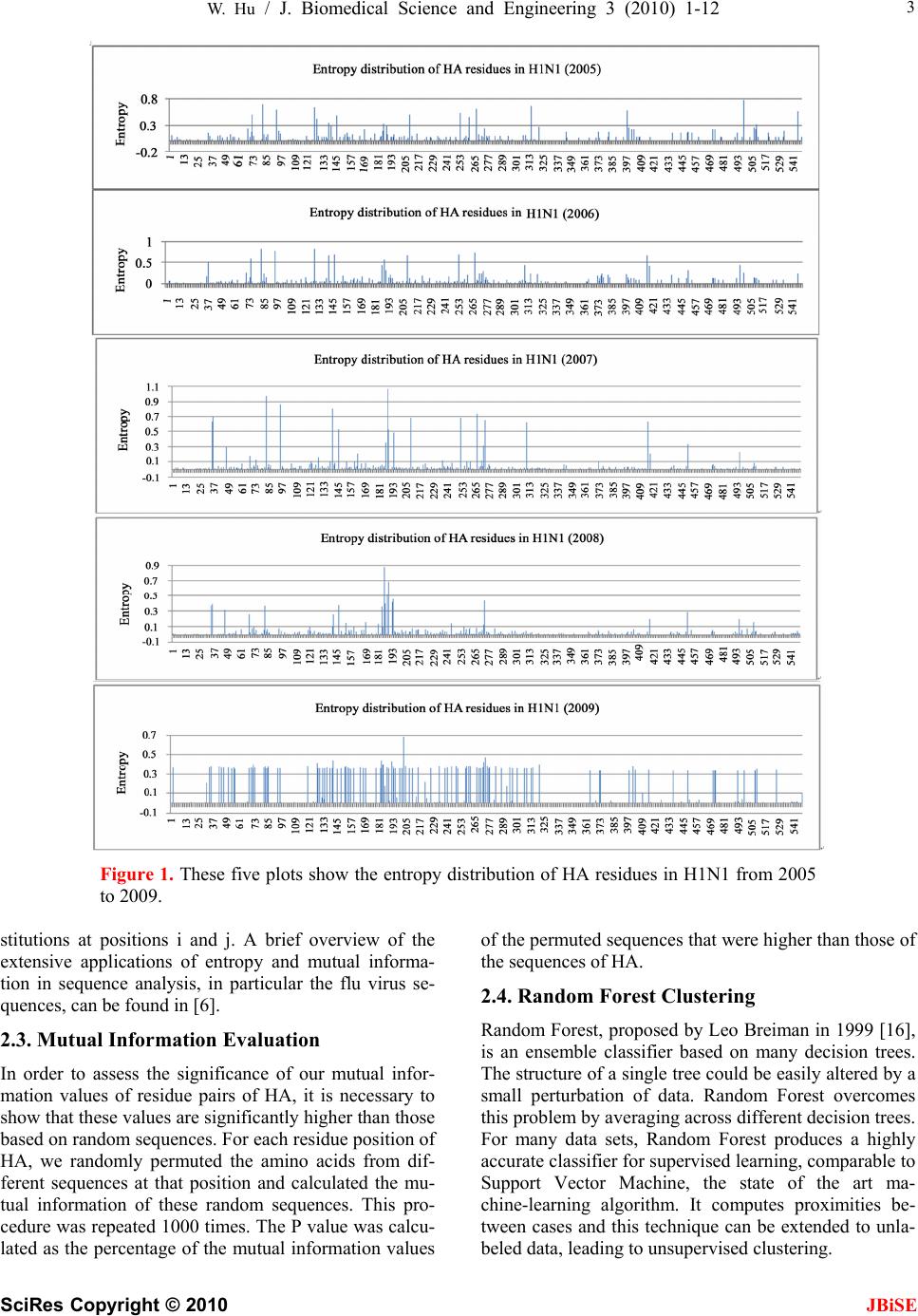 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE 3 Figure 1. These five plots show the entropy distribution of HA residues in H1N1 from 2005 to 2009. stitutions at positions i and j. A brief overview of the extensive applications of entropy and mutual informa- tion in sequence analysis, in particular the flu virus se- quences, can be found in [6]. 2.3. Mutual Information Evaluation In order to assess the significance of our mutual infor- mation values of residue pairs of HA, it is necessary to show that these values are significantly higher than those based on random sequences. For each residue position of HA, we randomly permuted the amino acids from dif- ferent sequences at that position and calculated the mu- tual information of these random sequences. This pro- cedure was repeated 1000 times. The P value was calcu- lated as the percentage of the mutual information values of the permuted sequences that were higher than those of the sequences of HA. 2.4. Random Forest Clustering Random Forest, proposed by Leo Breiman in 1999 [16], is an ensemble classifier based on many decision trees. The structure of a single tree could be easily altered by a small perturbation of data. Random Forest overcomes this problem by averaging across different decision trees. For many data sets, Random Forest produces a highly accurate classifier for supervised learning, comparable to Support Vector Machine, the state of the art ma- chine-learning algorithm. It computes proximities be- tween cases and this technique can be extended to unla- beled data, leading to unsupervised clustering.  4 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE Table 1. Amino acids on epitopes A, B, C, D, and E of H1 (A/California/04/2009 numbering) from [13]. Epi- tope Amino acids Number of amino acids A 118,120,121,122,126,127,128,129,132,133,134,135,137,139,140,141,142,143,146,147,149,165,252,253 24 B 124,125,152,153,154,155,156,157,160,162,183,184,185,186,187,189,190,191,193,194,195,196 22 C 34,35,36,37,38,40,41,43,44,45,269,270,271,272,273,274,276,277,278,283,288,292,295,297,298,302,303, 305,306,307,308,309,310 32 D 170,171,172,173,174,176,179,198,200,202,204,205,206,207,208,209,210,211,212,213,214,215,216,222,2 23,224,225,226,227,235,237,239,241,243,244,245 48 E 47,48,50,51,53,54,56,57,58,66,68,69,70,71,72,73,74,75,78,79,80,82,83,84,85,86,102,257,258,259,260,26 1,263,267 34 Table 2. 2009 H1N1 HA mutations on the five epitopes. Epitope Mutation Residues Number of Mutations A 126, 127, 128, 129,132,134,137,140,141,165, 252 11 B 152, 154,155,156, 183,184,185,189,193,194,195 11 C 35, 44, 269, 271,272,273,276,277,297,307 10 D 95, 167, 169, 204,206,207, 208, 210,215,223,226,244 12 E 50, 53, 56, 68, 70, 71, 72, 73, 82, 83, 84, 85, 257,259,260 15 To view the clusters formed by Random Forest, mul- tidimensional scaling [17] was utilized to project high- dimensional data down into a low-dimensional space while preserving the distances between them. First the proximities between cases i and j form a symmetric and positive definite matrix {prox(i,j)}. Then a second posi- tive definite and symmetric matrix {cv(i,j)} is con- structed using the entries of {prox(i,j)}. Random Forest extracts a few largest eigenvalues of the cv matrix and their corresponding eigenvectors. The values of () ()eivi are referred to as the ith scaling coordinate, where and are the ith eigenvalue and ei- genvector of matrix cv. In this study, the first and second scaling coordinates were utilized to visualize the data. ()ei ()vi 2.5. Important Sites in HA and NA The NA active site is a shallow pocket constructed from conserved residues, some of which contact the substrate directly and participate in catalysis, while others provide a structural framework [18]. According to the numbering in [7], these residues of N1 are 118, 119, 151, 152, 156, 179, 180, 223, 225, 228, 247, 277, 278, 293, 295, 368, and 402. The antigenic sites of N1 are residues 83 – 143, 156 – 190, 252 – 303, 330, 332, 340 –345, 368, 370,387 – 395, 431 – 435, 448 – 468. The HA active site located in a cleft is composed of the residues 91, 150, 152, 180, 187, 191, and 192. The active site cleft of HA is formed by its right edge (131_GVTAA) and left edge (221_RGQAGR) [19]. The human immune system responds primarily to the five epitope regions, A, B, C, D, and E, of the HA protein in H1N1. Table 1 presents the 160 amino acids on the five eiptope regions of HA in H1N1 as discovered in [13]. 