Journal of Cancer Therapy
Vol.4 No.2(2013), Article ID:29755,8 pages DOI:10.4236/jct.2013.42070
Molecular Pathology of Hereditary Diffuse Gastric Cancer
![]()
1Department of Pathology, Stanford University School of Medicine, Stanford, USA; 2Department of Pediatrics, Stanford University School of Medicine, Stanford, USA.
Email: ischrijver@stanfordmed.org
Copyright © 2013 Justin I. Odegaard, Iris Schrijver. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received December 29th, 2012; revised January 31st, 2013; accepted February 9th, 2013
Keywords: Hereditary Diffuse Gastric Cancer; CDH1; Genetic Testing
ABSTRACT
Hereditary diffuse gastric cancer is a rare, autosomal dominant hereditary cancer syndrome associated with germline mutations in CDH1 in which 60% - 80% of affected individuals develop advanced diffuse gastric cancer, many as young adults. At clinical presentation, ~90% of these malignancies represent advanced, surgically incurable disease. As such, pre-symptomatic identification of germline CDH1 mutation carriers followed by early prophylactic total gastrictomy is the sole effective management strategy available. DNA sequence analysis of the CDH1 gene to identify the affected germline allele is the diagnostic standard of care; however, CDH1’s relatively high frequency of polymorphisms and the limited amount of experience available regarding them dictate that many identified variants are, as yet, of unknown clinical significance. Given the dramatic consequences of inappropriately offered or withheld treatment, careful clinical selection of at-risk individuals is critical. To facilitate this, multiple groups have published screening criteria recommendations, and while there is disagreement regarding the optimal diagnostic approach, the most widely-used overlap to a considerable degree.
1. Introduction
Gastric adenocarcinoma is one of the leading causes of cancer-related mortality worldwide, accounting for ~700,000 deaths per annum [1]. While its presentation varies significantly, the disease may be divided into two predominant types, each characterized by distinct histologic features, etiologies, and clinical behavior. Intestinal-type gastric adenocarcinoma, so named for its histologic resemblance to adenocarcinomas of the lower gastrointestinal tract, comprises ~74% of cases worldwide [2] and is characterized by a highly stereotyped progression from chronic gastritis (often Helicobacter-associated) to intestinal metaplasia, low-grade dysplasia, high-grade dysplasia, and on to invasive carcinoma. Invasive tumors then proceed from well-differentiated to moderately-differentiated (the most common prevalent histology at diagnosis) and finally to poorly-differentiated adenocarcinoma, which is associated with advanced stage, biologically aggressive disease. This progression’s endoscopic stigmata—mass lesions, ulcers, tinctorial and textural alterations—are readily appreciable and targetable by biopsy, forming the basis for gastric cancer surveillance in high-incidence populations.
In contrast, diffuse-type gastric adenocarcinoma, so named for the diffuse manner in which the characteristic signet ring-shaped malignant cells permeate the stomach wall, comprises ~16% of cases [2] and is generally unassociated with endoscopic findings (until advancedstage disease), background dysplasia, or Helicobacter infection. While the innate biological aggressiveness of the two tumor types is similar stage for stage, the absence of any endoscopically-identifiable precursor lesions means diffuse-type disease most often presents in an advanced stage with diffuse thickening of the gastric wall (linitis plastica) and nodal metastases. Beyond staging associations, however, tumor type is of little therapeutic or prognostic use at this time. Despite this, discriminating between intestinal and diffuse gastric adenocarcinoma is of great importance for assessing familial risk.
While the majority of gastric adenocarcinoma cases of both types are sporadic, 5% - 10% demonstrate familial clustering [1]. The varied clinical classification schemes for these clusters are often loosely defined and overlapping; however, two basic approaches are commonly used. Histologic approaches segregate cases into familial intestinal-type gastric cancer, familial diffuse-type gastric cancer, and—when histologic data are unavailable—familial gastric cancer based on the other characteristics discussed above. However, this grouping, while inclusive, is of relatively little clinical utility. Conversely, syndromic approaches segregate cases into known clinical cancer syndromes associated with/defined by clinical and genetic characteristics (e.g. Li Fraumeni, Peutz-Jeghers, familial adenomatous polyposis, and Lynch syndromes). This approach, while providing more clinically useful information for some cases, leaves many “unassigned”— only ~50% of familial gastric cancer cases are thought to be attributable to autosomal dominant cancer syndromes [1].
Of all patients with gastric cancer, 1% - 3% will be diagnosed with hereditary diffuse gastric cancer (HDGC) [3], an autosomal dominant gastric cancer syndrome associated with germline mutations in the CDH1 gene [4]. Without treatment, 60% - 80% of individuals carrying these mutations will develop advanced diffuse gastric cancer, many as young adults (reported ages at diagnosis range from 14 to 85 with the majority presenting before 40 [5]), with 40% - 60% of surviving females developing lobular breast cancer (LBC) [5,6]. While this syndrome accounts for only a small fraction of total cases, its devastating phenotype and successes in its biological characterization and treatment serve as models for this classification effort. In this review, we summarize the current understanding of HDGC with emphasis on appropriate utilization of screening and diagnostic testing.
