Journal of Cancer Therapy
Vol.4 No.6(2013), Article ID:35274,16 pages DOI:10.4236/jct.2013.46128
Role of Cholinergic Receptors in Colorectal Cancer: Potential Therapeutic Implications of Vagus Nerve Stimulation?
![]()
1Centre de Recherche Inserm U836, Institute of Neurosciences, Grenoble, France; 2University of Grenoble, Grenoble, France; 3Hospitalo-Universitary Center, CHU of Grenoble, Grenoble, France.
Email: *jacquier-sarlin@ujf-grenoble.frt
Copyright © 2013 Marjolaine Pelissier-Rota et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 28th, 2013; revised June 30th, 2013; accepted July 8th, 2013
Keywords: Cell Adhesion; Colorectal Cancer; Tumor Progression; Cholinergic Receptors; Inflammation; Vagus Nerve
ABSTRACT
Inflammatory Bowel Disease (IBD) patients, such as Crohn’s disease or ulcerative colitis suffer from chronic and relapsing intestinal inflammation that favours the development of colitis associated cancer (CAC). This inflammation is initiated by aberrant activations of the innate immune responses associated to intestinal barrier defects. The conventional medical therapies consist to decrease the inflammatory response, which also decrease the risk of colon carcinoma but lead to severe side-effects. Recently, a number of animal studies have demonstrated that innate immune responses are attenuated by stimulation of the efferent arm of vagus nerve (VN) through its neurotransmitter acetylcholine (ACh), that acts on resident macrophages α7 nicotinic receptor (α7 nAChR). ACh also acts as a signalling molecule in epithetlial cells through cholinergic receptors such as nAChR or muscarinic (mAChR) receptors. In the current study, we aimed to extend these findings to CAC prevention by treating human adenocarcinoma cell lines through targeting cholinergic receptors with nicotine (which binds nAChR) and ACh (which binds both cholinergic receptors). Using HT-29 and Caco-2 cell lines, we demonstrated that ACh-induced activation of mAChR results in cell dissociation together with changes in expression and localization of intestinal tight and adherens junction proteins. ACh-induced modulation of cell adhesion proprieties correlates with the acquisition of invasive potential. By contrast, nicotine-mediated activation of nAChR maintains epithelial cell organisation. ACh-released by VN stimulation (VNS) could effectively preserve epithelium integrity thus limiting inflammatory response and tumor development. However, attention should be paid on the nature of the cholinergic receptor solicited. Indeed, regarding to the protective effects of nAChR signalling on epithelial cells, activation of mAChR would worsen the disease and led to increase inflammation. These data have important repercussions on the therapeutic potential of VNS in IBD and CAC, which may represent “the yin and yang” of the intestinal homeostasis.
1. Introduction
Inflammatory Bowel Diseases (IBD), such as Crohn’s disease (CD) or ulcerative colitis (UC), are multifactorial chronic inflammatory diseases of the gastrointestinal tract for which the exact causative mechanism is still unclear. According to a current hypothesis, an increased intestinal permeability due to an epithelial barrier defect, coupled with a dysfunctional immune response participate to the development of chronic intestinal inflammation [1].
In the long-term, IBD can induce further complication and patients have an increased risk developing other pathologies such as colorectal cancer (CRC). About 20% of CRC cases can be genetically attributed to a family history. Involvement and mechanisms by which environmental factors contribute to the disease are still unclear. Colitis associated cancer (CAC) is a type of colon cancer which is preceded by clinically detectable IBD. CAC develops from dysplasia, which is stimulated by chronic inflammation, rather than polyps. Even though CRC does not always develop after IBD, its high frequency in patients with IBD represents a paradigm for the connection between inflammation and cancer in terms of epidemicology and mechanistic studies in preclinical models (for review [2]). UC increases cumulative risk of CRC by up to 18% - 20%, while CD by up to 8% after 30 years of active disease. The increase in prevalence of CAC in IBD patients seems to correlate with the chronic inflamematory conditions of the intestinal mucosa, in particular with the degree [3], duration [4,5] and anatomical extent of colonic inflammation [6], as well as the presence of primary sclerosing cholangitis, and IBD management such as efficacy of anti-inflammatory therapies [7]. In animal models, intraperitoneal injection of the carcinogen azoxymethane (AOM) followed by repeated cycles of dextran sulfate sodium (DSS) or mice lacking the gene for the IL-10 cytokine, chronic inflammation also results in an increased frequency of intestinal tumors [8,9]. Epidemiological studies report that the long-term administration of anti-inflammatory drugs decreases the risk of colon carcinoma [10]. The conventional medical therapy for IBD consists to reduce the inflammatory response using 5-aminosalicylates, corticosteroids, immunosuppressives (azathioprine/mercaptopurine), and biological therapies (anti-TNFα), but these treatments have severe side-effects [11,12].
Vagus nerve (VN) is the main nerve of the parasympathic division of the autonomic nervous system for the thoracic and abdomino-pelvic viscera that controls heart rate, hormone secretion and gastrointestinal motility/secretion [13]. Recent studies indicate that VN is also an immunomodulator [14]. In experimental models of inflammatory disease, vagus nerve stimulation (VNS) attenuates the production of pro-inflammatory cytokines and inhibits the inflammatory process [15,16]. The inflammatory reflex is a centrally neuro-endocrine-immune integrated physiological mechanism that maintains homeostasis through: 1) the activation of the hypothalamic pituitary adrenal axis by VN afferent fibers and 2) the cholinergic anti-inflammatory pathway (CAP) by VN efferent fibers [17,18]. At the molecular level, the CAP relies on the effects of acethylcholine (ACh), the VN principal neurotransmitter, on macrophages [18]. ACh signals through either muscarinic (G-protein-coupled) receptors (mAChR) or nicotinic (ligand-gated ion channels receptors (nAChR) [19]. There are five subtypes of mAChR (M1-M3) which are products of distinct genes [20]. They present similar structure constituted of seven transmembrane helice (TM1-TM7) and three extracellular and three intracellular loops [21]. nAChRs are transmembrane proteins composed of five subunits arranged around an axis perpendicular to the membrane. Therefore, nAChR is a homo-(α7 or α9) or a hetero-pentamer composed from various subunits (α2 - α10; β2 - β4, d, e, g). These cholinergic receptors are present in the central and peripheral nervous systems, in immunocompetent cells (monocyte, lymphocyte, and macrophage). Activation of α7 nAChR, either directly through interaction with selective nicotinic agonist (such as nicotine) or indirectly through the activation of the autonomic nervous systems (e.g. VN), inhibits adaptive and innate immune responses [22-24]. The affinity of nicotine for this receptor is greater than its physiological agonist, ACh [25]. nAChR signaling also reduces macrophage cytokine production and inflammation in animal models of pancreatitis [26], DSS-induced colitis [27], 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis [28] and intestinal ileus [29]. Thus, VNS appears as an alternative therapy to conventional treatment, for digestive disorders such as IBD. Currently, VNS is used in treatment of patient with seizure disorders and depression through the stimulation of VN afferent fibers [30] or metabolism disorders such as obesity (for review [31]).
Inflammation could contribute to carcinogenesis by increasing the level of reactive oxygen species that have a mutagenic effect on DNA (tumor initiation) [32] or by generating an environment in favour of sustained growth, angiogenesis, migration and invasion of tumor cells (tumor progression and metastasis). Various components of the inflammatory environment in IBD are key elements in the different steps of cancer [33]. Recent works have elucidated the role of various immune cells and mediators in all the steps of colon carcinogenesis with the dissection of some molecular pathways [2,34,35]. In this paper, we will focus on the therapeutic potential of VNS in CAC, not only by regulating inflammatory response at the immune level but also by preventing or limiting intestinal barrier breakdown associated to the inflammation process. Indeed, epithelial cells also express cholinergic receptors [36]. While the role of α7 nAChR in intestinal cells is less understood, cholinergic receptors regulate ion transport across cell membrane and thereby affect intestinal water movement [37]. The main subtype of mAChR in rat and human intestinal epithelial cells is the M3 and to a lesser extends the M1 receptor [38-40].
Epithelia form a barrier constituted of specialized cells characterized by structural features including polarized morphology and cell-cell contacts. They lie on a basement membrane, which is organized into a complex structure containing collagen type IV (COIV), various laminin isoforms, and proteoglycans [41-43]. Interactions between cells and this specialized extracellular matrix (ECM) are crucial for essential biological processes such as migration, proliferation, differentiation, and cell survival. The cell-cell or cell-ECM interactions are mediated through various transmembrane receptors, which are linked intracellularly to cytoskeleton components and signal transduction molecules [44,45]. Tight junctions (TJ) are composed of transmembrane proteins (claudins, occludins and junctional adhesion molecules), scaffold proteins like zona occludens (ZOs) that link the actin cytoskeleton, and intracellular regulatory molecules including kinases [46]. Directly beneath TJ are the adherens junctions (AJ) with E-cadherin connected to the actin cytoskeleton via α/β catenins and regulated by p120ctn [47]. Disorganization of these cellular junctions could participate in the infection process but also in the cellular dedifferentiation preceding carcinogenesis and cell migration [48]. These epithelial alterations are more pronounced in UC tissues in which the development of malignancies is apparently more frequent than in CD tissues, suggesting that disturbances of junction-associated molecules are likely to be involved in carcinogenesis from IBD patients. Using HT-29 and Caco-2 cells, two human colonic adenocarcimoma cell lines, we demonstrated that VNS could preserve epithelium integrity thus limiting inflammatory response and tumor development. However, attention should be paid on the nature of the cholinergic receptor solicited. Indeed, regarding to the protective effects of nicotine-induced activation of nAChR on epithelial cells, mAChR activation by ACh favours the acquisition of invasive potential with a modulation of cell adhesion proprieties. These data have important repercussions on the therapeutic potential of VNS in IBD and CAC.
2. Materials and Methods
2.1. Cell Culture
The human colon adenocarcinoma cell lines HT-29 and Caco-2/TC7 were cultured at 37˚C in a 5% CO2 atmosphere in DMEM containing 25 mM glucose (Invitrogen, Cergy Pontoise, France) and supplemented with 10% FCS, 5% penicillin and streptomycin. The medium was changed every day to avoid glucose exhaustion, which leads to differentiation. The differentiation of HT-29 cells was initiated by replacing standard medium by glucose free DMEM (Invitrogen) supplemented with 10% dialyzed foetal calf serum, 5 mM galactose, 15 mM HEPES, selenous acid (10−2 mg/ml), penicillin, and streptomycin [49]. This medium (Gal-medium or differentiating medium) was changed every day. The differentiation of Caco-2 occurs after 15 days of culture postconfluence. The cells were harvested in phosphate-buffered saline (PBS) supplemented with 1 mM EDTA and 0.05% trypsin (w/v).
2.2. Chemical Reagents and Antibodies
Polyclonal antibodies directed against M3 mAChR (Cat. N˚. AB41169) provided from Abcam (12964, Abcam, Paris, France). Anti-human E-cadherin (Clone HECD1) monoclonal antibody was obtained from Takara Biochemicals (Cambrex Bio Science, Paris, France). Monoclonal antibody against p120ctn (clone 98) was purchased from BD Biosciences/Transduction Laboratories (Pont de Claix, France). Anti-actin (Cat. N˚. A2066), and anti-β catenin (Cat. N˚. C2206) polyclonal antibodies and a7 nAChR (clone 306) monoclonal antibody were obtained from Sigma Aldrich (L’Isle d’Abeau, France). Polyclonal antibody directed against Src-PTyr418 (AT- 7135) was purchased from MBL Calbiochem (VWR International, Fontenay-sous-Bois, France), monoclonal anti-Src (Clone GD11) was from Millipore (Molsheim, France). Occludine clone (OC-3F10) and ZO-1(clone ZO1-1412) monoclonal antibodies were from Invitrogen (Cergy Pontoise, France). Alexa-conjugated goat antimouse secondary antibody was obtained from Molecular Probes (Eugene, OR). Horse Radish Peroxydase-conjugated goat anti-mouse was from Bio-Rad (Marnes-la-Coquette, France), donkey anti-rabbit antibodies were from Jackson Immunoresearch (Immunotech, Marseille, France).