3. RESULTS 3.1. Unusually High Entropy Activities of HA in 2009 H1N1 HA is the primary target for neutralizing antibodies, and the gradual accumulation of substitutions at the antibody sites of HA is the main cause for flu virus to resist hu- man immunity. As entropy measures the disorder of amino acid frequency at each residue of HA, we sought to compare the entropy activities of 2009 H1N1 HA with those in the previous years. Due to the rapid spread of the 2009 H1N1 A virus around the world, unusual en- tropy patterns of its sequences are anticipated. The se- quence variation within the 2009 H1N1 strains as re- flected by its entropy distribution along with other H1N1 HA sequences from 2005 to 2008 are illustrated in Fig- ure 1, where the high entropy activities of 2009 H1N1 HA were observed, especially in the HA1 domain, indi- cating the 2009 H1N1 strains are under high immune pressure. 3.2. Mutations of HA in 2009 H1N1 To find the sequence variation of HA in 2009 H1N1, three strains (A/California/04/2009(H1N1), A/South Carolina/1/1918(H1N1), and A/Mississippi/UR06-0537/ 2007(H1N1)) were aligned with MAFFT [14] and the resulting multi-sequence alignment was visualized in Jalview [20] (Figure 1). There were 118 mutations, 59 of which (50%) were mutations on the five epitopes, implying that HA in particular has a high amino acid substitution rate in its epitope regions. More precisely, 11 mutations were on epitope A, 11 mutations on B, 10 mutations on C, 12 mutations on D, and 15 mutations on E. The detailed distribution of these mutations on the  W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE 5 Table 3. Comparison of amino acid residues of 2009 H1N1 HA near the receptor-binding sites. The numbers in paren- thesis indicate the entropy of the amino acids of HA at that position. Residues/Amino Acids Right edge 131 132 133 134 135 A/California/04/2009 G (0.0) V (0.06)T (0.36)A (0.04)A (0.36) A/Mississippi/UR06-0537/2007 G V S A S A/South Carolina/1/1918 G V T A A Left edge 221 222 223 224 225 226 A/California/04/2009 R (0.0) D (0.22)Q (0.05)E (0.01)G (0.0) R (0.0) A/Mississippi/UR06-0537/2007 R D Q E G R A/South Carolina/1/1918 R D Q A G R Receptor binding 91 150 152 180 187 191 192 A/California/04/2009 Y (0.0) W (0.0)V (0.37)H (0.0)D (0.18)L (0.03) Y (0.0) A/Mississippi/UR06-0537/2007 Y W T H D L Y A/South Carolina/1/1918 Y W T H D L Y Figure 2. Three sequence alignment using MAFFT and visualized by Jalview: the top sequence is A/California/04/ 2009(H1N1), the middle one is A/Mississippi/UR06-0537/2007(H1N1), and the bottom one is A/South Caro- lina/1/1918(H1N1). five epitopes is in Table 2. Epitopes A and B are the dominant ones as they have the highest mutation rate among the five, suggesting that A and B are under the most pressure from the immune system. The information about the dominant epitopes can be used to calculate the Pepitope, a specific measure of antigenic distance be- tween two strains of influenza to estimate the vaccine efficacy [21]. We first displayed the five epitope regions of 2009 H1N1 HA in the homology 3D model built in [13] in Figure 3 and then mapped these 118 mutations of 2009 H1N1 HA to this 3D model (Figure 4). To learn the sequence variation at or around the active site of 2009 H1N1 HA, we built Table 3 to show that the amino acids at these sites were highly conserved, which was in agreement with the previous findings in [22]. There were three residues, 133 and 135 on the left edge and 152 on the active site, that had high entropy and amino acid substitution. In [19] residue 152 was found to allow the