2. CDH1 and HDGC Development
The CDH1 gene encodes E-cadherin, the transmembrane glycoprotein that anchors the epithelial adherens junction to the actin-based cytoskeleton through interactions with various catenin proteins. Loss of E-cadherin function disrupts cell-cell adhesion and mitotic spindle orientation, thus impairing the cell’s ability to establish and maintain polarity, which is critical for tissue patterning, epithelial barrier maintenance, cellular differentiation, and regulation of proliferation [7-10]. While somatic CDH1 inactivation has been described in numerous different carcinomas, it is most often a late event presaging tumor progression and the acquisition of an invasive mesenchymal phenotype [11]. In HDGC, where inactivation of one CDH1 allele occurs in the germline, loss of E-cadherin appears to have an entirely different role in carcinogenesis. These individuals demonstrate multiple, clonallydistinct tumors in which biallelic loss of E-cadherin expression is the first demonstrable genetic event [12], strongly suggesting CDH1 inactivation acts here as an initiating event rather than one of tumor progression.
In individuals bearing germline CDH1 mutations, loss of E-cadherin expression is first detected in proliferating cells in the upper isthmus of the gastric gland, the presumed location of the gastric epithelium stem cell [4,13]. Without properly aligned mitotic spindles, these E-cadherin-deficient progenitor cells propagate abnormal cells out of the epithelial plane [9], forming subepithelial deposits of characteristic signet ring-shaped neoplastic cells that comprise the earliest histologically identifiable stigmata of HDGC (Figures 1 (a) and (b)). Indeed, careful histologic reconstruction of entire prophylactic gastrectomy specimens demonstrates that carriers of germline CDH1 mutations typically bear anywhere from several to several hundred minute foci of signet ring carcinoma (SRC),
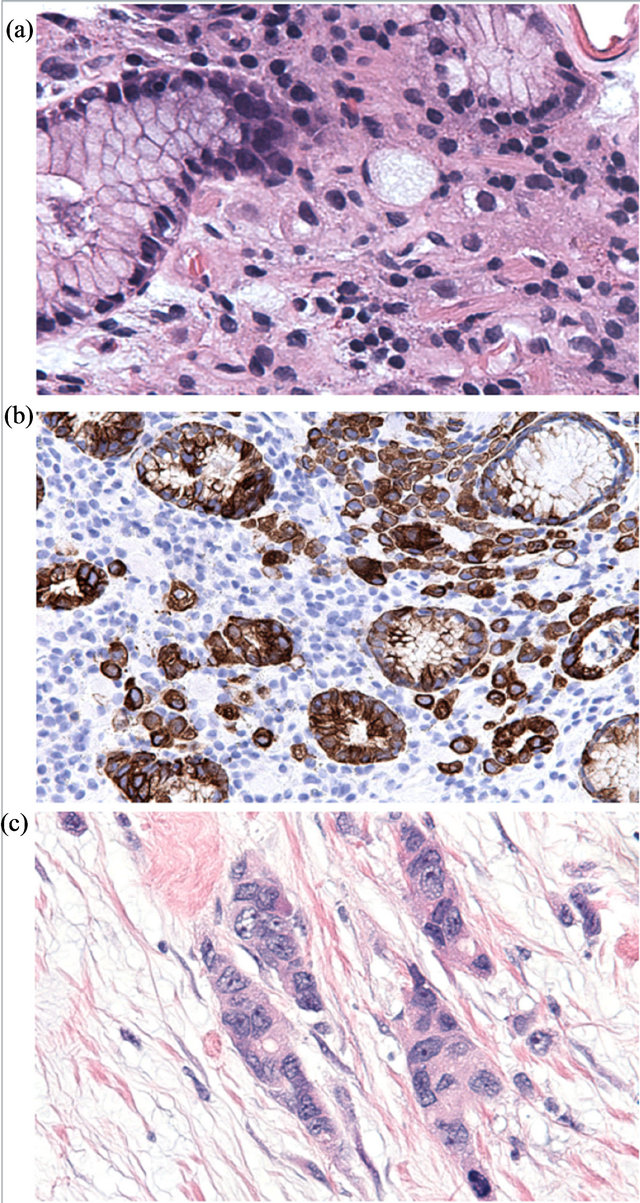
Figure 1. Histologic features of HDGC. Early HDGC-associated SRC comprise bland signet ring cells infiltrating benign gastric epithelium (a) H&E stain; (b) cytokeratin immunohistochemistry). Tumor progression (c) is accompanied by loss of typical signet ring morphology, increased cytologic atypia and mitotic activity, deep invasion, and metastasis.
most with early invasion into the superficial lamina propria (occasional SRC foci remain entirely superficial to the basal lamina [SRC in situ] or are seen to spread through surrounding non-neoplastic epithelium in a pagetoid manner) [6,12,14]. While early reports from Maori kindreds suggested a predilection for the antral/antral transition zone [15,16], studies of North American and European families have failed to generalize this finding across larger populations [17-19]. Importantly, early SRCs are relatively indolent when compared to the clinicallyapparent SRCs that appear later: mitoses are observed only rarely outside of the upper isthmus (less often than in surrounding non-neoplastic epithelium) as is staining for Ki-67 and other proliferation markers [13]. Interestingly, the number of SRC foci correlates neither with patient age (a phenomenon also observed in familial adenomatous polyposis [20]) nor with development of advanced disease—with appropriate treatment, the prognosis of patients even with large T1a disease burdens appears to remain excellent (>90% 5-year survival [21,22]), though long-term experience is not yet available.