2.3. RT-PCR
Total RNA extractions were performed using TrizolTM reagent and 1 µg of total RNA was denaturized and subsequently processed for reverse transcription using MMLV (Invitrogen) according to manufacturer’s instructions and run on thermocycler (Eppendorf). Primer sequences and probes are:
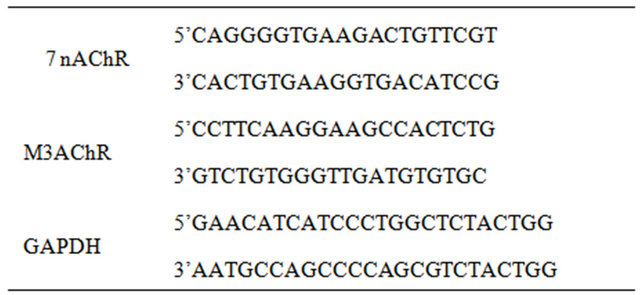
PCR conditions are: 5 min at 92˚C followed by 35 cycles (40 sec at 92˚C, 40 sec at 60˚C and 1 min at 72˚C) and 10 min at 72˚C. PCR were analyzed on 1.5% agarose gel. Quantification was performed using Image J (NIH software). GAPDH were used as housekeeping gene.
2.4. Immunoblot
Cells were grown to confluence and lysed with a buffer made of 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and supplemented with 1 mM PMSF, 2 mg/mL aprotinin, 10 mg/mL leupeptin, 10 mM pepstatin, 2 mM CaCl2 and MgCl2, for 15 min on ice. Protein concentrations in lysates were determined using the copper reduction/bicinchoninic acid (BCA) assay (Pierce Chemical Co) according to the manufacturer’s instructions. Proteins (30 mg in SDS-b mercaptoethanol sample buffer) were resolved on 10% polyacrylamide gels, transferred into PVDF membranes (Hybond-C super; Amersham), and blocked in 5% bovine serum albumin in 0.1% Tween 20 in TBS for 1 hr at room temperature. After overnight incubation at 4˚C with primary antibodies diluted in the blocking solution, blots were washed in TBS, 0.1% Tween 20 and then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (dilution of 1:10000) for 1 hr at room temperature before extensive washes. The blots were revealed by chemiluminescence (Amersham ECL reagents) and quantified with Image J software from NIH. Primary antibodies were used at the following dilutions: antihuman a7 nAChR and M3 mAChR, E-cadherin, p120ctn and b-catenin (1:1000), ZO-1 (1:2000), and anti-actin (1:2000).
2.5. Immunofluorescent Staining
Cells were grown on glass coverslips and were treated as described previously [50]. After fixation with PFA-4% sucrose, non-specific sites were blocked for 1 hr at 37˚C with 3% BSA-0.5% Tween20. Then cells were incubated for 1 hr at 37˚C with specific antibodies which were diluted in the blocking solution at 1:100 for primary and 1:500 for secondary antibodies. Fluorescence photomicrographs were taken with a confocal microscope at the x100 objective (Leica TCS SPE) or an epifluorescence microscope at the x100 objective (ZEISS, Avio Vert 200 M).
2.6. Extracellular Matrix Preparation and Cell Adhesion
Tissue culture dishes were coated with LM-332 using the following methods: A431 epidermoid cells were cultured to confluence on various surfaces at 37˚C to allow for the deposit of LM-332, then cells were removed as previously described [51,52]. Briefly, confluent monolayers were sequentially extracted with 1% (v/v) Triton X-100 in PBS, followed by 2M urea in 1M NaCl. All extraction buffers contained protease inhibitors (1mM phenyl-methylsulfonyl fluoride and 2 mM N-ethylmaleimide). Plates were washed in PBS, incubated with 1%BSA, and stored at −20˚C. Human collagen type IV from placenta was obtained from Sigma Aldrich. Coating of plastic Petri dishes (Microtiter plates 96-well, Nunclone; Nunc, Roskilde, Denmark) was performed by overnight incubation with extracellular matrix proteins (10 µg/mL) at 4˚C. Plates were saturated with 3% (w/v) BSA in PBS for 2 hrs at 37˚C to block nonspecific adhesion. HT-29 cells were harvested and pre-treated or not with 100 nM nicotine or ACh for 30 min before to be plated (5 ´ 104 cells/ well) in triplicate in coated 96-well microtiter plates and incubated from 0 to 60 min at 37˚C. Non adherent cells were removed by washing three times with PBS, and cell adhesion was estimated by a colorimetric cell proliferation assay (CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay; Promega).
2.7. Cell Invasion Assay
Total HT-29 cells (2.5 ´ 10−5/mL) were placed in the top compartment of a 24-multiwell insert plate (BD Falcon), which was separated from the bottom compartment by BD-Matrigel Matrix membrane, with 0.4-µm pore size. Serum-free RPMI with or without nicotine or ACh (100 nM) were added into the top compartment and 10% FCS into the bottom compartment. After 48 hrs at 37˚C in a 5% CO2 atmosphere, cells that had invaded through the Matrigel were analyzed: cotton swabs were used to remove cells on the upper surface of inserts. After fixation with PFA 4%, migratory cells were stained with Hematoxylin Gill’s formula (Vector Laboratories) and manually counted under the microscope. The mean values of the readings and their SEM were calculated, and statistical differences were analyzed using Student’s t-test for non-paired samples.
2.8. Assay for Cell Proliferation
Cell were plated in 96-well plates (2000 cells/well) at day 0 and cultured in complete DMEM medium without or with nicotine and ACh (100 nM). Cell proliferation was evaluated from day 1 to 3 using the CellTiter 96 Kit (Promega) according to the manufacturer’s instructions.
2.9. Densitometric Analysis and Statistics
Immunoblots shown are representative of at least three independent experiments. All graphs represent the mean value ± SD of protein expression levels measured by densitometric analysis in “Image J” software (NIH). Statistics were performed with unpaired t-test and statistical significance was given by the number of asterisks (*P < 0.05; **P < 0.01; ***P < 0.001).
3. Results
3.1. Expression and Regulation of Cholinergic Receptors Expression in Human Colon Cancer Cell Lines
It has been described that human colon cancer cell lines expressed mainly M3AChR and α4, α5, α7 and β1 nAChR subunits but only α7 subunits form a functional receptor [53]. First, we analyzed whether α7 nAChR and M3AChR, the two major cholinergic receptors susceptible to be activated by ACh released by VNS are present tors was visualized by staining HT-29 cells and then imaging under confocal microscopy (Figure 1(a)). Without treatment, immunofluorescence staining demonstrated that these receptors produced varying intensities of labelling in the cells, seemingly localized at the membrane. Some cytoplasmic dots are observed probably matching with cholinergic receptor neosynthesis or endocytosis. There is also an accumulation of vesicles enriched in α7
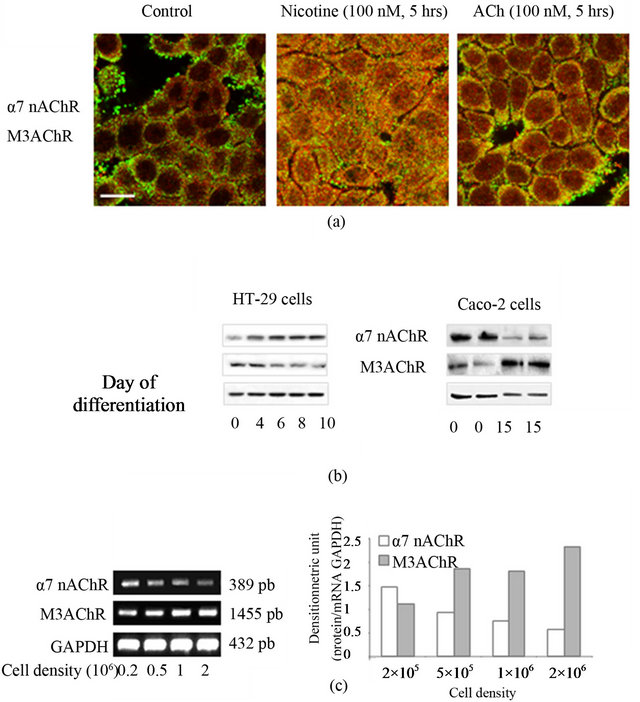
Figure 1. Functional cholinergic receptors: expression, distribution and regulation. (a) Expression and distribution of functional cholinergic receptors in HT-29 cells. Confocal immunofluoresence analysis of α7 nAChR (green) and M3AChR (red) in HT-29 cells incubated (or not) with nicotine or ACh (100 nM, 5 hrs). Co-localization is illustrated by yellow staining. Scale bar, 20 µm. (b) Cholinergic recaptor expression during cell differentiation. HT-29 cells are differentiated during 10 days by glucose starvation. Caco-2 cells are differentiated during 15 days after these cells formed a monolayer. Expressions of α7 nAChR and M3AChR in HT-29 cells and Caco-2 TC7 cell lines are quantified from immunoblot. Actin is used as loading control. C: Influence of cell density on cholinergic receptor expression. HT-29 cells are seeded at different concentration. Expression of cholinergic receptors are analysed by RT-PCR with specific primers. GAPDH are used as keeping house gene.
nAChR at the extern edge of cells not engaged in cellcell contacts. In presence of cholinergic ligands (nicotine or ACh, 100 nM, 5 hrs) we observed cell dissociation (black spaces appeared between adjacent cells) associated with a modification of the cell shape (more spreading cells) in presence of ACh. Cholinergic treatment induces a hypercholinergic response (desensitization process) which is illustrated by a decrease of cholinergic receptor density at the membrane of epithelial cells with a concomitant increase of cytoplasmic labelling which indicate that they receptors are functional. Then their expression was analyzed at the protein level and according to the status of differentiation of epithelial cell lines (Figure 1(b)). For this purpose we chose two adenocarcinomas cell lines (HT-29 and Caco-2) able to differentiate in vitro under specific culture conditions. The differentiated state of HT-29 cells is obtained by substitution of glucose for galactose (Gal medium) in culture medium [54]. A variety of changes in the adhesive properties of HT-29 cells is observed during the first ten days of culture in the Gal medium which corresponds to the initial step of their differentiation [55,56]. Enterocyte differentiation of Caco-2 cells occurs after they reached confluence. They must be grown in confluent monolayer at least for 15 days [57]. Western blot also confirmed the presence of α7 nAChR and M3AChR in both cell lines. According to the differentiation status of cells, it appeared that α7 nAChR increases during the differentiation of HT-29 cells and stays constant in Caco-2 cells. In contrast M3AChR expression decreases in HT-29 cells while it increases in Caco-2 cells. AChR expression according to the cell density was analyzed by RT-PCR (Figure 1(c). α7nAChR are present at low cell density and their expression decreases at higher cell concentration. In contrast the expression of M3AChR slightly increases with the cell density. These observations could explain the discrepancy noticed during the differentiation of the two cell lines. Indeed during the differentiation process, the substitution of glucose by galactose leads to cell death and thus lost of HT-29 cells. In contrast Caco- 2 cells are still confluent.