substitution for a hydrophobic residue based on an investigation of 191 different sequences of 15 subtypes of HAs, a discovery illustrated in our analysis as well (Table 3). A recent study [23] indicated that substitutions F71S, T128S, E302K, M314L in HA1 of 2009 H1N1 are es- sential for the interaction between swine and humans; and residues 94, 196 and 274 are predicted to be “hot spots” for mutations that may increase infectivity of the virus (Figure 2). It also found that the highly conserved 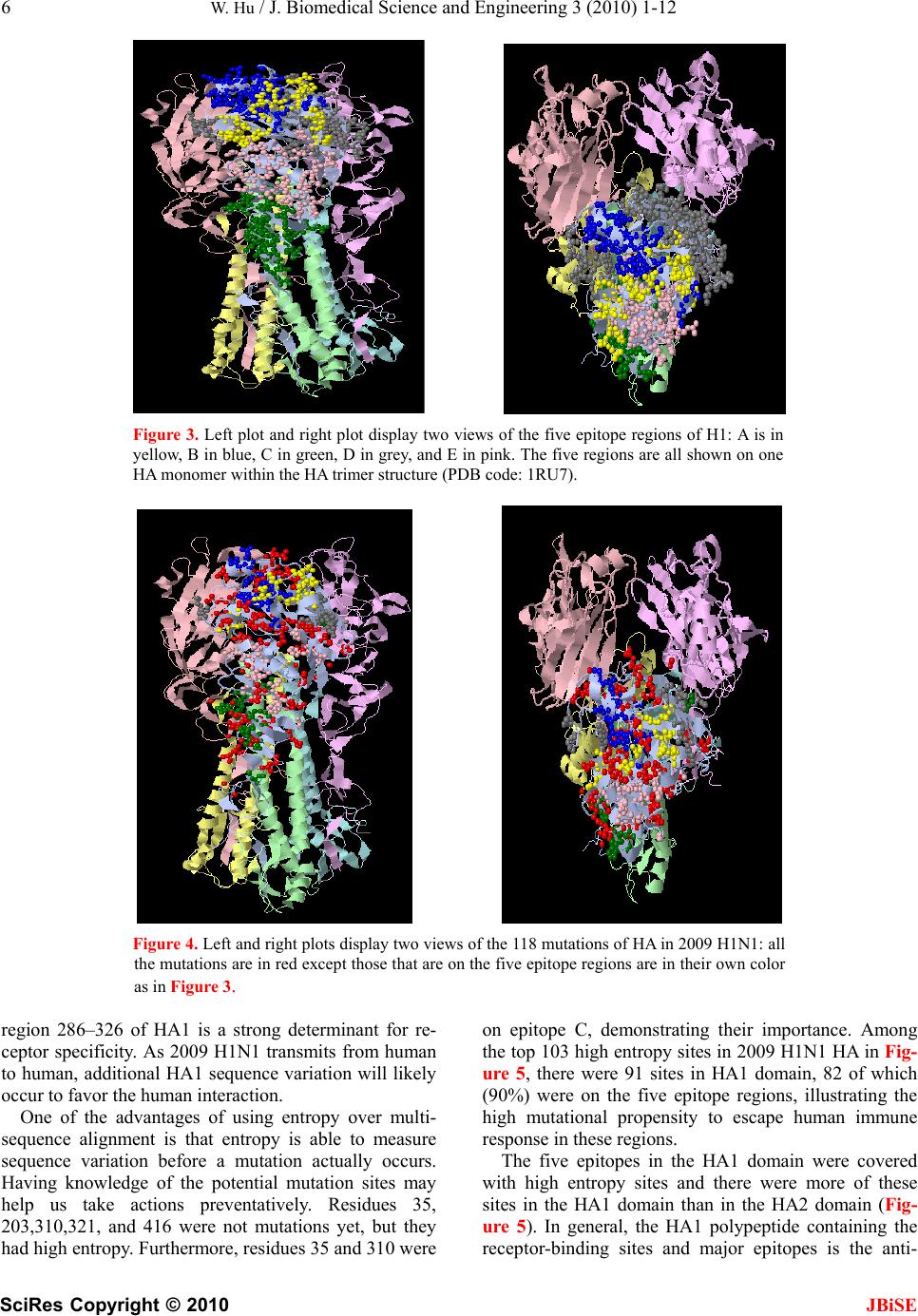 6 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE Figure 3. Left plot and right plot display two views of the five epitope regions of H1: A is in yellow, B in blue, C in green, D in grey, and E in pink. The five regions are all shown on one HA monomer within the HA trimer structure (PDB code: 1RU7). Figure 4. Left and right plots display two views of the 118 mutations of HA in 2009 H1N1: all the mutations are in red except those that are on the five epitope regions are in their own color as in Figure 3. region 286–326 of HA1 is a strong determinant for re- ceptor specificity. As 2009 H1N1 transmits from human to human, additional HA1 sequence variation will likely occur to favor the human interaction. One of the advantages of using entropy over multi- sequence alignment is that entropy is able to measure sequence variation before a mutation actually occurs. Having knowledge of the potential mutation sites may help us take actions preventatively. Residues 35, 203,310,321, and 416 were not mutations yet, but they had high entropy. Furthermore, residues 35 and 310 were on epitope C, demonstrating their importance. Among the top 103 high entropy sites in 2009 H1N1 HA in Fig- ure 5, there were 91 sites in HA1 domain, 82 of which (90%) were on the five epitope regions, illustrating the high mutational propensity to escape human immune response in these regions. The five epitopes in the HA1 domain were covered with high entropy sites and there were more of these sites in the HA1 domain than in the HA2 domain (Fig- ure 5). In general, the HA1 polypeptide containing the receptor-binding sites and major epitopes is the anti- 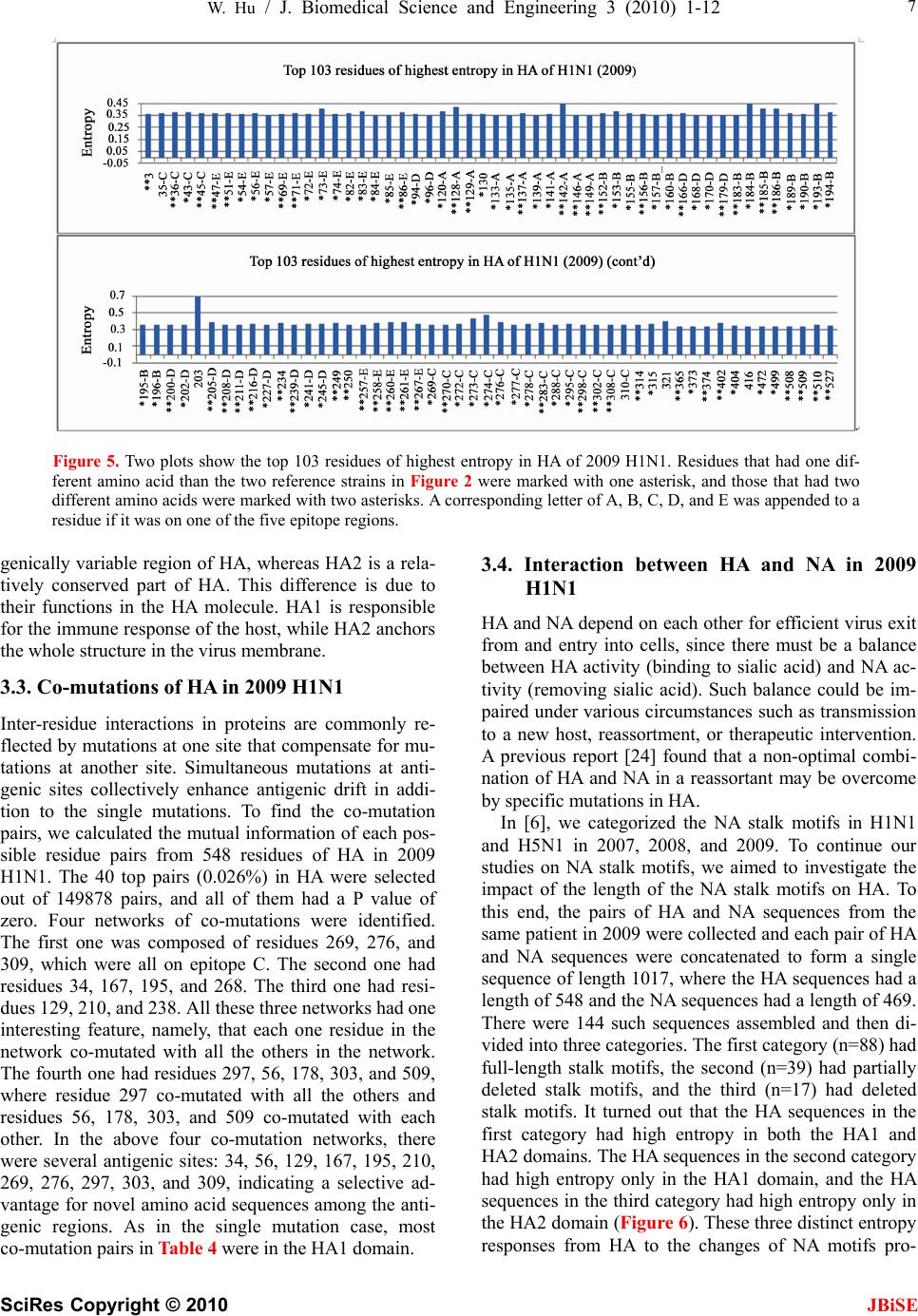 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE 7 Figure 5. Two plots show the top 103 residues of highest entropy in HA of 2009 H1N1. Residues that had one dif- ferent amino acid than the two reference strains in Figure 2 were marked with one asterisk, and those that had two different amino acids were marked with two asterisks. A corresponding letter of A, B, C, D, and E was appended to a residue if it was on one of the five epitope regions. genically variable region of HA, whereas HA2 is a rela- tively conserved part of HA. This difference is due to their functions in the HA molecule. HA1 is responsible for the immune response of the host, while HA2 anchors the whole structure in the virus membrane. 3.3. Co-mutations of HA in 2009 H1N1 Inter-residue interactions in proteins are commonly re- flected by mutations at one site that compensate for mu- tations at another site. Simultaneous mutations at anti- genic sites collectively enhance antigenic drift in addi- tion to the single mutations. To find the co-mutation pairs, we calculated the mutual information of each pos- sible residue pairs from 548 residues of HA in 2009 H1N1. The 40 top pairs (0.026%) in HA were selected out of 149878 pairs, and all of them had a P value of zero. Four networks of co-mutations were identified. The first one was composed of residues 269, 276, and 309, which were all on epitope C. The second one had residues 34, 167, 195, and 268. The third one had resi- dues 129, 210, and 238. All these three networks had one interesting feature, namely, that each one residue in the network co-mutated with all the others in the network. The fourth one had residues 297, 56, 178, 303, and 509, where residue 297 co-mutated with all the others and residues 56, 178, 303, and 509 co-mutated with each other. In the above four co-mutation networks, there were several antigenic sites: 34, 56, 129, 167, 195, 210, 269, 276, 297, 303, and 309, indicating a selective ad- vantage for novel amino acid sequences among the anti- genic regions. As in the single mutation case, most co-mutation pairs in Table 4 were in the HA1 domain. 