While the number of SRC foci does not seem to increase with age, older patients do demonstrate larger tumors, reflecting a proportionate increase in their probability of developing advanced-stage disease. As SRC foci grow larger (>3 mm), poorly-differentiated cells with a mesenchymal phenotype begin to appear along the deep aspect of the tumor, presaging more aggressive behavior [12,13,15]. With disease progression, these poorlydifferentiated cells come to dominate the tumor, especially in the more deeply-invasive areas, with significant numbers of signet ring cells found only in the superficial portions of the tumor in advanced (stage T3 or higher) disease [13,15] (Figure 1(c)).
3. Mutational Spectrum of CDH1
As in other autosomal dominant cancer syndromes, HDGC is associated with “first hit” germline inactivation of one CDH1 allele, with a “second hit” event heralding the onset of carcinogenesis. Despite the presence of functionally critical domains within E-cadherin, no mutation “hot spots” have been identified for germline inactivation, though some (e.g. c.1003C > T [p.R335X], c.1137G > A [splice site mutation], c.1901C > T [p.A634V]) have been reported in multiple unrelated families [23-25]. (In contrast, somatic CDH1 mutations detected in sporadic diffuse gastric cancer are largely splice site mutations involving exons 8 and 9 [26].) To date, over 100 different germline mutations have been described and are found scattered throughout the 2.6 kb coding sequence as well as in regulatory sequences [22]. Mutations are most often small insertion/deletion events (35%); however, missense (28%), nonsense (16%), and splice site (16%) mutations as well as large deletions (5%) have all been described [27]. Irrespective of their specific classification, the majority of these events result in a frameshift and a premature stop codon, often with activation of nonsense-mediated mRNA decay [28].
In contrast to the diversity of germline mutations, the majority of second hit events in early tumors involve promoter hypermethylation [29], though numerous mutations, deletions, and epigenetic modifications have also been described. In early second hit events, however, catastrophic loss of gene function, as occurs in the majority of germline mutations, is almost never seen [30,31]. Rather, these events result in partial loss of E-cadherin function either through decreased expression (as in promoter hypermethylation, for example) or hypomorphic mutation. Interestingly, second hit events detected in metastatic disease (commonly loss of heterozygosity) are often distinct from those in the primary tumor and often do result in complete loss of E-cadherin expression, perhaps reflecting the reduced susceptibility of advanced tumors to anoikis [30-32].
While the germline CDH1 mutational spectrum is quite varied, HDGC demonstrates a surprising absence of genotype-phenotype correlation [22]. As such, it is unclear if the variation in reported penetrance and average age of diagnosis [24] reflects population-specific differences in contributing genetic or environmental risk factors or true differences between mutations. Despite the apparent variance in penetrance, the risk of gastric cancer remains high regardless of the specific germline CDH1 allele, with no evidence of significant allele-specific disease attenuation as seen in other cancer syndromes, e.g. familial adenomatous polyposis [20].
4. CDH1 Testing
HDGC presents a formidable diagnostic challenge. Pathologic sequence variants have been reported throughout the CDH1 coding sequence and associated regulatory sequences, necessitating full sequencing of all 16 exons and nearby flanking intronic regions [22]. Moreover, large deletion events undetectable by standard Sanger sequencing technology comprise ~5% of described events, which supports the added value of quantitative copy number variation analysis (e.g. by multiplex ligation-dependent probe amplification or next-generation sequencing) [27]. Irrespective of its necessity, such a broad approach will inevitably identify previously unreported variants and variants that are otherwise of uncertain clinical significance. This challenge is compounded by CDH1’s relatively high frequency of polymorphisms [33] and the limited experience with them—diagnostic sequencing of CDH1 is a relatively recent and low-volume pursuit. Even polymorphisms outside of the coding sequence may complicate testing as those lying within exon amplification primer binding sites can disrupt primer annealing, causing allele drop-out and false negative sequencing results [33]. Given the rarity of HDGC, most of these variants in the general population will be unassociated with gastric cancer; however, the significant consequences of either withholding needed or providing unnecessary treatment render management of these individuals fraught.
Whereas there is widespread agreement that low pretest probability and high error penalties render CDH1 testing of all cases of gastric cancer or the general population inappropriate, there is no universally agreed upon set of criteria by which to define populations of sufficient pre-test probability [34]. Rather, multiple sets of overlapping and often conflicting guidelines are used across institutions (Table 1). The first and still most widely accepted [34] criteria were initially proposed by the International Gastric Cancer Linkage Consortium (IGCLC) in 1999 and recommended testing only kindreds in which 1) two or more cases of diffuse gastric cancer had been identified in firstor second-degree relatives with at least one diagnosed before the age of 50, or 2) three or more cases of diffuse gastric cancer had been identified regardless of age at diagnosis [35]. While these criteria are still the most widely accepted and stringent, their sensitivity was subsequently felt to be insufficient, especially in light of new observations concerning lobular breast cancer incidence and the importance of tumor histology. In response, the Stanford Cancer Center, the New Zealand HDGC Group, and the British Columbia Cancer Agency, as well as individual members of the IGCLC and other groups, separately released their own updated guidelines expanding the original criteria to cover what each felt to be the most at-risk populations (Table 1).