3.2. Only mAChR Induces a Loss and Redistribution of TJ and AJ Proteins
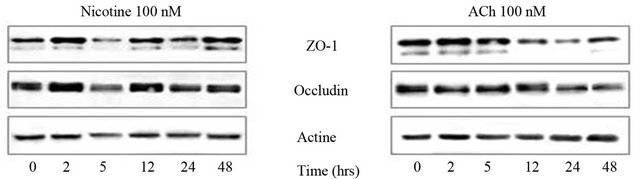
Disruption of epithelial barrier integrity is identified as one of the pathologic mechanisms in IBD and cancer development. Adequate intestinal TJ protein function determines intestinal barrier integrity. We measured changes in expression and localization of occluding and ZO-1 to determine whether modulation of these TJ proteins correlates with changes observed with cholinergic treatment [58,59]. For this purpose, we use differentiated Caco-2 cells which established “mature” TJ compared to HT-29 cells. Confluent monolayers of Caco-2 cells differentiated for 15 days were harvested and immunoblotted (Figure 2(a)). Western blots indicated that while nicotine didn’t modify the expression of ZO-1 and occludin, ACh induces a time dependent loss of expression of these two TJ proteins. Fluorescence microscopy analysis confirmed the changes observed in western blots (Figure 2(b)). Membrane polarity was estimated by the actin cytoskeletal organization in microvilli at the apical side and actomyosin ring behind. In contrast to nicotine which preserves the actin cytoskeleton structures, ACh induces a reorganization of actin cytoskeleton with a loss of actomyosin ring and microvilli. At the same time, there is a redistribution of ZO-1 and to a lesser extend occludin from the intercellular junctions. Effect of cholinergic treatment was then evaluated on subjacent AJ proteins (Figure 3). Similar results were obtained with HT-29 and Caco-2 cells. In HT-29 cells, ACh also induces a loss of E-cadherin and catenins protein expres
 (a)
(a)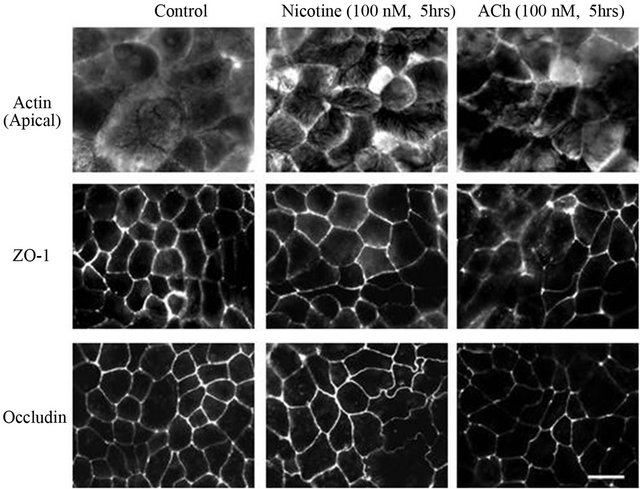 (b)
(b)
Figure 2. Cholinergic signaling modifies TJ protein expression and distribution. (a) Differentiated Caco-2 cells, which express TJ, are treated (or not) with nicotine or ACh (100 nM) during 0 to 48 hrs. Expressions of ZO-1 and occludin are analyzed by immunoblotting. Actin is used as a loading control. B: Immunofluorescence images of actin cytoskeleton organization at the apical pole and TJ protein distribution in Caco-2 incubated (or not) with nicotine or ACh (100 nM, 5 hrs). Scale bar: 20 µm.
sion determined by western blots (Figure 3(a)). Confocal microscopy analysis showed that nicotine induces slight cell dissociation but E-cadherin was still at the membrane together with p120ctn/β-ctn. Both catenins were co-localized (Figure 3(b)). In presence of ACh, cells completely dissociated and E-cadherin staining was cytoplasmic. Membrane labelling of catenins also disappeared and less co-localization between the two proteins was observed.
Localization and stability of AJ proteins were controlled by phosphorylation/dephosphorylation events. The kinase Src is one of the kinases involved in this process and it can be activated by cholinergic ligands. We then compared the ability of nicotine and ACh to activate Src by measuring its phosphorylation on tyr418 (Figure 4). Exposure of HT-29 cells to nicotine doesn’t induce any Src activation during the time course experiment. By contrast Ach treatment leads to a time-dependent activation of Src with a maximum at 5 hrs.
Altogether these data indicate that ACh but not nicotine induces an alteration of epithelial cell integrity characterized by a loss of TJ and AJ proteins subsequently to cell dissociation and protein endocytosis and degradation. This phenomenon could be address by the activation of
 (a)
(a)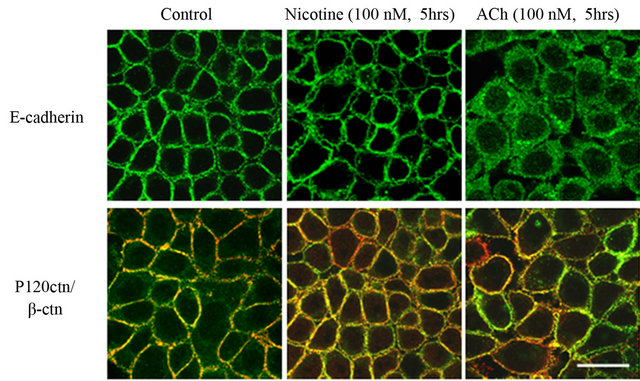 (b)
(b)
Figure 3. Cholinergic signaling modifies AJ protein expression and distribution. (a) HT-29 cells are treated (or not) with nicotine or ACh (100 nM) during 0 to 48 hrs. Expressions of E-cadherin, b-catenin and p120ctn are analyzed by immunoblotting. Actin is used as a loading control. (b) Confocal immunofluorescence images of AJ proteins. HT- 29 cells which express AJ are incubated (or not) with nicotine or ACh (100 nM, 5 hrs. Upper panel: E-cadherin (green), lower panel: p120ctn (green), β-ctn (red). Co-localization of p120ctn and β-catenin is illustrated by yellow staining. Scale bar: 20 µm.

Figure 4. Src-phosphorylation following AChR activation. Westernblots showing Src-pTyr418 and Src total expression following nicotine and ACh treatment (100 nM) during 0 to 24 hrs in HT-29 cells.
the Src kinase.
3.3. ACh Modifies Cell-ECM Adhesion and Stimulates Human Cancer Cell Invasion
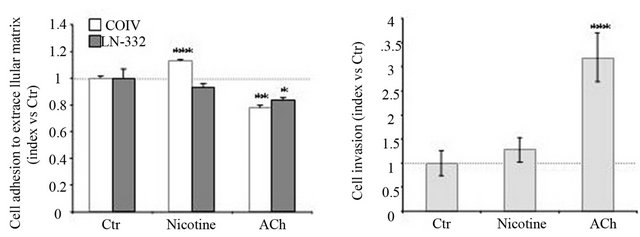
The AJ’s disassembly correlates with a loss of cell-cell adhesion and an acquisition of migratory potential [60]. This event is associated to a modification of cell-ECM adhesive properties. We then compared the adhesive potential of HT-29 cells treated or not with cholinergic receptors on collagen IV (COIV) and laminin-332 (LN- 332), two major proteins of the basal membrane (Figure 5(a)). Nicotinic treatment enhances cell adhesion to COIV and had no effect on LN-332. By contrast, exposure of HT-29 cells to ACh diminishes adhesion to both ECM proteins. These modifications of ACh-induced cellECM interactions were correlated to an enhanced capacity of HT-29 cell invasion (Figure 5(b)). Using Matrigel chamber assays, we found that ACh induced a 3-fold increase in HT-29 cell invasion. Nicotine had no effect on HT-29 cell invasion. These results indicate that activation of mAChR by ACh modifies cell adhesive proprieties in favour of invasion.
3.4. Ach Enhances Cell Proliferation
ACh is an autocrine/paracrine factor in various non neuronal cells. Consistent with previous observations, we found that ACh reproducibly induced a 1.7-fold increase in HT-29 cell proliferation at 48 hrs and 72 hrs after seeding (Figure 6). Nicotine also significantly stimulated HT-29 cell proliferation (1.3-fold increase).
In this present work, data indicate that ACh-induced mAChR signaling plays a key role in colon cancer cell polarity and adhesion, proliferation and invasion compared to nAChR.
4. Discussion
IBD patients suffer from chronic and relapsing intestinal inflammation that favours the development of CAC. This
 (a) (b)
(a) (b)
Figure 5. Cholinergic signaling modulates migration and invasion potential in HT-29 cells. (a) Contrary to mAChR signaling, nAChR activation promotes cell adhesion. HT-29 cells are treated (or not) with nicotine or ACh (100 nM, 5 hrs) 30 min before adhesion and then plated on either type IV collagen (COIV) or laminin-332 (LN-332) at 10 µg/mL. Cell adhesion is evaluated using a Cell Titer Aqueous MTT reagent kit. Results are expressed relative to the untreated cells which are fixed at 1. Data represent the mean +/- SEM of 4 experiments. (b) Contrary to nAChR signaling, mAChR activation favors cell migration. Effect of nicotine and ACh (100 nM, 48 hrs) on HT-29 cell transmigration is determined using a transwell assay. Cells which migrated through the matrigel are stained with hematoxylin and visualized and counted by phase contrast microscopy. Results are expressed according to the control which is fixed at 1. Data (n > 3) represent the mean +/− SEM.p
inflammation is initiated by aberrant activations of the innate immune responses associated to intestinal barrier defects. Recently, a number of animal studies have demonstrated that innate immune responses are attenuated by stimulation of the efferent arm of VN through its neurontransmitter ACh, that acts on resident macrophages a7 nAChR, [15,16]. It appeared that ACh is not only a neurotransmitter but it also acts as a signaling molecule in non-neuronal tissues. In the current study, we aimed to extend these findings to CAC prevention by treating human adenocarcinoma cell lines through targeting cholinergic receptors with nicotine and ACh. nAChRs and mAChRs might affect the inflammatory response in an opposite manner and may represent “the yin and yang” of the intestinal homeostasis [61]. Our data suggest that nAChR and mAChR might also affect differently cancer progression (Figure 7).
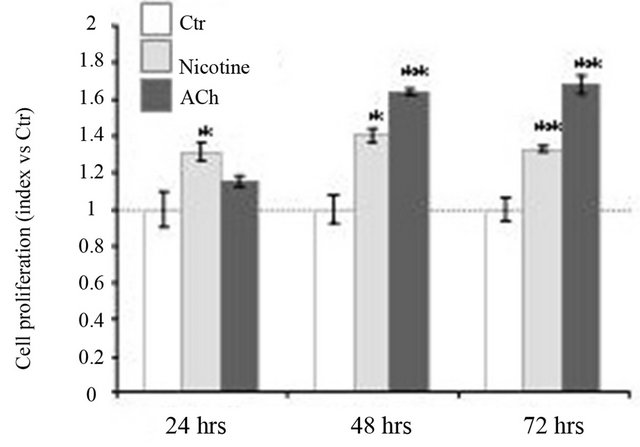
Figure 6. Cholinergic signaling modulates HT-29 cell proliferation. To determine proliferation potential, HT-29 cells are incubated (or not) with nicotine or ACh (100 nM) during 24, 48 and 72 hrs. Cell proliferation is determined by MTT assays. Results are expressed relative to the proliferation of untreated cells, which is fixed at 1. Data represent the mean +/− SEM of 3 experiments.
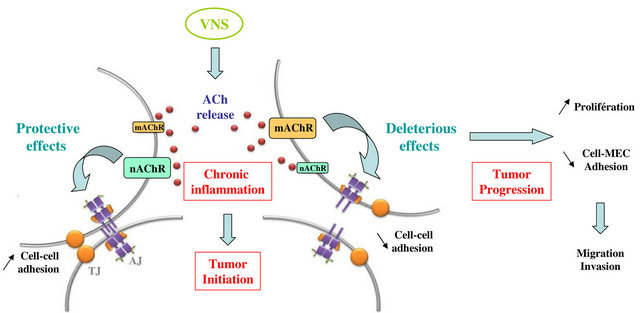
Figure 7. “The ying and the yang” effect of VNS. In IBD patients, VNS would have protective effects on epithelial barrier integrity via nAChR. This effect relies on a positive modulation of intercellular junction proteins in term of expression and localization. In an opposite manner, VNSmediated activation of mAChR leads to cell-cell contact disruption with intercellular junction proteins degradation and delocalization together with a decrease of cell-ECM adhesion. Altogether these effects favor cell migration and invasion as observed in tumor progression.