3.4. Interaction between HA and NA in 2009 H1N1 HA and NA depend on each other for efficient virus exit from and entry into cells, since there must be a balance between HA activity (binding to sialic acid) and NA ac- tivity (removing sialic acid). Such balance could be im- paired under various circumstances such as transmission to a new host, reassortment, or therapeutic intervention. A previous report [24] found that a non-optimal combi- nation of HA and NA in a reassortant may be overcome by specific mutations in HA. In [6], we categorized the NA stalk motifs in H1N1 and H5N1 in 2007, 2008, and 2009. To continue our studies on NA stalk motifs, we aimed to investigate the impact of the length of the NA stalk motifs on HA. To this end, the pairs of HA and NA sequences from the same patient in 2009 were collected and each pair of HA and NA sequences were concatenated to form a single sequence of length 1017, where the HA sequences had a length of 548 and the NA sequences had a length of 469. There were 144 such sequences assembled and then di- vided into three categories. The first category (n=88) had full-length stalk motifs, the second (n=39) had partially deleted stalk motifs, and the third (n=17) had deleted stalk motifs. It turned out that the HA sequences in the first category had high entropy in both the HA1 and HA2 domains. The HA sequences in the second category had high entropy only in the HA1 domain, and the HA sequences in the third category had high entropy only in the HA2 domain (Figure 6). These three distinct entropy responses from HA to the changes of NA motifs pro-  8 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE Table 4. Top 40 pairs of co-mutations in HA of 2009 H1N1. All have a P value of zero. A corresponding letter of A, B, C, D, and E was appended to a residue if it was on one of the five epitope regions. (34-C,167-D) (34-C,195-B) (34-C,268) (44-C,449) (46,248) (56-E,297-C) (68-E,156-B) (71-E,132-A) (71-E,287) (81,277-C) (84-E,182) (95-D,165-A)(127-A,189-B)(129-A,210-D)(129-A,238) (132-A,287) (134-A,492) (145,207-D) (145,307-C) (155-B,259-E)(167-D,195-B)(167-D,268) (167-D,471) (178,294) (178,297-C) (178,509) (195-B,268) (195-B,372) (195-B,471) (167-D,471) (207-D,307-C) (207-D,401) (210-D,238) (268,372) (269-C,276-C) (269-C,309-C)(276-C,309-C)(294,297-C) (297-C,509) (307-C,401) Table 5. Top 53 pairs of co-mutations in between HA and NA of 2009 H1N1. All have a P value of zero. (46,13) (248,13) (320,13) (226,15) (269,16) (276,16) (309,16) (226,19) (55,21) (403,23) (95,34) (165,34) (55,42) (373,47) (240,48) (55,59) (314,75) (44,173) (155,189) (259,189) (44,220) (314,232) (271,234) (46,241) (248,241) (44,257) (56,263) (178,263) (297,263) (509,263) (151,264) (46,257) (256,269) (56,288) (178,288) (297,288) (509,288) (507,289) (56,321) (178,321) (297,321) (509,321) (154,336) (275,341) (472,341) (154,382) (55,385) (509,389) (209,427) (207,432) (307,432) (401,432) (249,453) vided another support of the notion that HA and NA need to maintain a functional balance. In addition to the study of the impact of the NA stalk motifs on HA, we also discovered co-mutation pairs, one mutation in HA and one in NA, that correlated each other across the two proteins. We calculated the mutual information of each possible residue pairs from 1017 residues of HA and NA (as a single sequence) in 2009 H1N1. The top 57 pairs (0.011%) were selected out of 516636 pairs in the sequences of HA and NA, and they all had a P value of zero. Discarding the pairs in HA or NA, and retaining only those with one residue in HA and one residue in NA resulted in 53 pairs (Table 5). Among the pairs found, there were two co-mutation networks, one in HA consisting of residues 56, 178, 297, and 509 and one in NA