Although expansion of the original IGCLC criteria was undoubtedly warranted, the profusion of overlapping and at times conflicting guidelines has fostered confusion regarding the diagnostic standard of care. Complicating this, performance characteristics for the various recommendations vary widely at multiple levels. Firstly, the various guidelines perform very differently in the reported literature. For example, 30% - 50% of patients selected under the Modified IGCLC Criteria [36] (perhaps the most widely used currently) are positive on CDH1 screening [37,38], compared with 7% of those tested under criteria used by Roviello et al. [39]. Furthermore, the individual criteria that comprise published guidelines perform very differently; patients meeting the first initial IGCLC criterion (two or more cases of diffuse gastric cancer in close relatives with at least one diagnosed before the age of 50) have a 53% chance of testing positive [23,36], while isolated cases of diffuse gastric cancer diagnosed at a young age have a <10% chance [23,36,40, 41]. Lastly, the performance of an individual criterion
Table 1. Selected recommendations for CDH1 testing in HDGC. DGC: diffuse gastric cancer; LBC: lobular breast cancer.

may vary widely between reports. For example, the same first initial IGCLC criterion was associated with a 53% prevalence of CDH1 mutations in one study [36] while another found no positives in this group [39]. Much of this variation may be explained by small sample sizes due to the rarity of HDGC; however, incidence variance between populations must also be considered. Regardless of its source, this variation has lead to controversy even among experts [34] and frustrated efforts to achieve consensus on a single set of patient selection criteria.
Despite the current variation between guidelines, criteria, and reports, the larger series to date have reported similar overall rates of detection—31% (13/42) (Table 1 of Reference [36]), 39% (15/38) [23], 29% (46/160) [41], and 29% (9/31) [25] with a recent meta-analysis reporting 29% (94/322) [40]—despite variation in criteria used and the definitions there of. These results are somewhat lower than the historical aggregate of CDH1 testing results (38.4%, 417/1085, many published without reference to how patients were selected for testing) [40], perhaps reflecting the relatively recent relaxation of screening guidelines and the higher rate of detection in families with known CDH1 mutations (62.8%) [40]. Nevertheless, it is clear that even with the most stringent clinical criteria, CDH1 testing will fail to yield a causative variant in many cases. In this context, variants of unknown significance assume an even greater importance. In such cases, we recommend using family studies that ideally include all available affected individuals and an assessment of residue conservation across species. In silico analyses (e.g. SIFT, PolyPhen, and other prediction scores [42-44]) can also be used in an attempt to subcategorize variants as likely silent or likely deleterious [45], although such predictions are not always possible and must be interpreted with caution, as these tools are not validated for clinical use and, at times, contradict one another.
5. Clinical Management of Germline CDH1 Mutations
Without intervention, germline CDH1 mutations are associated with a 60% - 80% lifetime risk of advanced diffuse gastric cancer [6,46], ~90% of which represent surgically incurable disease at the time of diagnosis [47]. As such, pre-symptomatic identification of at-risk patients is the cornerstone of clinical management. While most experts recommend restricting testing to individuals over the age of consent [34], HDGC may present as early as 14 years of age in some kindreds [4,5], requiring testing of children at younger ages than might otherwise be appropriate. There is general agreement that CDH1 testing should ideally not be delayed past 20 years of age [34].
The standard of care (and indeed the only effective treatment) for an identified germline CDH1 mutation is prophylactic complete gastrectomy [34,40]. Meticulous removal of gastric tissue (including heterotopia like Meckel’s diverticulum, if present) entirely eliminates the risk of gastric cancer, though patients remain at risk for other CDH1-associated malignancies, primarily lobular breast cancer. Whereas the ages of diagnosis of the individual’s affected family members should be taken into account, the general consensus is that the operation should take place after 20 years of age [34], where the risk of advanced HDGC, ~1%, approximates total gastrectomy’s perioperative mortality at experienced centers [5,37,38]. While useful as an adjunct or in cases where gastrectomy is not an available or exercised option, active endoscopic surveillance is inappropriate for first-line management of known CDH1 mutation carriers due to inadequate sensitivity, even with endoscopists specializing in HDGC [34]; only ~12% - 16% of early invasive disease (defined as pT2 or less) is endoscopically detectable despite expert surveillance [6,40,48]. Indeed, even advanced disease (pT3 and higher) may be endoscopically occult due to HDGC’s penchant for diffusely permeating surrounding non-neoplastic tissue [17,19,48]. Highlighting these findings, the collected experience from 220 patients with germline CDH1 mutations (77% of whom elected to undergo prophylactic gastrectomy) demonstrated that only 2.5% had symptoms and 12.4% had pre-operative findings on endoscopy or radiology at the time of surgery [40]. Despite this, 87% of those who underwent gastrectomy demonstrated invasive SRC on histologic examination [40] (even this is likely an underestimate as not all cases were sampled appropriately; in another study, SRC was detected in 2/3 of “negative” prophylactic gastrectomy specimens upon expert review [6]).
While no definitive data exist, breast and colorectal cancer risk in carriers of germline CDH1 mutations is managed by extrapolation from experience with carriers of germline mutations in BRCA1/2 or DNA mismatch repair genes, respectively. The IGCLC recommends female CDH1 mutation carriers to undergo biannual clinical breast exams with annual MRI and traditional mammography starting at age 35, while all CDH1 mutation carriers are recommended to start standard colorectal cancer screening programs at age 40 or 10 years before the earliest family member’s diagnosis [6].