4.1. Acetylcholine, Neurotransmitter and Autocrine/Paracrine Mediator
Emerging evidences indicate that normal and neoplastic non-neuronal cells can produce and release ACh in sufficient quantity to modulate cell function [62]. Neuronal and non-neuronal (epithelial, endothelial and immune cells) ACh is synthesized by the enzyme choline acetyltransferase (ChAT) using acetyl-CoA and choline as substrates. The later is taken up from the extracellular site by a specific high affinity choline transport-system (CHT1) [62,63]. In contrast to the situation in nerves where ACh is stored in specific vesicles, there is no storage compartment in non-neuronal cells and ACh appeared to be released directly after synthesis. In both conditions, ACh is rapidly hydrolysed by acetylcholinesterase (AChE), thereby limiting its action to immediately neighbouring cells. ACh targets mAChR and nACR with a greater affinity for the former. Both receptors are widely expressed in different non-neuronal cell types (for review [64,65]. Different mAChRs couple differentially to multiple G proteins that are describe to mediate distinct cytoplasmic signaling pathways implicated in various cellular functions. Activation of nAChR leads to Ca2+ influx that directly modulates these signaling pathways.
4.2. Regulation of the Cholinergic System in Cancer
All components of the cholinergic system such as synthesis, storage, release, inactivation as well as the expression and function of various cholinergic receptors can be affected in pathophysiological conditions. Some substantial alterations of the non-neuronal cholinergic system have already described in human colon cancers. The cholinergic system for ACh synthesis and degradation is modified in human colon cancer cell lines compared to normal tissues. The activity of AChE was measured in 55 human samples of healthy and malignant colon, sigmoid colon and rectum. It appeared that cancer decreases the average of AChE activity value, such increasing the rate of ACh [66]. Moreover, Cheng and colleagues demonstrated that ChAT staining is undetectable to weak in normal enterocytes but is moderate to strong in 50% of colon cancer examined and colon cancer cell lines [67]. This data show that colon cancer cells possess the ability to produce and release ACh, which could act as an autocrine/paracrine factor. While there is few data on the regulation of ACh release during inflammation, it has been shown that corticoids negatively modulate the non-neuronal cholinergic system by regulating transport and synthesis of ACh in particular at the surface epithetlium of trachea and intestine [68].
In gastrointestinal tissue, the primary mAChR subunits are M1AChR, M2AChR and M3AChR, but colon epithelial cells mainly express M1AChR and M3AChR [38-40,69]. Frutcht et al. reported that human colon cancer cell lines and colon cancer tissues express M3AChR. Their expression is increased up to 8-fold in cancer compared to normal tissue [70]. This over-expression is observed in 60% of colon cancer examined [71]. The over-expression of M3AChR is associated to a drop of degradation and an increasing of ACh synthesis. Corticoids also modulate the expression of mAChR, in a cell specific manner: glucocorticoids decrease M2AChR and M3AChR density in airway smooth muscles [72] while they up-regulated M2AChR in parasympathic nerves [73].
Since the discovery of ubiquitous presence of nAChR in mammalian cells, studies from many laboratories have linked nAChR with various pathological conditions including cancer [74]. These studies were performed with nicotine and concern tobacco-related carcinogenesis such as lung cancer. However it is clear that various nAChR subtypes are expressed in non-neuronal cells but their pattern of expression and their functional roles is under debate [65,75]. nAChRs are expressed in various human cancers with subunit expression altered except the a7-subunit, which is constantly expressed and functional (reviewed in cardinale et al., 2012). It has been proposed that looking at the differentiation status of the tumor, there is a tendency of a major expression of α7 nAChR protein levels in more differentiated human lung tumors [76]. We found that the M3AChR and α7 nAChR are expressed in HT-29 and Caco-2. Their expression is not correlated to the differentiation status of these cell lines but rather to the cellular density. This result was confirmed using various human colonic cell lines characterized by different stage of differentiation (data not shown).
4.3. How mAChR May Participate in CAC?
Muscarinic receptor activation stimulates proliferation, migration and invasion of human colon cancer cells [77-79]. These effects have been attributed to M3AChRinduced secretion of matrix metalloprteinases (MMPs) such as MMP7 that releases EGF ligands or MMP1. Most of M3AChR effects seem to be mediated by transactivation of the EGFR. Co-expression of M3AChR and EGFR in many colon cancer cell lines associated with over expression of these receptors in the majority of colon cancer suggests that the functional interaction between M3AChR and EGFR is important for colon cancer regulation. In human colon cancer cell lines, we found that ACh alters the assembly of intercellular junctions (AJ and TJ), a process associated to epithelial barrier failure. This correlates with the observation that mAChR (in particular M3) increases epithelial cell permeability [80]. This ACh–induced alteration of cellular contacts, decreases cell-ECM adhesion and stimulates invasion of HT-29 cells. Recent research demonstrated that the molecular mechanism of cholinergic control of keratinocyte adhesion involves an ACh receptor-dependant changes in the phosphorylation status of adhesion [81,82]. The phosphorylation of adhesion molecules plays an important role in assembly/disassembly of intercellular junctions controlling their structural integrity and the adhesive capacity [83-86]. Tyrosine phosphorylation of p120ctn or serine phosphorylation of α-ctn correlate with a decrease of cell adhesion and represent the initial event for their degradation [87,88]. The phosphorylation status of a given adhesion molecule is determined by both protein kinase and phosphatase activities. Various kinases have been described to phosphorylate proteins among which the Src kinase [89,90]. We found that ACh but not nicotine induces Src phosphorylation on Tyr 418.
Strong evidences for non-neuronal ACh production are reported in human keratinocytes and small lung cancer cells where this transmitter acts as an autocrine/paracrine growth factor stimulating cell proliferation [91,92]. Similarly, ACh produced and released by H508 colon cancer cells interacts with M3AChR and acts as an autocrine growth factor [93]. In HT-29 cells, we found that ACh also stimulates cell proliferation. An increasing number of evidences suggests that cyclooxygenase-2 (Cox-2) expression is associated with colon cancer induction and progression by stimulating cell proliferation, suppressing apoptosis, inducing tumor angiogenesis and promoting invasiveness [93]. However, M3AChR and Cox-2 share the same signaling cascade. M3AChR activation is coupled to the phospholipase C (PLC) signaling cascade and PLC-activated protein PKC is an inducer of Cox-2. In HT-29 cells, Yang and Frucht showed that stimulation of M3AChR by carbachol up-regulates COX-2 protein expression in a concentration and timedependent manner [71]. So, Cox-2 could constitute the molecular bridge between inflammation and cancer, and mediate in part the effect of M3AChR.
Together, these finding indicate that M3AChR expression plays a strong role in intestinal tumor promotion by modulating key process of carcinogenesis. Using different in vivo models, Raufman et al., showed that M3AChR gene ablation decreases both colon tumor number and size and the degree of dysplasia [94]. Indeed, only lowgrade adenomas are detected from Apcmin/+ M3AChR−/− mice while in Apcmin/+ M3AChR+/+ mice both low and high grade adenomas are observed [95]. Furthermore, M3AChR selective inhibitors have been proven to reduced the size of small cell lung cancer xenografts in nude mice [96] suggesting that M3AChR are key promoters of tumor growth.
4.4. Protective Effects of nAChR
As mAChR, nAChR have been described to modulate epithelial cell-cell contacts, adhesion and motility of respiratory epithelial cells, lung cancer and in a variety of human cancer cell lines [97-99]. Nicotine can induce the transition from a well-differentiated epithelial cell to a highly invasive carcinoma cell involving different signal transduction cascades. Long-term treatment of lung cancer and breast cancer cells is required for these effects. In these models it has been proposed that nicotine, acting via α7 nAChR, stimulates mRNA and protein expression of fibronectin with a concomitant down regulation of junctional protein expression and/or localisation [98,100, 101]. Evidence of nAChR mediated activation of integrin-dependent signaling pathway has also been obtained in colon and gastric cancer [102]. Regarding the gastrointestinal tract, Cho’s laboratory showed that nicotine induced activation of α7 nAChR and stimulated the growth and angiogenesis of gastric and colon cancer by systemic as well as cellular increase in stress neurotransmitters (adrenaline, noradrenaline) leading to β-adrenergic signaling, transactivation of the EGFR and release of EGF [103-105].These finding strengthen the hypothesis that modulation of nAChR upon chronic exposition to tobacco may contribute to the development and progression of cancer.
However in some cases, nicotine and tobacco could exert protective effects. UC patients with a history of smocking usually developed their disease after they had stopped smocking [106-108]. Smoking has been found to reduce development and severity of UC and to worsen the disease in CD patients [106,109,110]. Similarly, nicotine administration ameliorates disease in DSS experimental colitis [111]; while worsened the course in TNBS colitis [112]. In patients, [113] treatment with transdermal nicotine was effective at inducing disease remission in UC patients but patients suffered from adverse effects [114]. Thus the effects of nicotine and a7 nAChR agonists may depend on many factors such as disease model and severity, the expression and the subtype of nAChRs. According to these data and our observations, it appeared that cholinergic receptor expression must be determined before VNS therapy.
Using a concentration similar to those observed in the blood of smokers (0.01 - 1 mM), we found that nicotine-mediated activation of α7 nAChR exert protective effect on epithelial morphology that can preserve from failure of barrier function. Nicotine increased or maintained the expression of various intercellular proteins such as p120ctn, Z0-1, Occludin. This mechanism may contribute to maintain barrier integrity. McGilligan has described similar results, showing that nicotine decrease Caco-2 permeability by regulating the expression of TJ proteins [115]. This process is important to reduce inflammation but also cancer progression. In a rodent model of intestinal inflammation after severe burn injury, Costantini and collaborators showed that nicotine administration prevent burn-induced intestinal permeability and limited histological injury by controlling intercellular protein expression and localisation [116]. These effects could also be mediated by VNS of enteric nervous system [117].
Many mechanisms are responsible for the up-regulation of junction proteins by nicotine. One possibility is the activation of extracellular signal related kinase (ERK) or mitogen activated kinase (MAPK). Nicotine is described to increase the expression of these kinases [118, 119]. Another possibility is a nicotine-mediated decrease of the nuclear factor kappa B (NFkB). This factor has been described to regulate TJ permeability [120,121] and Cox-2 expression [122,123], a CRC tumor promoting factor [93]. Finally, α7 nAChR activation can regulate the stability of the junction proteins by controlling their phosphorylation status. Indeed, in keratinocytes, Chernyavsky et al., showed that α7 nAChR activates adhesion molecules by both inhibiting Src family kinases and activating protein tyrosine phosphatases (PTP) [124]. nAChRs can be associated with both Src and PTP in large multimeric complexes controlling their activation [125].
Recently, nAChR has been implicated in regulating the release of the neurotransmitter g-aminobuturic acid (GABA), which may act as a tumor suppressor in particular, in colon carcinoma and lung adenocarcinoma cells [126,127]. The heterotrimeric α4b2 nAChR, which binds ACh and nicotine with higher affinity than α7 nAChR, stimulates the release of GABA. This higher affinity for nicotine is thought to cause the long-term inactivation (or desensitization) of the heteromeric nAChR that has been observed after chronic exposition to nicotine such as smokers. By contrast the sensibility of α7 nAChr remains unchanged, its expression is up-regulated by nicotine and its biological effect is increased [128].