consisting of residues 263, 288, and 321, where each residue in the network co-mutated with all residues in the other network. NA residues 263 and 321 were a part of a co-mutation network in NA consisting of residues 149, 263, 321, and 389 discovered in [6], which had a property that each of the residues in this network co-mutated with all the other three. NA residues 263 and 288 were also antigenic sites in NA. NA residue 288 was located in a cluster of muta- tion sites consisting of residues 285, 286, 287, 288, and 289 in NA. The fact that NA residue 288 was part of a NA network of residues 263, 288, and 321 that co-mutated with a HA network of residues 56, 178, 297, and 509 suggested there might be a link between the mutation cluster in NA near residue 288 and the co-mu- tation network of residues 149, 263, 321, and 389 found in [6]. This conclusion could not be inferred if we were only using the information from NA along to study NA. 3.5. Phylogenetic Analysis of HA in 2009 H1N1 The HA and NA genes of 2009 H1N1 are in the classical swine lineage and the Eurasian swine genetic lineage respectively [25]. In [6], it was shown that the eight rep- resentatives of the novel NA sequences in 2009 H1N1 were diverse enough to cover the major branches of the phylogenetic tree of past NA strains. With more HA se- quences available to date than in May or June 2009, we constructed the phylogenetic tree of the representative HA sequences from 1918 to 2009 with the neighbor- joining method using MEGA 4 software [26]. Seven HA sequences in 2009 were selected using cd-hit with iden- tity removed at 98.5% from all the HA sequences in 2009. HA sequences of 29 were selected using cd-hit with identity removed at 98% from all the HA sequences in the years prior to 2009. In contrast to the diversity of NA sequences in 2009 H1N1 [6], the seven representa- tive HA sequences were mainly clustered together in the phylogenetic tree in Figure 7, which implied that the HA sequences had remained the same diversity as they were on 16 May 2009 when a similar phylogenetic tree was constructed from a collection of HA sequences in [25]. As demonstrated in the study in [6], Random For- est-based clustering can reveal some subtle features of sequence clusters that a phylogenetic tree built with the neighbor-joining method cannot. We attempted to em- ploy the Random Forest-based clustering technique to cluster the same sequences used in Figure 7 and the re- sults are in Figure 8. Due to the space limitation, we used a number from 1 to 36 to represent a sequence in Figure 8, where the same number is attached to the start of the corresponding sequence name in Figure 7. The numbers 1 to 7 were assigned to the seven representative HA sequences in 2009 H1N1. In Figure 8, the sequences numbered 1, 2, 4, 5, 6, and 7 were close in the second scaling coordinate, and those numbered 1, 4, 5, and 7 were also close in the first scaling coordinate. This de- tailed clustering information about the seven sequences 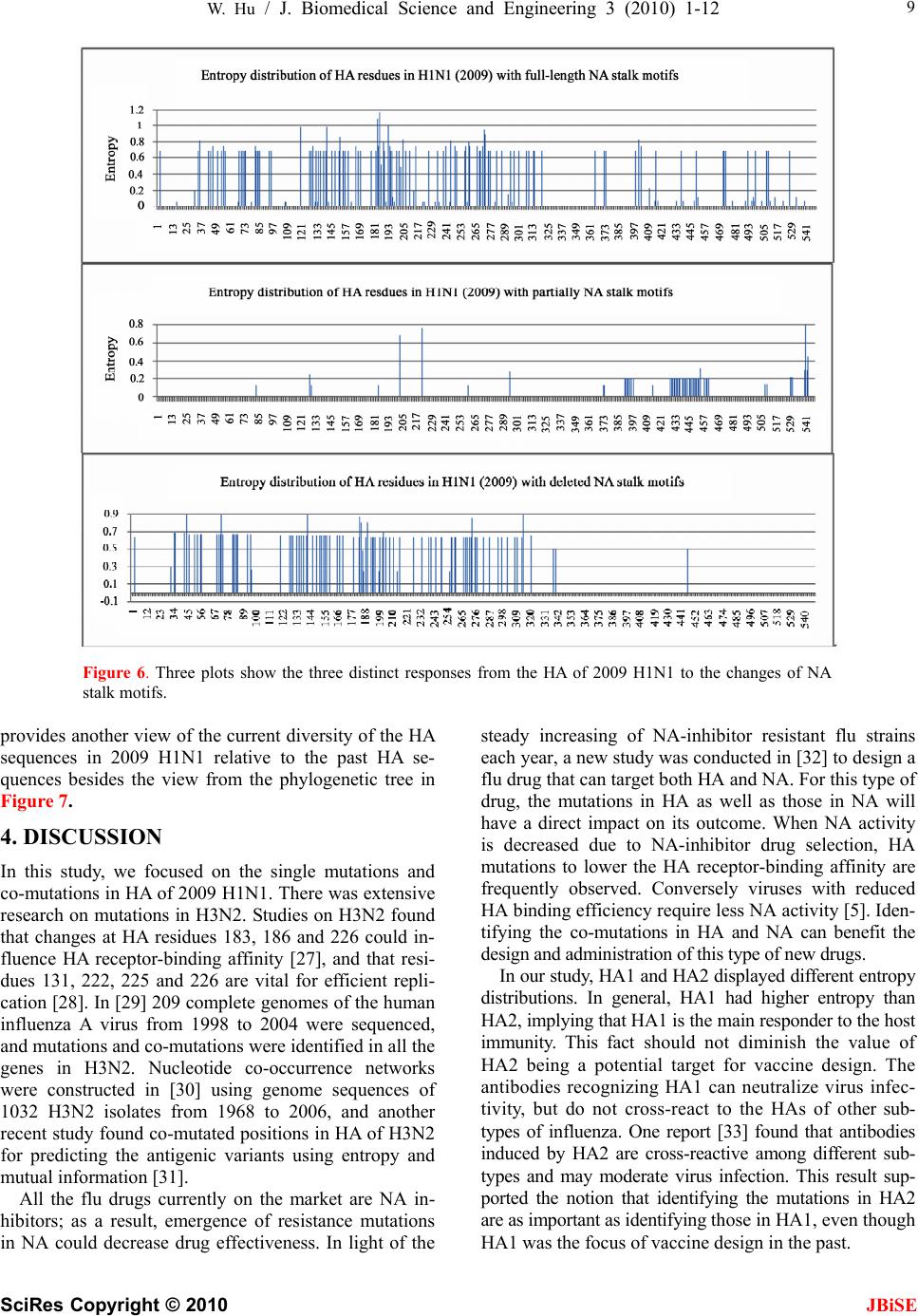 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE 9 Figure 6. Three plots show the three distinct responses from the HA of 2009 H1N1 to the changes of NA stalk motifs. provides another view of the current diversity of the HA sequences in 2009 H1N1 relative to the past HA se- quences besides the view from the phylogenetic tree in Figure 7. 4. DISCUSSION In this study, we focused on the single mutations and co-mutations in HA of 2009 H1N1. There was extensive research on mutations in H3N2. Studies on H3N2 found that changes at HA residues 183, 186 and 226 could in- fluence HA receptor-binding affinity [27], and that resi- dues 131, 222, 225 and 226 are vital for efficient repli- cation [28]. In [29] 209 complete genomes of the human influenza A virus from 1998 to 2004 were sequenced, and mutations and co-mutations were identified in all the genes in H3N2. Nucleotide co-occurrence networks were constructed in [30] using genome sequences of 1032 H3N2 isolates from 1968 to 2006, and another