It is very much unclear what constitutes proper clinical management for individuals that meet clinical criteria for HDGC but bear no demonstrable germline CDH1 abnormality. Though its appropriateness is controversial [34], some institutions offer prophylactic gastrectomy to all patients with high clinical suspicion for HDGC regardless of CDH1 status, whereas others offer only surveillance endoscopy, the benefits of which are equally uncertain. While published data are scarce, Seevaratnam et al. [40] describe a small cohort of patients with suspected HDGC that elected prophylactic gastrectomy despite negative CDH1 testing. In all nine patients, the resulting prophylactic gastrectomy specimens failed to demonstrate any evidence of carcinoma (carriers of CDH1 mutations in the same study demonstrated SRC in 87% of specimens). Further sequencing analyses are also of dubious value; large scale sequencing of the CDH1-interacting catenin family of genes in a cohort of HDGC families without demonstrable CDH1 abnormalities yielded only two non-synonymous variants of uncertain significance in JUP and none in CTNNA1, CTNNB1, or CTNND1 [49]. While mutations in other tumor suppressor genes such as TP53, BRCA1/2, and APC can explain some instances of hereditary gastric cancer, affected individuals demonstrate a diversity of cancers of which gastric cancer is a minority [1] and are thus unlikely to be confused clinically with HDGC.
6. Conclusions
HDGC is a rare and lethal autosomal dominant cancer syndrome with a grim prognosis diverted only by prophylactic complete gastrectomy. The consequences of even a flawlessly-performed gastrectomy, however, are significant; 100% of patients suffer marked long-term morbidity from gastrointestinal symptoms, nutritional disorders, and major lifestyle restrictions. The conesquences of withholding gastrectomy, however, are even more grievous; up to 80% of affected individuals will develop advanced diffuse gastric cancer, most as young adults, and most will die of this disease. Flanked by such weighty error penalties, clinicians must approach any suspected HDGC patient with great care. While demonstrating inactivation of a CDH1 allele in the germline unambiguously establishes the diagnosis, the majority of sequence variants detected in the general population will be of uncertain clinical significance and, ultimately, unassociated with gastric cancer. To avoid unnecessary gastrectomy, multiple sets of screening criteria have been developed to help clinicians select patients for CDH1 gene testing. While these do disagree on specifics, the more well-established guidelines (e.g. modified IGCLC, Stanford Cancer Center) perform similarly in practice, identifying germline CDH1 abnormalities in ~30% of selected individuals [40]. As such, while criteria enrich for mutation carriers, CDH1 testing will still be negative in the majority of individuals or will return variants of uncertain clinical significance. Optimal management of such patients is an evolving pursuit with the current plurality of experts suggesting surveillance endoscopy [34].
HDGC represents a bench-to-bedside success story in which linkage of disease to a specific genetic abnormality allows prospective identification of high-risk individuals, thus enabling life-saving intervention. Despite this, our understanding of the syndrome remains rudimentary. What defects underlie the ~70% of clinically-affected individuals that do not carry demonstrable CDH1 mutations, and how are they best managed? Why diffuse gastric cancer and not other types or sites? With patients now living into older age since the advent of prophylactic gastrectomy, will we see new cancer predilections emerge? Why do the vast majority of HDGC-related tumors (the multitude of minute SRC foci observed in prophylactic gastrectomy specimens) lie indolent for decades, and what drives a small subset of them to evolve to lethal malignancies? Can we identify when this transformation will take place so as to delay gastrictomy or even prevent it altogether? The answers to these questions and others will shape future diagnostic and therapeutic approaches to this rare disorder.
REFERENCES
- N. Chun and J. M. Ford, “Genetic Testing by Cancer Site,” The Cancer Journal, Vol. 18, No. 4, 2012, pp. 355-363. doi:10.1097/PPO.0b013e31826246dc
- H. Wu, J. A. Rusiecki, K. Zhu, J. Potter and S. S. Devesa, “Stomach Carcinoma Incidence Patterns in the United States by Histologic Type and Anatomic Site,” Cancer Epidemiology, Biomarkers, and Prevention, Vol. 18, No. 7, 2009, pp. 1945-1952. doi:10.1158/1055-9965.EPI-09-0250