The effect induced by nicotine may be different in various cell types, differentiated or “totipotent”, and having diverse metabolic properties to convert nicotine in its metabolite such as cotinine which is more toxic. Moreover, these effects may be related to different factors such as: 1) length of exposure (short versus chronic); 2) concentration (high or low); 3) anatomical distribution and expression of receptor subtypes. Finally, the α7 nAChR effects could be modulated by the secreted mammalian LY-6/urokinase plasminogen activator receptor-related protein-1 (SLURP-1) that has been recently identified by an endogenous ligand for α7 nAChR which exerts protective effect [59].
4.5. Potential Therapeutic Use of VNS
To date different approaches have been done targeting nAChR in cancer therapy in particular tobacco-related carcinogenesis. In this case, the challenge is to develop compound capable of inhibiting α7 nAChR or potentiating α4b2 nAChR to counterbalance the deleterious effects of chronic nicotine (for review [129]. Most promising compounds have side-effects in particular on the central nervous system since they can cross the blood-brain barrier. In improving the strategy for treating IBD patients, VNS could represent an attractive therapeutic approach to prevent CAC. VNS (afferent fibers) at high frequency (30 Hz) is already successfully used in humans in the treatment of depression, seizure and more recently metabolism troubles [30,31]. Now this technique could be extended to gastrointestinal disorders such as IBD using a low frequency stimulation (5 - 10 Hz) which is known to activate efferent fibers (for review [12]). In this condition, VNS uses a physiological anti-inflammatory pathway and represents a safe technique with even less side-effects. A reduction of inflammation is a key step for decrease the risk of CAC. However we should keep in mind that the nature and the subtype of cholinergic receptors on target cells should be carefully investigated before evaluating the effectiveness of nAChR as a drug target in these patients. Indeed, stimulation of mAChR play key role in colon cancer cell invasion.
5. Acknowledgements
The authors gratefully acknowledge grant support from Association pour la Recherche sur le Cancer, Ligue Nationale contre le Cancer, GEFLUC and ESPOIR. B. Ducarouge and M. Pelissier-Rota are the recipient of a fellowship from the Ministère de la Recherche et de l’Enseignement Supérieur. We thank Pierre-Emmanuel Buyse for his technical support.
REFERENCES
- M. Scharl and G. Rogler, “Inflammatory Bowel Disease: Dysfunction of Autophagy?” Digestive Disease, Vol. 30, No. 3, 2012, pp. 12-19. doi:10.1159/000342588
- S. Danese and A. Mantovani, “Inflammatory Bowel Disease and Intestinal Cancer: A Paradigm of the Yin-Yang Interplay between Inflammation and Cancer,” Oncogene, Vol. 29, No. 23, 2010, pp. 3313-3323. doi:10.1038/onc.2010.109
- M. Rutter, B. Saunders, K. Wilkinson, S. Rumbles, G. Schofield, M. Kamm, C. Williams, A. Price, I. Talbot and A. Forbes, “Severity of Inflammation Is a Risk Factor for Colorectal Neoplasia in Ulcerative Colitis,” Gastroenterology, Vol. 126, No. 2, 2004, pp. 451-460. doi:10.1053/j.gastro.2003.11.010
- J. A. Eaden, K. R. Abrams and J. F. Mayberry, “The Risk of Colorectal Cancer in Ulcerative Colitis: A Meta-Analysis,” Gut, Vol. 48, No. 4, 2001, pp. 536-535. doi:10.1136/gut.48.4.526
- M. D. Rutter, B. P. Saunders, K. H. Wilkinson, S. Rumbles, G. Schofield, M. A. Kamm, C. B. Williams, A. B. Price, I. C. Talbot and A. Forbes, “Thirty-Year Analysis of a Colonoscopic Surveillance Program for Neoplasia in Ulcerative Colitis,” Gastroenterology, Vol. 130, No. 4, 2006, pp. 1030-1038. doi:10.1053/j.gastro.2005.12.035
- A. Ekbom, M. Zack and H. O. Adami, “The Epidemiology of Inflammatory Bowel Disease: A Large, PopulationBased Study in Sweden,” Gastroenterology, Vol. 100, No. 2, 1991, pp. 350-358.
- S. I. Grivennikov, “Inflammation and Colorectal Cancer: Colitis-Associated Neoplasia,” Seminars in Immunopathology, Vol. 35, No. 2, 2013, pp. 229-244. doi:10.1007/s00281-012-0352-6
- H. S. Cooper, S. Murthy, K. Kido, H. Yoshitake and A. Flanigan, “Dysplasia and Cancer in the Dextran Sulfate Sodium Mouse Colitis Model. Relevance to Colitis-Associated Neoplasia in the Human: A Study of Histopathology, B-Catenin and p53 Expression and the Role of Inflammation,” Carcinogenesis, Vol. 21, No. 4, 2000, pp. 757-768. doi:10.1093/carcin/21.4.757
- R. L. Shattuck-Brandt, G. W. Varilek, A. Radhika, F. Yang, M. K. Washington and R. N. DuBois, “Cyclooxygenase 2 Expression Is Increased in the Stroma of Colon Carcinomas from IL-10(-/-) Mice,” Gastroenterology, Vol. 118, No. 2, 2000, pp. 337-345. doi:10.1016/S0016-5085(00)70216-2
- P. Sansone, G. Piazzi, P. Paterini, A. Strillacci, C. Ceccarelli, F. Minni, G. Biasco, P. Chieco and M. Bonafè, “Cyclooxygenase-2/Carbonic Anhydrase-IX Up-Regulation Promotes Invasive Potential and Hypoxia Survival in Colorectal Cancer Cells,” Journal of Cellular and Molecular Medicine, Vol. 13, No. 9, 2009, pp. 3876-3887. doi:10.1111/j.1582-4934.2008.00580.x
- K. Nys and S. Vermeire, “Autophagy: A New Target or an Old Strategy for the Treatment of Crohn’s Disease?” Nature Reviews Gastroenterology & Hepatology, 2013, in Press.
- B. Bonaz, C. Picq, V. Sinniger, J. F. Mayol and D. Clarençon, “Vagus Nerve Stimulation: From Epilepsy to the Cholinergic Anti-Inflammatory Pathway,” Neurogastroenterology & Motility, Vol. 25, No. 3, 2013, pp. 208-221. doi:10.1111/nmo.12076
- C. Cheyuo, A. Jacob, R. Wu, M. Zhou, G. F. Coppa and P. Wang, “The Parasympathetic Nervous System in the Quest for Stroke Therapeutics,” Journal of Cerebral Blood Flow & Metabolism, Vol. 31, 2011, pp. 1187-1195. doi:10.1038/jcbfm.2011.24
- V. A. Pavlov and K. J. Tracey, “The Cholinergic AntiInflammatory Pathway,” Brain, Behavior, and Immunity, Vol. 19, No. 6, 2005, pp. 493-499.
- L. V. Borovikova, M. Zhang, H. Yang, G. I. Botchkina, L. R. Watkins, H. Wang, N. Abumrad, J. W. Eaton and K. J. Tracey, “Vagus Nerve Stimulation Attenuates the Systemic Inflammatory Response to Endotoxin,” Nature, Vol. 405, No. 6785, 2000, pp. 458-462. doi:10.1038/35013070
- H. Wang, M. Yu, M. Ochani, C. A. Amella, M. Tanovic, S. Susarla, J. H. Li, H. Wang, H. Yang, L. Ulloa, Y. AlAbed, C. J. Czura and K. J. Tracey, “Nicotinic Acetylcholine Receptor Alpha7 Subunit Is an Essential Regulator of Inflammation,” Nature, Vol. 421, No. 6921, 2003, pp. 384- 388. doi:10.1038/nature01339
- K. J. Tracey, “The Inflammatory Reflex,” Nature, Vol. 420, No. 6917, 2002, pp. 853-859. doi:10.1038/nature01321
- K. J. Tracey, “Physiology and Immunology of the Cholinergic Antiinflammatory Pathway,” Journal of Clinical Investigation, Vol. 117, No. 2, 2007, pp. 289-296. doi:10.1172/JCI30555
- A. Miyazawa and N. Unwin, “Structure and Gating Mechanism of the Acetylcholine Receptor Pore,” Nature, Vol. 423, No. 6943, 2003, pp. 949-955. doi:10.1038/nature01748
- J. Wess, “Molecular Biology of Muscarinic Acetylcholine Receptors,” Critical Reviews in Neurobiology, Vol. 10, No. 1, 1996, pp. 69-99. doi:10.1615/CritRevNeurobiol.v10.i1.40
- L. G. Paleari, A. Cesario and P. Russo, “The Cholinergic System and Cancer,” Seminars in Cancer Biology, Vol. 18, No. 3, 2008, pp. 211-217. doi:10.1016/j.semcancer.2007.12.009
- M. Sopori, “Effects of Cigarette Smoke on the Immune System,” Nature Reviews Immunology, Vol. 2, No. 5, 2002, pp. 372-377. doi:10.1038/nri803
- C. J. Czura, S. G. Friedman and K. J. Tracey, “Neural Inhibition of Inflammation: The Cholinergic Anti-Inflammatory Pathway,” Journal of Endotoxin Research, Vol. 9, No. 6, 2003, pp. 409-413.
- L. Ulloa, “The Vagus Nerve and the Nicotinic Anti-Inflammatory Pathway,” Nature Reviews Drug Discovery, Vol. 4, No. 8, 2005, pp. 673-684. doi:10.1038/nrd1797
- H. M. Schuller, “Nitrosamines as Nicotinic Receptor Ligands,” Life Sciences, Vol. 80, No. 24-25, 2007, pp. 2274-2280. doi:10.1016/j.lfs.2007.03.006
- D. J. van Westerloo, I. A. Giebelen, S. Florquin, M. J. Bruno, G. J. Larosa, L. Ulloa, K. J. Tracey and T. van der Poll, “The Vagus Nerve and Nicotinic Receptors Modulate Experimental Pancreatitis Severity in Mice,” Gastroenterology, Vol. 130, No. 6, 2006, pp. 1822-1830. doi:10.1053/j.gastro.2006.02.022
- J. E. Ghia, P. Blennerhassett, H. Kumar-Ondiveeran, E. F. Verdu and S. M. Collins, “The Vagus Nerve: A Tonic Inhibitory Influence Associated with Inflammatory Bowel Disease in a Murine Model,” Gastroenterology, Vol. 131, No. 4, 2006, pp. 1122-1130. doi:10.1053/j.gastro.2006.08.016
- J. Meregnani, D. C, M. Vivier, A. Peinnequin, C. Mouret, V. Sinniger, C. Picq, A. Job, F. Canini, M. Jacquier-Sarlin and B. Bonaz, “Anti-Inflammatory Effect of Vagus Nerve Stimulation in a Rat Model of Inflammatory Bowel Disease,” Autonomic Neuroscience: Basic and Clinical, Vol. 160, No. 1, 2011, pp. 82-89. doi:10.1016/j.autneu.2010.10.007
- F. The, C. Cailotto, J. van der Vliet, W. J. de Jonge, R. J. Bennink, R. M. Buijs and G. E. Boeckxstaens, “Central Activation of the Cholinergic Anti-Inflammatory Pathway Reduces Surgical Inflammation in Experimental PostOperative Ileus,” British Journal of Pharmacology, Vol. 163, No. 5, 2011, pp. 1007-1016. doi:10.1111/j.1476-5381.2011.01296.x
- A. H. Milby, C. H. Halpern and G. H. Baltuch, “Vagus Nerve Stimulation for Epilepsy and Depression,” Neurotherapeutics, Vol. 5, No. 1, 2008, pp. 75-85. doi:10.1016/j.nurt.2007.10.071
- V. A. Pavlov and K. J. Tracey, “The Vagus Nerve and the Inflammatory Reflex—Linking Immunity and Metabolism,” Nature Reviews Endocrinology, Vol. 8, 2012, pp. 743-754. doi:10.1038/nrendo.2012.189
- S. P. Hussain, L. J. Hofseth and C. C. Harris, “Radical Causes of Cancer,” Nature Reviews Cancer, Vol. 3, No. 6, 2003, pp. 267-285. doi:10.1038/nrc1046
- G. K. Zupanc, I. Horschke and D. A. Lovejoy, “Corticotropin Releasing Factor in the BRain of the Gymnotiform Fish, Apteronotus leptorhynchus: Immunohistochemical Studies Combined with Neuronal Tract Tracing,” General and Comparative Endocrinology, Vol. 114, No. 3, 1999, pp. 349-364.doi:10.1006/gcen.1999.7273
- P. M. O’Connor, T. K. Lapointe, P. L. Beck and A. G. Buret, “Mechanisms by Which Inflammation May Increase Intestinal Cancer Risk in Inflammatory Bowel Disease,” Inflammatory Bowel Diseases, Vol. 16, No. 8, 2010, pp. 1411-1420. doi:10.1002/ibd.21217
- J. Terzić, S. Grivennikov, E. Karin and M. Karin, “Inflammation and Colon Cancer,” Gastroenterology, Vol. 138, No. 6, 2010, pp. 2101-2114.