recent study found co-mutated positions in HA of H3N2 for predicting the antigenic variants using entropy and mutual information [31]. All the flu drugs currently on the market are NA in- hibitors; as a result, emergence of resistance mutations in NA could decrease drug effectiveness. In light of the steady increasing of NA-inhibitor resistant flu strains each year, a new study was conducted in [32] to design a flu drug that can target both HA and NA. For this type of drug, the mutations in HA as well as those in NA will have a direct impact on its outcome. When NA activity is decreased due to NA-inhibitor drug selection, HA mutations to lower the HA receptor-binding affinity are frequently observed. Conversely viruses with reduced HA binding efficiency require less NA activity [5]. Iden- tifying the co-mutations in HA and NA can benefit the design and administration of this type of new drugs. In our study, HA1 and HA2 displayed different entropy distributions. In general, HA1 had higher entropy than HA2, implying that HA1 is the main responder to the host immunity. This fact should not diminish the value of HA2 being a potential target for vaccine design. The antibodies recognizing HA1 can neutralize virus infec- tivity, but do not cross-react to the HAs of other sub- types of influenza. One report [33] found that antibodies induced by HA2 are cross-reactive among different sub- types and may moderate virus infection. This result sup- ported the notion that identifying the mutations in HA2 are as important as identifying those in HA1, even though HA1 was the focus of vaccine design in the past.  10 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE 1 H1N1 Washington 01 2009 gi|243031572|g 7 H1N1 St Louis 746 2009 gi|260100660|gb 6 H1N1 St Louis 690 2009 gi|260100658|gb 2 H1N1 PaisVasco RR3427 2009 gi|25459738 5 H1N1 Busan 01 2009 gi|259023925|gb|ACV 32 H1N1 Pennsylvania 16 2007 gi|18807643 31 H1N1 Maldonado FMD1033 2007 gi|183223 4 H1N1 North Carolina 02 2009 gi|2559608 34 H1N1 HongKong 1052 2008 gi|206575146| 25 H1N1 TW 4845 1999 gi|89033083|gb|ABD5 17 H1N2 Stockholm 13 2002 gi|22859335|gb 29 H1N1 New Caledonia V77245 2007 gi|148 30 H1N1 Tehran 30 2006 gi|162956892|gb|A 11 H1N1 human Taiwan 3355 97 gi|14571962 12 H1N1 Switzerland 5389 95 gi|14587045| 33 H1N1 Taiwan VGHYM0725 08 1988 gi|2020 16 H1N1 Fiji 15899 83 gi|11595850|gb|CAC 8 H1N1 Lepine 1948 gi|8096357|gb|BAA9611 9 H1N1 TF 15 1951 gi|8096359|gb|BAA96111 21 H1N1 Fort Worth 50 gi|89114296|gb|ABD 15 H1N1 Rhodes 47 gi|21717604|gb|AAM7668 22 H1N1 Cameron 1946 gi|89903075|gb|ABD7 13 H1N1 Puerto Rico 8 34 Mount Sinai gi| 20 H1N1 Melbourne 35 gi|89148077|gb|ABD6 14 H1N1 Weiss 1943 gi|21717600|gb|AAM766 19 H1N1 Bellamy 1942 gi|89152215|gb|ABD6 23 H1N1 Wilson Smith 33 gi|89782156|gb|A 10 H1N1 South Carolina 1 1918 gi|4325039 18 H1N1 Switzerland 8808 2002 gi|3842252 36 H1N1 Alma Ata 1417 84 gi|385579|gb|AA 3 H1N1 Taiwan T1339 2009 gi|255689237|gb 35 H1N1 Ohio 01 2007 gi|229473302|gb|ACQ 24 H1N1 New Jersey 1976 gi|146759952|gb| 27 H1N1 Philippines 344 2004 gi|11793578 26 H1N1 Iowa CEID23 2005 gi|112456302|gb 28 H1N1 Thailand 271 2005 gi|117935791|g 46 62 17 31 62 47 39 94 90 41 33 27 74 38 43 45 60 38 53 67 93 78 86 66 35 59 32 35 40 16 46 13 0.05 Figure 7. Phylogenetic tree of the HA protein sequences of the H1 subtype family. 5. CONCLUSIONS There is a great interest in gaining more understanding of the 2009 H1N1 virus given the urgency of the current 2009 flu pandemic. In our previous study on 2009 H1N1 [6], three aspects of NA were investigated: the mutations and co-mutations, the stalk motifs, and the phylogenetic analysis. In this study, we focused on HA and the inter- action between HA and NA. The118 mutations of 2009 H1N1 HA were uncovered and mapped to the 3D ho- mology model of H1, and the mutations on the five epi- tope regions on H1 were identified. This information is essential for the development of new drugs and vaccine. With entropy and mutual information analysis, we were able to locate several antigenic sites in