- J. Stone, S. Bevan, D. Cunningham, A. Hill, N. Rahman, J. Peto, A. Marossy and R. S. Houlston, “Low Frequency of Germline E-Cadherin Mutations in Familial and Nonfamilial Gastric Cancer,” British Journal of Cancer, Vol. 79, No. 11-12, 1999, pp. 1935-1937. doi:10.1038/sj.bjc.6690308
- P. Guilford, J. Hopkins, J. Harraway, N. McLeod, P. Harawira, H. Taite, R. Scoular, A. Miller and A. E. Reeve, “E-Cadherin Germline Mutations in Familial Gastric Cancer,” Nature, Vol. 392, No. 6674, 1998, pp. 402-405. doi:10.1038/32918
- P. D. Pharoah, P. Guilford and C. Caldas, “Incidence of Gastric Cancer and Breast Cancer in CDH1 (E-Cadherin) Mutation Carriers from Hereditary Diffuse Gastric Cancer Families,” Gastroenterology, Vol. 121, No. 6, 2001, pp. 1348-1353. doi:10.1053/gast.2001.29611
- R. C. Fitzgerald, R. Hardwick, D. Huntsman, F. Carneiro, P. Guilford, V. Blair, D. C. Chung, J. Norton, K. Ragunath, J. H. Van Krieken, S. Dwerryhouse and C. Caldas, “Hereditary Diffuse Gastric Cancer: Updated Consensus Guidelines for Clinical Management and Directions for Future Research,” Journal of Medical Genetics, Vol. 47, No. 7, 2010, pp. 436-444. doi:10.1136/jmg.2009.074237
- P. Carneiro, M. S. Fernandes, J. Figueiredo, J. Caldeira, J. Carvalho, H. Pinheiro, M. Leite, S. Melo, P. Oliveira, J. Simoes-Correia, M. J. Oliveira, F. Carneiro, C. Figueiredo, J. Paredes, C. Oliveira and R. Seruca, “E-Cadherin Dysfunction in Gastric Cancer—Cellular Consequences, Clinical Applications, and Open Questions,” FEBS Letters, Vol. 586, No. 18, 2012, pp. 2981-2989. doi:10.1016/j.febslet.2012.07.045
- M. Al-Hajj and M. F. Clarke, “Self-Renewal and Solid Tumor Stem Cells,” Oncogene, Vol. 23, No. 43, 2004, pp. 7274-7282. doi:10.1038/sj.onc.1207947
- B. Humar and P. Guilford, “Hereditary Diffuse Gastric Cancer: A Manifestation of Lost Cell Polarity,” Cancer Science, Vol. 100, No. 7, 2009, pp. 1151-1157. doi:10.1111/j.1349-7006.2009.01163.x
- N. den Elzen, C. V. Buttery, M. P. Maddugoda, G. Ren and A. S. Yap, “Cadherin Adhesion Receptors Orient the Mitotic Spindle during Symmetric Cell Division in Mammalian Epithelia,” Molecular Biology of the Cell, Vol. 20, No. 16, 2009, pp. 3740-3750. doi:10.1091/mbc.E09-01-0023
- C. D’Souza-Schorey, “Diassembling Adherens Junctions: Breaking up Is Hard to Do,” Trends in Cellular Biology, Vol. 15, No. 1, 2005, pp. 19-26. doi:10.1016/j.tcb.2004.11.002
- P. Guilford, V. Blair, H. More and B. Humar, “A Short Guide to Hereditary Diffuse Gastric Cancer,” Hereditary Cancer in Clinical Practice, Vol. 5, No. 4, 2007, pp. 183- 194. doi:10.1186/1897-4287-5-4-183
- B. Humar, R. Fukuzawa, V. Blair, A. Dunbier, H. More, A. Charlton, H. K. Yang, W. H. Kim, A. E. Reeve, I. Martin and P. Guilford, “Destabilized Adhesion in the Gastric Proliferative Zone and c-Src Kinase Activation Mark the Development of Early Diffuse Gastric Cancer,” Cancer Research, Vol. 67, No. 6, 2007, pp. 2480-2489. doi:10.1158/0008-5472.CAN-06-3021
- Y. Chen, K. Kingham, J. M. Ford, J. Rosing, J. Van Dam, R. B. Jeffrey, T. A. Longacre, N. Chun, A. Kurian and J. A. Norton, “A Prospective Study of Total Gastrectomy for CDH1-Positive Hereditary Diffuse Gastric Cancer,” Annals of Surgical Oncology, Vol. 18, No. 9, 2011, pp. 2594-2598. doi:10.1245/s10434-011-1648-9
- V. Blair, I. Martin, D. Shaw, I. Winship, D. Kerr, J. Arnold, P. Harawira, M. McLeod, S. Parry, A. Charlton, M. Findlay, B. Cox, B. Humar, H. More and P. Guilford, “Hereditary Diffuse Gastric Cancer: Diagnosis and Management,” Clinical Gastroenterology and Hepatology, Vol. 4, No. 3, 2006, pp. 262-275. doi:10.1016/j.cgh.2005.12.003
- A. Charlton, V. Blair, D. Shaw, S. Parry, P. Guilford and I. G. Martin, “Hereditary Diffuse Gastric Cancer: Predominance of Multiple Foci of Signet Ring Carcinoma in Distal Stomach and Transition Zone,” Gut, Vol. 53, No. 6, 2004, pp. 814-820. doi:10.1136/gut.2002.010447
- F. Carneiro, D. G. Huntsman, T. C. Smyrk, D. A. Owen, R. Seruca, P. Pharoah, C. Caldas and M. Sobrinho-Simoes, “Model of the Early Development of Diffuse Gastric Cancer in E-Cadherin Mutation Carriers and Its Implications for Patient Screening,” Journal of Pathology, Vol. 203, No. 2, 2004, pp. 681-687. doi:10.1002/path.1564
- M. E. Barber, V. Save, F. Carneiro, S. Dwerryhouse, P. Lao-Sirieix, R. H. Hardwick, C. Caldas and R. C. Ritzgerald, “Histopathological and Molecular Analysis of Gastrectomy Specimens from Hereditary Diffuse Gastric Cancer Patients Has Implications for Endoscopic Surveillance of Individuals at Risk,” Journal of Pathology, Vol. 216, No. 3, 2008, pp. 286-294. doi:10.1002/path.2415