- O. M. Kalashnyk, G. L. Gergalova, S. V. Komisarenko and M. V. Skok, “Intracellular Localization of Nicotinic Acetylcholine Receptors in Human Cell Lines,” Life Sciences, Vol. 91, No. 21-22, 2012, pp. 1033-1037. doi:10.1016/j.lfs.2012.02.005
- C. L. Hirota and D. M. McKay, “Cholinergic Regulation of Epithelial Ion Transport in the Mammalian Intestine,” British Journal of Pharmacology, Vol. 149, No. 5, 2006, pp. 463-479. doi:10.1038/sj.bjp.0706889
- R. Kopp, G. Lambrecht, E. Mutschler, U. Moser, R. Tacke and A. Pfeiffer, “Human HT-29 Colon Carcinoma Cells Contain Muscarinic M3 Receptors Coupled to Phosphoinositide Metabolism,” European Journal of Pharmacology: Molecular Pharmacology, Vol. 172, No. 4-5, 1989, pp. 397-405. doi:10.1016/0922-4106(89)90021-7
- K. E. J. Dickinsona, R. A. Frizzellb and M. C. Sekar, “Activation of T84 Cell Chloride Channels by Carbachol Involves a Phosphoinositide-Coupled Muscarinic M3 Receptor,” European Journal of Pharmacology: Molecular Pharmacology, Vol. 225, No. 4, 1992, pp. 291-298. doi:10.1016/0922-4106(92)90102-2
- K. E. O’Malley, C. B. Farrell, K. M. O’Boyle and A. W. Baird, “Cholinergic Activation of Cl-Secretion in Rat Colonic Epithelia,” European Journal of Pharmacology, Vol. 275, No. 1, 1995, pp. 83-89.
- P. Simon-Assmann, M. Kedinger, A. De Arcangelis, V. Rousseau and P. Simo, “Extracellular Matrix Components in Intestinal Development,” Experientia, Vol. 51, No. 9-10, 1995, pp. 883-900. doi:10.1007/BF01921739
- M. Aumailley and T. Krieg, “Laminins: A Family of Diverse Multifunctional Molecules of Basement Membranes,” Journal of Investigative Dermatology, Vol. 106, No. 2, 1996, pp. 209-214. doi:10.1111/1523-1747.ep12340471
- J.-F. Beaulieu, “Extracellular Matrix Components and Integrins in Relationship to Human Intestinal Epithelial Cell Differentiation,” Progress in Histochemistry and Cytochemistry, Vol. 31, No. 4, 1997, pp. 1-76. doi:10.1016/S0079-6336(97)80001-0
- S. Miyamoto, S. K. Akiyama and K. M. Yamada, “Synergistic Roles for Receptor Occupancy and Aggregation in Integrin Transmembrane Function,” Science, Vol. 267, No. 5199, 1995, pp. 883-885. doi:10.1126/science.7846531
- M. A. Schwartz, M. D. Schaller and M. H. Ginsberg, “Integrins: Emerging Paradigms of Signal Transduction,” Annual Review of Cell and Developmental Biology, Vol. 11, No. 1, 1995, pp. 549-599. doi:10.1146/annurev.cb.11.110195.003001
- Z. Hossain and T. Hirata, “Molecular Mechanism of Intestinal Permeability: Interaction at Tight Junctions,” Molecular BioSystems, Vol. 4, No. 12, 2008, pp. 1181-1185. doi:10.1039/b800402a
- C. M. Niessen, “Tight Junctions/Adherens Junctions: Basic Structure and Function,” Journal of Investigative Dermatology, Vol. 127, 2007, pp. 2535-2532. doi:10.1038/sj.jid.5700865
- J. A. Jankowski, F. K. Bedford and Y. S. Kim, “Changes in Gene Structure and Regulation of E-Cadherin during Epithelial Development, Differentiation, and Disease,” Progress in Nucleic Acid Research and Molecular Biology, Vol. 57, 1997, pp. 187-215. doi:10.1016/S0079-6603(08)60281-0
- A. Zweibaum, M. Pinto, G. Chevalier, E. Dussaulx, N. Triadou, B. Lacroix, K. Haffen, J.-L. Brun and M. Rousset, “Enterocytic Differentiation of a Subpopulation of the Human Colon Tumor Cell Line HT-29 Selected for Growth in Sugar-Free Medium and Its Inhibition by Glucose,” Journal of Cellular Physiology, Vol. 122, No. 1, 1985, pp. 27-29. doi:10.1002/jcp.1041220105
- N. T. Chartier, M. Laine, S. Gout, G. Pawlak, C. A. Marie, P. Matos, et al., “Laminin-5-Integrin Interaction Signals through PI 3-Kinase and Rac1b to Promote Assembly of Adherens Junctions in HT-29 Cells,” Journal of Cell Science, Vol. 119, 2006, pp. 31-46. doi:10.1242/jcs.02698
- E. A. Wayner, S. G. Gil, G. F. Murphy, M. S. Wilke and W. G. Carter, “Epiligrin, a Component of Epithelial Basement Membranes, Is an Adhesive Ligand for Alpha 3 Beta 1 Positive T Lymphocytes,” The Journal of Cell Biology, Vol. 121, No. 5, 1993, pp. 1141-1152. doi:10.1083/jcb.121.5.1141
- J. B. Weitzman, R. Pasqualini, Y. Takada and M. E. Hemler, “The Function and Distinctive Regulation of the Integrin VLA-3 in Cell Adhesion, Spreading, and Homotypic Cell Aggregation,” The Journal of Biological Chemistry, Vol. 268, No. 12, 1993, pp. 8651-8657.
- A. E. Summers, C. J. Whelan and M. E. Parsons, “Nicotinic Acetylcholine Receptor Subunits and Receptor Activity in the Epithelial Cell Line HT29,” Life Sciences, Vol. 72, No. 18-19, 2003, pp. 2091-2094. doi:10.1016/S0024-3205(03)00089-4
- C. Huet, C. Sahuquillo-Merino, E. Coudrier and D. Louvard, “Absorptive and Mucus-Secreting Subclones Isolated from a Multipotent Intestinal Cell Line (HT-29) Provide New Models for Cell Polarity and Terminal Differentiation,” The Journal of Cell Biology, Vol. 105, No. 1, 1987, pp. 345-357. doi:10.1083/jcb.105.1.345
- S. P. Gout, M. R. Jacquier-Sarlin, L. Rouard-Talbot, P. Rousselle and M. R. Block, “RhoA-Dependent Switch between Alpha2Beta1 and alpha3beta1 Integrins Is Induced by Laminin-5 during Early Stage of HT-29 Cell Differentiation,” Molecular Biology of the Cell, Vol. 12, No. 10, 2001, pp. 3268-3281.
- S. Gout, C. Marie, M. Lainé, G. Tavernier, M. R. Block and M. Jacquier-Sarlin, “Early Enterocytic Differentiation of HT-29 Cells: Biochemical Changes and Strength Increases of Adherens Junctions,” Experimental Cell Research, Vol. 299, No. 2, 2004, pp. 498-510. doi:10.1016/j.yexcr.2004.06.008
- J. Fogh, “Human Tumor Cells in Vitro,” Plenum Press, New York, 1975, pp. 115-141.
- H. P. S. Wong, L. Yu, E. K. Y. Lam, E. K. K. Tai, W. K. K. Wu and C. H. Cho, “Nicotine Promotes Cell Proliferation via Alpha7-Nicotinic Acetylcholine Receptor and Catecholamine-Synthesizing Enzymes-Mediated Pathway in Human Colon Adenocarcinoma HT-29 Cells,” Toxicology and Applied Pharmacology, Vol. 221, No. 3, 2007, pp. 261-267. doi:10.1016/j.taap.2007.04.002
- A. Pettersson, L. Nilsson, G. Nylund, A. KhorramManesh, S. Nordgren and D. S. Delbro, “Is Acetylcholine an Autocrine/Paracrine Growth Factor via the Nicotinic Alpha7-Receptor Subtype in the Human Colon Cancer Cell Line HT-29?” European Journal of Pharmacology, Vol. 609, No. 1-3, 2009, pp. 27-33. doi:10.1016/j.ejphar.2009.03.002
- [61] C. D’Souza-Schorey, “Disassembling Adherens Junctions: Breaking Up Is Hard to Do,” Trends in Cell Biology, Vol. 15, No. 1, 2005, pp. 19-26. doi:10.1016/j.tcb.2004.11.002
- [62] S. Razani-Boroujerdi, M. Behl, F. F. Hahn, J. C. PenaPhilippides, J. Hutt and M. L. Sopori, “Role of Muscarinic Receptors in the Regulation of Immune and Inflammatory Responses,” Journal of Neuroimmunology, Vol. 194, No. 1, 2008, pp. 83-88. doi:10.1016/j.jneuroim.2007.11.019
- [63] I. Wessler, C. J. Kirkpatrick and K. Racké, “Non-Neuronal Acetylcholine, a Locally Acting Molecule, Widely Distributed in Biological Systems: Expression and Function in Humans,” Pharmacology & Therapeutics, Vol. 77, No. 1, 1998, pp. 59-79. doi:10.1016/S0163-7258(97)00085-5
- [64] T. Okuda and T. Haga, “High-Affinity Choline Transporter,” Neurochemical Research, Vol. 28, No. 3-4, 2003, pp. 483-488. doi:10.1023/A:1022809003997
- [65] K. Racké, U. R. Juergens and S. Matthiesen, “Control by Cholinergic Mechanisms,” European Journal of Pharmacology, Vol. 533, No. 1-3, 2006, pp. 57-68. doi:10.1016/j.ejphar.2005.12.050
- [66] I. Wessler and C. J. Kirkpatrick, “Acetylcholine beyond Neurons: The Non-Neuronal Cholinergic System in Humans,” British Journal of Pharmacology, Vol. 154, No. 8, 2008, pp. 1558-1571. doi:10.1038/bjp.2008.185
- [67] M. F. Montenegro, F. Ruiz-Espejo, F. J. Campoy, E. Muñoz-Delgado, M. Páez de la Cadena, F. J. RodríguezBerrocal and C. J. Vidal, “Cholinesterases Are DownExpressed in Human Colorectal Carcinoma,” Cellular and Molecular Life Sciences, Vol. 63, No. 18, 2006, pp. 2175-2182. doi:10.1007/s00018-006-6231-3
- [68] K. R. Cheng, R. Samimi, G. F. Xie, J. Shant, C. Drachenberg, M. Wade, R. J. Davis, G. Nomikos and J.-P. Raufman, “Acetylcholine Release by Human Colon Cancer Cells Mediates Autocrine Stimulation of Cell Proliferation,” American Journal of Physiology-Gastrointestinal and Liver Physiology, Vol. 295, No. 3, 2008, pp. G591-G597. doi:10.1152/ajpgi.00055.2008
- [69] I. Wessler, T. Reinheimer, H. Klapproth, F.-J. Schneider, K. Racké and R. Hammer, “Mammalian Glial Cells in Culture Synthesize Acetylcholine,” Naunyn-Schmiedeberg’s Archives of Pharmacology, Vol. 356, No. 5, 1997, pp. 694-697. doi:10.1007/PL00005107
- [70] J.-P. Raufman, R. Samimi, N. Shah, S. Khurana, J. Shant, C. Drachenberg, G. F. Xie, J. Wess and K. R. Cheng, “Genetic Ablation of M3 Muscarinic Receptors Attenuates Murine Colon Epithelial Cell Proliferation and Neoplasia,” Cancer Research, Vol. 68, 2008, pp. 3573-3578. doi:10.1158/0008-5472.CAN-07-6810
- [71] H. Frucht, R. T. Jensen, D. Dexter, W. L. Yang and Y. Xiao, “Human Colon Cancer Cell Proliferation Mediated by the M3 Muscarinic Cholinergic Receptor,” Clinical Cancer Research, Vol. 5, No. 9, 1999, pp. 2532-2539.