HA that could potentially become mutational sites. In addition to the identification of single mutations and co-mutations in 2009 H1N1 HA, we were also able with help from our previous results in [6] to find two co-mutation networks, one in HA and one in NA, where each mutation in one  W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE 11 Figure 8. Random Forest-based clusters of the same HA sequences used in Figure 7. Here a number from 1 to 36 is used to represent a sequence, which is attached to the start of the corresponding sequence name in Figure 7. network co-mutates with the mutations in the other net- work across the two proteins HA and NA. These two networks residing in HA and NA separately may provide a link between the mutations that can influence the drug binding sites in NA and those that can affect the host immune response or vaccine efficacy in HA. The distinct entropy responses from HA to the changes of NA stalk motifs were discovered, suggesting the functional de- pendence between them. Finally, our phylogenetic analysis indicated that the seven representative se- quences of HA in 2009 H1N1 were mainly clustered together in the phylogenetic tree made of past represen- tative HA sequences, quite contrary to the NA case [6]. The phylogenetic tree in Figure 7 was similar in struc- ture to the phylogenetic tree constructed from a collec- tion of HA sequences of 2009 H1N1 on 16 May 2009 in [25], which implied that the diversity of 2009 H1N1 HA sequences remained relatively the same. This view is also supported by the clusters of the same HA sequences created with the Random Forest-based clustering tech- nique (Figure 8). Taken together, our results highlighted the importance of conducting timely analysis on the 2009 H1N1 virus and of the integrated approach to studying both surface proteins HA and NA together to reveal their interdependence, which could not be accom- plished by studying them individually. 6. ACKNOWLEDGMENTS We thank Houghton College for its financial support. REFERENCES [1] Castrucci, M.R. and Kawaoka, Y. (1993) Biologic im- portance of neuraminidase stalk length in influenza A virus. J Virol, 67, 759-764. [2] Els, M.C., Air, G.M., Murti, K.G. et al. (1985) An 18-amino acid deletion in an influenza neuraminidase. Virology, 142, 241-247. [3] Zhou, H.B., Yu, Z.J., Hu, Y. Tu, J.G. et al. (2009) The special neuraminidase stalk-motif responsible for in- creased virulence and pathogenesis of H5N1 influenza A virus. PLoS One, 4(7), 6277. [4] Wagner, R., Matrosovich, M. and Klenk, H.D. (2002) Functional balance between haemagglutinin and neura- minidase in influenza virus infections. Rev. Med. Virol, 12, 159-166. [5] Lu, B., Zhou, H.L., Ye, D., Kemble, G. and Jin, H. (2005) Improvement of influenza A/Fujian/411/02 (H3N2) virus growth in embryonated chicken eggs by balancing the hemagglutinin and neuraminidase activities, using re- verse genetics. Journal of Virology, 79, 6763-6771. [6] Hu, W. (2009) Analysis of correlated mutations, stalk motifs, and phylogenetic relationship of the 2009 influ- enza A virus neuraminidase sequences. Journal of Bio- medical Science and Engineering, 2, 550-555 [7] Sebastian, M.S., Ma, J.M., Raphael, T.C.L., Fernanda, L. S. and Frank, E. (2009) Mapping the sequence mutations of the 2009 H1N1 influenza A virus neuraminidase relative to drug and antibody binding sites. Biol Direct, 4(18).  12 W. Hu / J. Biomedical Science and Engineering 3 (2010) 1-12 SciRes Copyright © 2010 JBiSE [8] Laurel, G., James, S., Dmitriy Z. et al. (2005) A single amino acid substitution in 1918 influenza virus hemag- glutinin changes receptor binding specificity. Journal of Virology, 79, 11533-11536. [9] Skehel, J.J., Stevens, D.J., Daniels, R.S., Douglas, A.R., Knossow, M. et al. (1984) A carbohydrate side chain on hemagglutinins of Hong Kong influenza viruses inhibits recognition by a monoclonal antibody. Proc Natl Acad Sci U S A, 81, 1779-1783. [10] Wiley, D.C., Wilson, I.A. and Skehel, J.J. (1981) Struc- tural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature, 