- D. G. Huntsman, F. Carneiro, F. R. Lewis, P. M. MacLeod, A. Hayashi, K. G. Monaghan, R. Maung, R. Seruca, C. E. Jackson and C. Caldas, “Early Gastric Cancer in Young Asymptomatic Carriers of Germ-Line E-Cadherin Mutations,” New England Journal of Medicine, Vol. 344, No. 25, 2001, pp. 1904-1909. doi:10.1056/NEJM200106213442504
- M. D. Crabtree, I. P. Tomlinson, I. C. Talbot and R. K. Phillips, “Variability in the Severity of Colonic Disease in Familial Adenomatous Polyposis Results from Differences in Tumor Initiation Rather than Progression and Depends Relatively Little on Patient Age,” Gut, Vol. 49, 2001, pp. 540-543. doi:10.1136/gut.49.4.540
- S. M. Everett and A. T. Axon, “Early Gastric Cancer in Europe,” Gut, Vol. 41, No. 2, 1997, pp. 142-150. doi:10.1136/gut.41.2.142
- P. Guilford, B. Humar and V. Blair, “Hereditary Diffuse Gastric Cancer: Translation of CDH1 Germline Mutations into Clinical Practice,” Gastric Cancer, Vol. 13, No. 1, 2010, pp. 1-10. doi:10.1007/s10120-009-0531-x
- P. Kaurah, A. MacMillan, N. Boyd, J. Senz, A. De Luca, N. Chun, et al., “Founder and Recurrent CDH1 Mutations in Families with Hereditary Diffuse Gastric Cancer,” Journal of the American Medical Association, Vol. 297, No. 21, 2007, pp. 2360-2372. doi:10.1001/jama.297.21.2360
- H. More, B. Humar, W. Weber, R. Ward, A. Christian, C. Lintott, F. Graziano, A. M. Ruzzo, E. Acosta, B. Boman, M. Harlan, P. Ferreira, R. Seruca, G. Suriano and P. Guilford, “Identification of Seven Novel Germline Mutations in the Human E-Cadherin Gene,” Human Mutation, Vol. 28, No. 2, 2007. p. 203. doi:10.1002/humu.9473
- G. Suriano, S. Yew, P. Ferreira, J. Senz, P. Kaurah, J. M. Ford, et al., “Characterization of a Recurrent Germ Line Mutation of the E-Cadherin Gene: Implications for Genetic Testing and Clinical Management,” Clinical Cancer Research, Vol. 11, No. 15, 2005. pp. 5401-5409. doi:10.1158/1078-0432.CCR-05-0247
- G. Berx, K.-F. Becker, H. Hoflier and F. Roy, “Mutations of the Human E-Cadherin (CDH1) Gene,” Human Mutation, Vol. 12, No. 4, 1998, pp. 226-237. doi:10.1002/(SICI)1098-1004(1998)12:4<226::AID-HUMU2>3.0.CO;2-D
- C. Oliveira, J. Senz, P. Kaurah, H. Pinheiro, R. Sanges, A. Haegert, et al., “Germline CDH1 Deletions in Hereditary Diffuse Gastric Cancer Families,” Human Molecular Genetics, Vol. 18, No. 9, 2009, pp. 1545-1555. doi:10.1093/hmg/ddp046
- R. Karam, J. Carvalho, I. Bruno, C. Graziado, J. Senz, D. Huntsman, F. Carneiro, R. Seruca, M. F. Wilkinson and C. Olveira, “The NMD mRNA Surveillance Pathway Downregulates Aberrant E-Cadherin Transcripts in Gastric Cancer Cells and in CDH1 Mutation Carriers,” Oncogene, Vol. 27, No. 30, 2008, pp. 4255-4260. doi:10.1038/onc.2008.62
- B. Humar, V. Blair, A. Charlton, H. More, I. Martin and P. Guilford, “E-Cadherin Deficiency Initiates Gastric Signet-Ring Carcinoma in Mice and Man,” Cancer Research, Vol. 69, No. 5, 2009, pp. 2050-2056. doi:10.1158/0008-5472.CAN-08-2457
- C. Olveira, J. de Bruin, S. Nabais, M. Ligtenberg, C. Moutinho, F. M. Nagengast, R. Seruca, H. van Krieken and F. Carneiro, “Intragenic Deletion of CDH1 as the Inactivating Mechanism of the Wild-Type Allele in an HDGC Tumour,” Oncogene, Vol. 23, No. 12, 2004. pp. 2236- 2240. doi:10.1038/sj.onc.1207335
- W. M. Grady, J. Willis, P. J. Guilford, A. K. Dunbier, T. T. Toro, H. Lynch, G. Wiesner, K. Ferguson, C. Eng, J. G. Park, S. J. Kim and S. Markowitz, “Methylation of the CDH1 Promoter as the Second Genetic Hit in Hereditary Diffuse Gastric Cancer,” Nature Genetics, Vol. 26, No. 1, 2000, pp. 16-17. doi:10.1038/79120
- P. W. Derksen, X. Liu, F. Saridin, H. van der Gulden, J. Zevenhoven, B. Evers, J. R. van Beijnum, A. W. Griffioen, J. Vink, P. Krimpenfort, J. L. Peterse, R. D. Cardiff, A. Berns and J. Jonkers, “Somatic Inactivation of E-Cadherin and p53 in Mice Leads to Metastatic Lobular Mammary Carcinoma through Induction of Anoikis Resistance,” Cancer Cell, Vol. 10, No. 5, 2006. pp. 437-449. doi:10.1016/j.ccr.2006.09.013
- F. M. Mullins, L. Dietz, M. Lay, J. L. Zehnder, J. Ford, N. Chun and I. Schrijver, “Identification of an Intronic SNP Leading to Allele Dropout during Validation of a CDH1 Sequencing Assay: Implications for Designing PCR-Based Assays,” Genetics in Medicine, Vol. 9, No. 11, 2007, pp. 752-760. doi:10.1097/GIM.0b013e318159a369
- M. Dixon, et al., “A RAND/UCLA Appropriateness Study of the Management of Familial Gastric Cancer,” Annals of Surgical Oncology, Vol. 20, No. 2, 2012, pp. 533- 541. doi:10.1245/s10434-012-2584-z
- C. Caldas, et al., “Familial Gastric Cancer: Overview and Guidelines for Management,” Journal of Medical Genetics, Vol. 36, No. 12, 1999, pp. 873-880.