- [72] W.-L. Yang and H. Frucht, “Cholinergic Receptor UpRegulates COX-2 Expression and Prostaglandin E(2) Production in Colon Cancer cells,” Carcinogenesis, Vol. 21, No. 10, 2000, pp. 1789-1793. doi:10.1093/carcin/21.10.1789
- [73] C. W. Emala, J. Clancy and C. A. Hirshman, “Glucocorticoid Treatment Decreases Muscarinic Receptor Expression in Canine Airway Smooth Muscle,” American Journal of Physiology, Vol. 272, No. 4, 1997, pp. L745-L751.
- [74] D. B. Jacoby, B. L. Yost, B. Kumaravel, Y. Chan-Li, H.-Q. Xiao, K. Kawashima and A. D. Fryer, “Glucocorticoid Treatment Increases Inhibitory M(2) Muscarinic Receptor Expression and Function in the Airways,” American Journal of Respiratory Cell and Molecular Biology, Vol. 24, No. 4, 2001, pp. 485-91. doi:10.1165/ajrcmb.24.4.4379
- [75] H. M. Schuller, “Cell Type Specific, Receptor-Mediated Modulation of Growth Kinetics in Human Lung Cancer Cell Lines by Nicotine and Tobacco-Related Nitrosamines,” Biochemical Pharmacology, Vol. 38, No. 20, 1989, pp. 3439-3442. doi:10.1016/0006-2952(89)90112-3
- [76] C. Gotti and F. Clementi, “Neuronal Nicotinic Receptors: From Structure to Pathology,” Progress in Neurobiology, Vol. 74, No. 6, 2004, pp. 363-396. doi:10.1016/j.pneurobio.2004.09.006
- [77] Russo P CA, Cardinale A., “Presentation to the Special Issue: ‘α7-Nicotinic Receptor (α7-nAChR): One Target Different Diseases’,” Current Drug Targets, Vol. 13, No. , 2012, 573.
- [78] J.-P. Raufman, J. Shant, G. F. Xie, K. R. Cheng, X.-M. Gao, B. Shiu, N. Shah, C. B. Drachenberg, J. Heath, J. Wess and S. Khurana, “Muscarinic Receptor Subtype-3 Gene Ablation and Scopolamine Butylbromide Treatment Attenuate Small Intestinal Neoplasia in Apcmin/+ Mice,” Carcinogenesis, Vol. 32, No. 9, 2011, pp. 1396-1402. doi:10.1093/carcin/bgr118
- [79] A. Belo, K. R. Cheng, A. Chahdi, J. Shant, G. F. Xie, S. Khurana and J.-P. Raufman, “Muscarinic Receptor Agonists Stimulate Human Colon Cancer Cell Migration and Invasion,” American Journal of Physiology-Gastrointestinal and Liver Physiology, Vol. 300, No. 5, 2011, pp. G749-G760. doi:10.1152/ajpgi.00306.2010
- [80] K. Cheng, P. Zimniak and J. P. Raufman, “Transactivation of the Epidermal Growth Factor Receptor Mediates Cholinergic Agonist-Induced Proliferation of H508 Human Colon Cancer Cells,” Cancer Research, Vol. 63, No. 20, 2003, pp. 6744-6750.
- [81] H. L. Cameron and M. H. Perdue, “Muscarinic Acetylcholine Receptor Activation Increases Transcellular Transport of Macromolecules across Mouse and Human Intestinal Epithelium in Vitro,” Neurogastroenterology & Motility, Vol. 19, No. 1, 2007, pp. 47-56. doi:10.1111/j.1365-2982.2006.00845.x
- [82] V. T. Nguyena, J. Arredondoa, A. I. Chernyavskya, Y. Kitajimab and S. A. Grando, “Keratinocyte Acetylcholine Receptors Regulate Cell Adhesion,” Life Sciences, Vol. 72, No. 18-19, 2003, pp. 2081-2085. doi:10.1016/S0024-3205(03)00087-0
- [83] Nguyen VT AJ, Chernyavsky AI, Kitajima Y, Pittelkow M, Grando SA., “Pemphigus Vulgaris IgG and Methylprednisolone Exhibit Reciprocal Effects on Keratinocytes,” The Journal of Biological Chemistry, Vol. 279, 2004, pp. 2135-2146. doi:10.1074/jbc.M309000200
- [84] S. Alemà and A. M. Salvatore, “p120 Catenin and Phosphorylation: Mechanisms and Traits of an Unresolved Issue,” Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, Vol. 1773, No. 1, 2007, pp. 47-58. doi:10.1016/j.bbamcr.2006.06.001
- [85] E. Calautti, M. Grossi, C. Mammucari, Y. Aoyama, M. Pirro, Y. Ono, J. Li and G. P. Dotto, “Fyn Tyrosine Kinase Is a Downstream Mediator of Rho/PRK2 Function in Keratinocyte Cell-Cell Adhesion,” The Journal of Cell Biology, Vol. 156, No. 1, 2002, pp. 137-148. doi:10.1083/jcb.200105140
- [86] J. Lilien, J. Balsamo, C. Arregui and G. Xu, “Turn-Off, Drop-Out: Functional State Switching of Cadherins,” Developmental Dynamics, Vol. 224, No. 1, 2002, pp. 18- 29. doi:10.1002/dvdy.10087
- [87] M. Serresa, O. Filholb, H. Lickertc, C. Grangeassed, E. M. Chambazb, J. Stappertc, C. Vincenta and D. Schmitt, “The Disruption of Adherens Junctions Is Associated with a Decrease of E-Cadherin Phosphorylation by Protein Kinase CK2,” Experimental Cell Research, Vol. 15, No. 2, 2000, pp. 255-264. doi:10.1006/excr.2000.4895
- [88] S. Dupre-Crochet, A. Figueroa, C. Hogan, E. C. Ferber, C. U. Bialucha, J. Adams, E. C. N. Richardson and Y. Fujita, “Casein Kinase 1 Is a Novel Negative Regulator of ECadherin-Based Cell-Cell Contacts,” Molecular and Cellular Biology, Vol. 27, No. 10, 2007, pp. 3804-3816. doi:10.1128/MCB.01590-06
- [89] H. Aberle, A. Bauer, J. Stappert, A. Kispert and R. Kemler, “Beta-Catenin Is a Target for the Ubiquitin-Proteasome Pathway,” The EMBO Journal, Vol. 16, 1997, pp. 3797-3804. doi:10.1093/emboj/16.13.3797
- [90] B. C. Elias, T. Suzuki, A. Seth, F. Giorgianni, G. Kale, L. Shen, J. R. Turner, A. Naren, Dominic M. Desiderio and Radhakrishna Rao, “Phosphorylation of Tyr-398 and Tyr-402 in Occludin Prevents Its Interaction with ZO-1 and Destabi-Lizes Its Assembly at the Tight Junctions,” The Journal of Biological Chemistry, Vol. 284, 2009, pp. 1559-1569. doi:10.1074/jbc.M804783200
- [91] A. Kourtidis, S. P. Ngok and P. Z. Anastasiadis, “p120 Catenin: An Essential Regulator of Cadherin Stability, Adhe-Sion-Induced Signaling, and Cancer Progression,” Progress in Molecular Biology and Translational Science, Vol. 116, 2013, pp. 409-432. doi:10.1016/B978-0-12-394311-8.00018-2
- [92] P. F. Song, H. S. Sekhon, Y. B. Jia, J. A. Keller, J. K. Blusztajn, G. P. Mark and E. R. Spindel, “Acetylcholine Is Synthesized by and Acts as an Autocrine Growth Factor for Small Cell Lung Carcinoma,” Cancer Research, Vol. 63, No. 1, 2003, pp. 214-221.
- [93] T. Schlereth., F. Birklein, K. an Haack, S. Schiffmann, H. Kilbinger, C. J. Kirkpatrick and I. Wessler, “In Vivo Release of Non-Neuronal Acetylcholine from the Human Skin as Measured by Dermal Microdialysis: Effect of Botulinum Toxin,” British Journal of Pharmacology, Vol. 147, No. 2, 2006, pp. 183-187. doi:10.1038/sj.bjp.0706451
- [94] R. Y. Huang and G. G. Chen, “Cigarette Smoking, Cyclooxygenase-2 Pathway and Cancer,” Biochimica et Biophysica Acta, Vol. 1815, No. 2, 2011, pp. 158-169.
- [95] J. P. Raufman, R. Samimi, N. Shah, S. Khurana, J. Shant, C. Drachenberg, G. Xie, J. Wess and K. Cheng, “Genetic Ablation of M3 Muscarinic Receptors Attenuates Murine Colon Epithelial Cell Proliferation and Neoplasia,” Cancer Research, Vol. 68, No. 10, 2008, pp. 3573-3578. doi:10.1158/0008-5472.CAN-07-6810
- [96] G. P. Boivin, K. Washington, K. Yang, J. M. Ward, T. P. Pretlow, R. Russell, D. G. Besselsen, V. L. Godfrey, T. Doetschman, W. F. Dove, H. C. Pitot, R. B. Halberg, S. H. Itzkowitz, J. Groden and R. J. Coffey, “Pathology of Mouse Models of Intestinal Cancer: Consensus Report and Recommendations,” Gastroenterology, Vol. 124, No. 3, 2003, pp. 762-77. doi:10.1053/gast.2003.50094
- [97] P. Song, H. S. Sekhon, A. Lu, J. Arredondo, D. Sauer, C. Gravett, G. P. Mark, S. A. Grando and E. R. Spindel, “M3 Muscarinic Receptor Antagonists Inhibit Small Cell Lung Carcinoma Growth and Mitogen-Activated Protein Kinase Phosphorylation Induced by Acetylcholine Secretion,” Cancer Research, Vol. 67, No. 8, 2007, pp. 3936- 3944. doi:10.1158/0008-5472.CAN-06-2484
- [98] S. Zia, A. Ndoye, V. T. Nguyen and S. A. Grando, “Nicotine Enhances Expression of the Alpha 3, Alpha 4, Alpha 5, and Alpha 7 Nicotinic Receptors Modulating Calcium Metabolism and Regulating Adhesion and Motility of Respiratory Epithelial Cells,” Research Communications in Molecular Pathology and Pharmacology, Vol. 97, No. 3, 1997, pp. 243-262.