289, 373-378. [11] Caton, A.J., Brownlee, G.G., Yewdell, J.W. and Gerhard, W. (1982) The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell, 31, 417-27. [12] Tsuchiya, E., Sugawara, K., Hongo, S., Matsuzaki, Y., Muraki, Y. et al. (2001) Antigenic structure of the hae- magglutinin of human influenza A/H2N2 virus. J Gen Virol, 82, 2475-2484. [13] Michael, W.D. and Pan, K.Y. (2009) The epitope regions of H1-subtype influenza A, with application to vaccine efficacy. Protein Engineering, Design & Selection, 22, 543-546. [14] Katoh, K., Kuma, K., Toh, H., Miyata, T. (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res, 33, 511-518. [15] David, M. (2003) Information theory, inference, and learning algorithms. Cambridge University Press. [16] Breiman, L. (2001) Random forests, machine learning, 45 (1), 5-32. [17] Cox, T.F. and Cox, M.A.A. (2001), Multidimensional scaling, chapman and hall. [18] Colman, P.M., Hoyne, P.A. and Lawrence, M.C. (1993) Sequence and structure alignment of paramyxovirus he- magglutinin-neuraminidase with influenza virus neura- minidase. J. Virol, 67, 2972-2980. [19] Andrea, K., Gabriel, R.N. and Ivan, K.H., Sccarontefan, J. (2002) Sequence similarities and evolutionary relation- ships of influenza virus A hemagglutinins, Virus Genes, 24, 57‐63. [20] Waterhouse, A.M., Procter, J.B., Martin, D.M.A., Clamp, M and Barton, G.J. (2009) Jalview version 2 – A multi- ple sequence alignment editor and analysis workbench. Bioinformatics, 5, 1189‐1191. [21] Enrique, T. and Mu˜noz, M.W.D. (2005) Epitope analy- sis for influenza vaccine design. Vaccine, 23, 1144‐1148. [22] Weis, W., Brown, J.H., Cusack, S., Paulson, J.C., Skehel, J.J. and Wiley, D.C. (1988) Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature, 333(6172), 426-31. [23] Veljko, V., Henry, L.N., Sanja G. et al. (2009) Identifi- cation of hemagglutinin structural domain and polymor- phisms which may modulate swine H1N1 interactions with human receptor. BMC Structural Biology, 9(62). [24] Nikolai, V.K., Mikhail, N.M. and Aleksandra, S.G. (2000) Intergenic HA–NA interactions in influenza A virus: postreassortment substitutions of charged amino acid in the hemagglutinin of different subtypes. Virus Research, 66,123-129. [25] Garten, R.J., Davis, C.T., Russell, C.A. et al. (2009) Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans. Science, 325, 197-201. [26] Kumar, S., Nei, M., Dudley, J. and Tamura. K., (2008) MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinfor- matics, 9, 299-306. [27] Lu, B., Zhou, H., Ye, D., Kemble, G. and Jin, H., (2005) Improvement of influenza A/Fujian/411/02 (H3N2) virus growth in embryonated chicken eggs by balancing the hemagglutinin and neuraminidase activities, using re- verse genetics. J. Virol, 79, 6763-6771. [28] Jin, H., Zhou, H., Liu, H., Chan, W.N., Adhikary, L. et al. (2005) Two residues in the hemagglutinin of A/Fujian/ 411/02-like influenza viruses are responsible for antigenic drift from A/Panama/2007/99. Virology, 336, 113- 119. [29] Elodie G., Naomi A.S., Martin S. et al. (2005) Large- scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature, 437, 1162-1166. [30] Du, X.J., Wang, Z., Wu, A.P., Song, L., Cao, Y., Hang, H.Y. and Jiang, T.J. (2008) Networks of genomic co-occurrence capture characteristics of human influenza A (H3N2) evolution. Genome Res, 18, 178-187. [31] Huang, J.W., King, C.C. and Yang, J.M. (2009) Co-evolution positions and rules for antigenic variants of human influenza A/H3N2 viruses. BMC Bioinformatics, 10(Suppl 1), S41. [32] Michel, W., Chen, C.C., Kemp, M.M. and Linhard, R.J. (2009) Synthesis and biological evaluation of non- hy- drolyzable 1,2,3-triazole-linked sialic acid derivatives as neuraminidase inhibitors. European Journal of Organic Chemistry, 2009(16), 2587. [33] Gocnı´k, M., Fislova´, T., Mucha, V., Sla´dkova´, T. et al. (2008) Antibodies induced by the HA2 glycopolypeptide of influenza virus haemagglutinin improve recovery from influenza A virus infection. Journal of General Virology, 89, 958-967.
|