- A. R. Brooks-Wilson, P. Kaurah, G. Suriano, S. Leach, J. Senz, N. Grehan, et al., “Germline E-Cadherin in Hereditary Diffuse Gastric Cancer: Assessment of 42 New Families and Review of Genetic Screening Criteria,” Journal of Medical Genetics, Vol. 41, No. 7, 2004, pp. 508- 517. doi:10.1136/jmg.2004.018275
- R. M. Cisco, J. M. Ford and J. A. Norton, “Hereditary Diffuse Gastric Cancer: Implications of Genetic Testing for Screening and Prophylactic Surgery,” Cancer, Vol. 113, No. 7, 2008, pp. 1850-1856. doi:10.1002/cncr.23650
- R. M. Cisco and J. A. Norton, “Hereditary Diffuse Gastric Cancer: Surgery, Surveillance, and Unanswered Questions,” Future Oncology, Vol. 4, No. 4, 2008. pp. 553-559. doi:10.2217/14796694.4.4.553
- F. Roviello, G. Corso, C. Pedrazzani, D. Marrelli, G. De Falco, A. Berardi, et al., “Hereditary Diffuse Gastric Cancer and E-Cadherin: Description of the First Germline Mutation in an Italian Family,” European Journal of Surgical Oncology, Vol. 33, No. 4, 2007, pp. 448-451. doi:10.1016/j.ejso.2006.10.028
- R. Seevaratnam, N. Coburn, R. Cardoso, M. Dixon, A. Bocicariu and L. Helyer, “A Systematic Review of the Indications for Genetic Testing and Prophylactic Gastrectomy among Patients with Hereditary Diffuse Gastric Cancer,” Gastric Cancer, Vol. 15 Supplement 1, 2012, pp. S153-S163. doi:10.1007/s10120-011-0116-3
- H. T. Lynch, E. Silva, D. Wirtzfeld, P. Hebbard, J. Lynch and D. G. Huntsman, “Hereditary Diffuse Gastric Cancer: Prophylactic Surgical Oncology Implications,” Surgical Clinics of North America, Vol. 88, No. 4, 2008, pp. 759- 778. doi:10.1016/j.suc.2008.04.006
- P. C. Ng and S. Henikoff, “SIFT: Predicting Amino Acid Changes That Affect Protein Function,” Nucleic Acid Research, Vol. 31, No. 13, 2003, pp. 3814-3814. doi:10.1093/nar/gkg509
- S. Sunyaev, V. Ramensky, I. Koch, W. Lathe, A. S. Kondrashov and P. Bork, “Prediction of Deleterious Human Alleles,” Human Molecular Genetics, Vol. 10, No. 6, 2001, pp. 591-597. doi:10.1093/hmg/10.6.591
- G. Suriano, S. Seixas, J. Rocha and R. Seruca, “A Model to Infer the Pathogenic Significance of CDH1 Germline Missense Variants,” Journal of Molecular Medicine, Vol. 84, No. 12, 2006, pp. 1023-1031. doi:10.1007/s00109-006-0091-z
- S. E. Plon, D. M. Eccles, D. Easton, W. D. Foulkes, M. Genuardi, M. S. Greenblatt, et al., “Sequence Variant Classification and Reporting: Recommendations for Improving the Interpretation of Cancer Susceptibility Genetic Test Results,” Human Mutation, Vol. 29, No. 11, 2008, pp. 1282-1291. doi:10.1002/humu.20880
- P. D. Pharoah, P. Guilford and C. Caldas, “Incidence of Gastric Cancer and Breast Cancer in CDH1 (E-Cadherin) Mutation Carriers from Hereditary Diffuse Gastric Cancer Families,” Gastroenterology, Vol. 121, No. 6, 2001, pp. 1348-1353. doi:10.1053/gast.2001.29611
- J. B. Koea, M. S. Karpeh and M. F. Brennan, “Gastric Cancer in Young Patients: Demographic, Clinicopathological, and Prognostic Factors in 92 Patients,” Annals of Surgical Oncology, Vol. 7, No. 5, 2000, pp. 346-351. doi:10.1007/s10434-000-0346-9
- M. Yamashina, “A Variant of Early Gastric Carcinoma. Histologic and Histochemical Studies of Early Signet Ring Cell Carcinomas Discovered Beneath Preserved Surface Epithelium,” Cancer, Vol. 58, No. 6, 1986. pp. 1333-1339. doi:10.1002/1097-0142(19860915)58:6<1333::AID-CNCR2820580625>3.0.CO;2-B
- J. M. Schuetz, S. Leach, P. Kaurah, J. Jeyes, Y. Butterfield, D. Huntsman and A. R. Brooks-Wilson, “Catenin Family Genes Are Not Commonly Mutated in Hereditary Diffuse Gastric Cancer,” Cancer Epidemiology, Biomarkers, and Prevention, Vol. 21, 2012, p. 2272. doi:10.1158/1055-9965.EPI-12-1110
- C. Caldas, F. Carneiro, H. T. Lynch, J. Yokota, G. L. Wiesner, S. M. Powell, et al., “Familial Gastric Cancer: Overview and Guidelines for Management,” Journal of Medical Genetics, Vol. 36, No. 12, 1999, pp. 873-880.