- [99] P. Dasgupta, W. Rizwani, S. Pillai, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, Chellappan S., “Nicotine induces Cell Proliferation, Invasion and Epithelial-Mesenchymal Transition in a Variety of Human Cancer Cell Lines,” International Journal of Cancer, Vol. 124, No. 1, 2009, pp. 35-36. doi:10.1002/ijc.23894
- [100] R. Davis, W. Rizwani, S. Banerjee, M. Kovacs, E. Haura, D. Coppola and S. Chellappan, “Nicotine Promotes Tumor Growth and Metastasis in Mouse Models of Lung Cancer,” PLoS One, Vol. 4, No. 10, 2009, Article ID: e7524. doi:10.1371/journal.pone.0007524
- [101] J. E. Michaelson, J. R. Ritzenthaler and J. Roman, “Regulation of Serum-Induced Fibronectin Expression by Protein Kinases, Cytoskeletal Integrity, and CREB,” American Journal of Physiology-Lung Cellular and Molecular Physiology, Vol. 282, 2002, pp. L291-L301.
- [102] Y. Zheng, J. D. Ritzenthaler, J. Roman and S. W. Han, “Nicotine Stimulates Human Lung Cancer Cell Growth by Inducing Fibronectin Expression,” American Journal of Respiratory Cell and Molecular Biology, Vol. 37, No. 6, 2007, pp. 681-690. doi:10.1165/rcmb.2007-0051OC
- [103] P. L. Wei, L. J. Kuo, M. T. Huang, W. C. Ting, Y. S. Ho, W. Wang, J. An and Y. J. Chang, “Nicotine Enhances Colon Cancer Cell Migration by Induction of Fibronectin,” Annals of Surgical Oncology, Vol. 18, No. 6, 2011, pp. 1782-1790. doi:10.1245/s10434-010-1504-3
- [104] H. P. S. Wong, L. Yu, E. K. Y. Lam, E. K. K. Tai, W. K. K. Wu and C. H Cho, “Nicotine Promotes Colon Tumor Growth and Angiogenesis through Beta-Adrenergic Activation,” Toxicological Sciences, Vol. 97, No. 2, 2007, pp. 279-287. doi:10.1093/toxsci/kfm060
- [105] V. Y. Shin, W. K. K. Wu, Y. N. Ye, W. H. L. So, M. W. L. Koo, E. S. L. Liu, et al., “Nicotine Promotes Gastric Tumor Growth and Neovascularization by Activating Extracellular Signal-Regulated Kinase and Cyclooxygenase-2,” Carcinogenesis, Vol. 25, No. 12, 2004, pp. 2487-2495. doi:10.1093/carcin/bgh266
- [106] V. Y. Shin, W. K. K. Wu, K. M. Chu, H. P. S. Wong, E. K. Y. Lam, E. K. K. Tai, et al., “Nicotine Induces Cyclooxygenase-2 and Vascular Endothelial Growth Factor Receptor-2 in Association with Tumor-Associated Invasion and Angiogenesis in Gastric Cancer,” Molecular Cancer Research, Vol. 3, 2005, pp. 607-615. doi:10.1158/1541-7786.MCR-05-0106
- [107] R. D. Pullan, J. Rhodes, S. Ganesh, V. Mani, J. S. Morris, G. T. Williams, R. G. Newcombe, M. Russell, C. Feyerabend, G. Thomas, et al., “Transdermal Nicotine for Active Ulcerative Colitis,” The New England Journal of Medicine, Vol. 330, 1994, pp. 811-815. doi:10.1056/NEJM199403243301202
- [108] J. Rhodes and G. Thomas, “Nicotine Treatment in Ulcerative Colitis,” Drugs, Vol. 49, No. 2, 1995, pp. 157- 160. doi:10.2165/00003495-199549020-00001
- [109] J. R. Ingram, P. Routledge, J. Rhodes, R. W. Marshall, D. C. Buss, B. K. Evans, et al., “Nicotine Enemas for Treatment of Ulcerative Colitis: A Study of the Pharmacokinetics and Adverse Events Associated with Three Doses of Nicotine,” Alimentary Pharmacology & Therapeutics, Vol. 20, No. 8, 2004, pp. 859-865. doi:10.1111/j.1365-2036.2004.02199.x
- [110] B. Tanja and B. Ulrich, “Inflammatory Bowel Disease and Smoking: A Review of Epidemiology, Pathophysiology, and Therapeutic Implications,” Inflammatory Bowel Diseases, Vol. 10, No. 6, 2004, pp. 848-859. doi:10.1097/00054725-200411000-00019
- [111] W. J. Sandborn, “Severe Ulcerative Colitis,” Current Treatment Options in Gastroenterology, Vol. 2, No. 2, 1999, pp. 113-118. doi:10.1007/s11938-999-0038-x
- [112] J. E. Ghia, P. Blennerhassett, H. Kumar-Ondiveeran, E. F. Verdu and S. M. Collins, “The Vagus Nerve: A Tonic Inhibitory Influence Associated with Inflammatory Bowel Disease in a Murine Model,” Gastroenterology, Vol. 131, No. 4, 2006, pp. 1122-1130. doi:10.1053/j.gastro.2006.08.016
- [113] S. A. Snoek, M. I. Verstege, E. P. van der Zanden, N. Deeks, D. C. Bulmer, M. Skynner, et al., “Selective Alpha7 Nicotinic Acetylcholine Receptor Agonists Worsen Disease in Experimental Colitis,” British Journal of Pharmacology, Vol. 160, No. 2, 2010, pp. 322-333. doi:10.1111/j.1476-5381.2010.00699.x
- [114] A. P. Sykes, C. Brampton, S. Klee, C. L. Chander, C. Whelan and M. E. Parsons, “An Investigation into the Effect and Mechanisms of Action of Nicotine in Inflammatory Bowel Disease,” Inflammation Research, Vol. 49, No. 7, 2000, pp. 311-319. doi:10.1007/s000110050597
- [115] J. McGrath, J. W. Mcdonald and J. K. Macdonald, “Transdermal Nicotine for Induction of Remission in Ulcerative Colitis,” The Cochrane Database of Systematic Reviews, Vol. 18, No. 4, 2004, Article ID: CD004722.
- [116] V. E. McGilligan, J. M. W. Wallace, P. M. Heavey, D. L. Ridley and I. R. Rowland, “The Effect of Nicotine in Vitro on the Integrity of Tight Junctions in Caco-2 Cell Monolayers,” Food and Chemical Toxicology, Vol. 45, No. 9, 2007, pp. 1593-1598. doi:10.1016/j.fct.2007.02.021
- [117] T. W. Costantini, M. Krzyzaniak, G. A. Cheadle, J. G. Putnam, A. M. Hageny, N. Lopez, et al., “Targeting α-7 Nicotinic Acetylcholine Receptor in the Enteric Nervous System: A Cholinergic Agonist Prevents Gut Barrier Failure after Severe Burn Injury,” The American Journal of Pathology, Vol. 181, No. 2, 2012, pp. 478-486. doi:10.1016/j.ajpath.2012.04.005
- [118] T. W. Costantini, V. Bansal, M. Krzyzaniak, J. G. Putnam, C. Y. Peterson, W. H. Loomis, et al., “Vagal Nerve Stimulation Protects against Burn-Induced Intestinal Injury through Activation of Enteric Glia Cells,” American Journal of Physiology, Gastrointestinal and Liver Physiology, Vol. 299, No. 6, 2010, pp. G1308-G1318. doi:10.1152/ajpgi.00156.2010
- [119] C. Bose, H. Zhang, K. B. Udupa and P. Chowdhury, “Activation of p-ERK1/2 by Nicotine in Pancreatic Tumor Cell Line AR42J: Effects on Proliferation and Secretion,” American Journal of Physiology, Gastrointestinal and Liver Physiology, Vol. 289, No. 5, 2005, pp. G926-G934. doi:10.1152/ajpgi.00138.2005
- [120] H. Nakayama, S. Ueno, T. Ikeuchi and H. Hatanaka, “Regulation of Alpha3 Nicotinic Acetylcholine Receptor Subunit mRNA Levels by Nerve Growth Factor and Cyclic AMP in PC12 Cells,” Journal of Neurochemistry, Vol. 74, No. 4, 2000, pp. 1346-1354. doi:10.1046/j.1471-4159.2000.0741346.x
- [121] T. Y. Ma, G. K. Iwamoto, N. T. Hoa, V. Akotia, A. Pedram, M. A. Boivin, et al., “TNF-Alpha-Induced Increase in Intestinal Epithelial Tight Junction Permeability Requires NF-Kappa B Activation,” American Journal of Physiology, Gastrointestinal and Liver Physiology, Vol. 286, No. 3, 2004, pp. G367-G376. doi:10.1152/ajpgi.00173.2003
- [122] E. Hollenbach, M. Neumann, M. Vieth, A. Roessner, P. Malfertheiner and M. Naumann, “Inhibition of p38 MAP Kinaseand RICK/NF-kappab-Signaling Suppresses Inflammatory Bowel Disease,” FASEB Journal, Vol. 18, No. 13, 2004, pp. 1550-1552.
- [123] C. C. Hsu, J. C. Lien, C. W. Chang, C. H. Chang, S. C. Kuo and T. F. Huang, “Yuwen02f1 Suppresses LPS-Induced Endotoxemia and Adjuvant-Induced Arthritis Primarily through Blockade of ROS Formation, NFkB and MAPK Activation,” Biochemical Pharmacology, Vol. 85, No. 3, 2013, pp. 385-395. doi:10.1016/j.bcp.2012.11.002
- [124] S. A. Benitah, P. F. Valerón and J. C. Lacal, “ROCK and Nuclear factor-KappaB-Dependent Activation of Cyclooxygenase-2 by Rho GTPases: Effects on Tumor Growth and Therapeutic Consequences,” Molecular Biology of the Cell, Vol. 14, No. 7, 2003, pp. 3041-3054. doi:10.1091/mbc.E03-01-0016
- [125] A. I. Chernyavsky, J. Arredondo, T. Piser, E. Karlsson and S. A. Grando, “Differential Coupling of M1 Muscarinic and Alpha7 Nicotinic Receptors to Inhibition of Pemphigus Acantholysis,” The Journal of Biological Chemistry, Vol. 283, 2008, pp. 3401-3408. doi:10.1074/jbc.M704956200
- [126] M. L. van Hoek, C. S. Allen and S. J. Parsons, “Phosphotyrosine Phosphatase Activity Associated with c-Src in Large Multimeric Complexes Isolated from Adrenal Medullary Chromaffin Cells,” The Biochemical Journal, Vol. 326, Pt. 1, 1997, pp. 271-277.
- [127] J. Joseph, B. Niggemann, K. S. Zaenker and F. Entschladen, “The Neurotransmitter Gamma-Aminobutyric Acid Is an Inhibitory Regulator for the Migration of SW 480 Colon Carcinoma Cells,” Cancer Research, Vol. 62, No. 22, 2002, pp. 6467-6469.
- [128] H. M. Schuller, H. A. N. Al-Wadei and M. Majidi, “GABAB Receptor Is a Novel Drug Target for Pancreatic Cancer,” Cancer, Vol. 112, No. 4, 2008, pp. 767-778. doi:10.1002/cncr.23231
- [129] H. Kawai and D. K. Berg, “Nicotinic Acetylcholine Receptors Containing Alpha 7 Subunits on Rat Cortical Neurons Do Not Undergo Long-Lasting Inactivation Even When Up-Regulated by Chronic Nicotine Exposure,” Journal of Neurochemistry, Vol. 78, No. 6, 2001, pp. 1367-1378. doi:10.1046/j.1471-4159.2001.00526.x
- [130] P. Ambrosi and A. Becchetti, “Targeting Neuronal Nicotinic Receptors in Cancer: New Ligands and Potential Side-Effects,” Recent Patents on Anti-Cancer Drug Discovery, Vol. 8, No. 1, 2013, pp. 38-52.
NOTES
*Corresponding author.