Journal of Cancer Therapy
Vol.4 No.2A(2013), Article ID:28504,14 pages DOI:10.4236/jct.2013.42A052
Molecular Targeted Therapy of Hepatocellular Carcinoma
![]()
1University of Nebraska College of Pharmacy, Omaha, USA; 2Saint Francis Cancer Treatment Center, Grand Island, USA.
Email: *mcopur@sfmc-gi.org
Received December 18th, 2012; revised January 19th, 2013; accepted January 28th, 2013
Keywords: Hepatocellular Carcinoma (HCC); Treatment; Molecular Therapies; Novel Agents
ABSTRACT
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third most common cause of cancer-related mortality worldwide. Despite decades of efforts by many investigators, systemic chemotherapy or hormonal therapy has notoriously failed to show an improvement in survival. With a median survival of 8 months, and 1- and 3-year survival rates of 20% and 5%, respectively, the effective treatment of HCC remains far from satisfactory. Better understanding of the pathogenesis of this disease, identification of molecular targets for therapeutic intervention and availability of promising molecularly targeted therapies may change this dismal picture. In this review we will focus on what is currently known about the molecular pathogenesis of HCC, and explore the currently available and future molecular based therapies targeting these pathways. Future research in this area will maximize clinical benefit while minimizing the toxicity and cost through utilization of novel targeted agents.
1. Introduction
The fifth most common cancer worldwide and the third most common cause of cancer-related deaths globally, HCC poses a great challenge [1,2]. The incidence of HCC in the United States has almost doubled in recent decades [3]. Due to multiple etiologies leading to the development of this disease, HCC patient populations are very diverse. It can occur in various underlying settings such as hepatitis B and C, nonalcoholic steatohepatitis, hemochromatosis, autoimmune hepatitis, alcohol-related liver disease, primary biliary cirrhosis, and primary sclerosing cholangitis. Systemic therapy with various classes of agents, including hormonal and cytotoxic agents, has provided little to no benefit. Improved understanding of the mechanism of hepatocarcinogenesis, coupled with the discovery of a multitude of molecular pathways leading to the development of HCC, has created a great opportunity to identify new targets and effective therapies (Figure 1). The initial demonstration of improved survival benefit by sorafenib in advanced HCC has opened a new era of molecularly targeted therapies, resulting in the discovery of a diverse spectrum of novel agents, many of which are currently under active clinical investigation and development. Targeted agents that inhibit angiogenesis, epidermal growth factor receptor, and mammalian target of rapamycin, insulin-like growth factor and many other are at different stages of clinical development (Figure 2). While these targeted therapies may offer hope as main treatments for patients who are not amenable to undergo surgical resection or transplant, they can constitute the basis for promising adjunctive therapies in combination with currently available treatment modalities. In this article we will review the pathways identified in the pathogenesis of HCC along with currently available and in development therapies targeting these pathways
2. Pathways and Targeted Therapeutic Agents in HCC
2.1. Vascular Endothelial Growth Factor, Fibroblast Growth Factor, and Platelet Derived Growth Factor and Receptors (VEGF/VEGFR, FGF/FGFR, PDGF/PDGFR) Pathway
Hepatocellular carcinoma (HCC) is one of the most vascular solid tumors. Vascular endothelial growth factor (VEGF) plays a critical role in mediating angiogenesis in HCC and is a potential therapeutic target. VEGF can function by inducing vascular promotion and growth on several cell types varying from hepatocytes, hepatic stellate cells, endothelial progenitor cells to hemangiocytes. Thus blockade of VEGF-mediated pathways, either by anti-VEGF neutralizing antibody or tyrosine kinase inhibitors distrupts carcinogenesis and angiogenesis [4]. In

Figure 1. Altered signaling pathways in HCC.
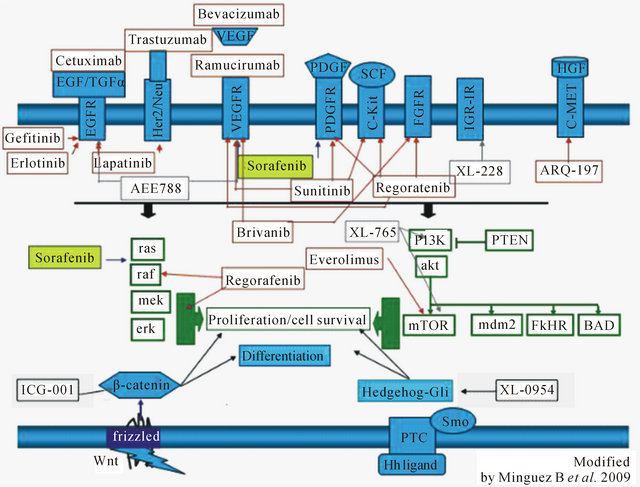
Figure 2. Molecularly targeted therapies in HCC.
addition to VEGF, several other angiogenic factors in HCC have recently been identified. These factors can also regulate angiogenic processes through interaction with VEGF or VEGF-independent pathways [5,6]. Increased levels of vascular endothelial growth factor (VEGF) and microvessel density have been shown to be associated with worsening survival in HCC [7-9]. A phase III clinical trial revealed that an anti-angiogenic agent “sorafenib” extended overall survival in patients with advanced HCC, as the first drug ever demonstrating a meaningful improvement in the treatment of tis deadly disease in 2008 [10]. Since then several antiangiogenic and other molecularly targeted agents have been and currently being evaluated in clinical trials of HCC (Table 1).
2.1.1. Bevacizumab
Bevacizumab, a recombinant humanized monoclonal antibody that targets VEGF has been studied either as a single agent or in combination with cytotoxic or other
Table 1. Molecular targets and therapeutic agents.

targeted agents. In a multicenter phase II study of 46 patients with compensated liver disease and unresectable HCC, bevacizumab led to a 13% partial response rate (PR) with a median progression-free survival (PFS) of 6.9 months and overall survival (OS) of 53% at 1 year, 28% at 2 years, and 23% at 3 years. Bevacizumab was associated with significant reductions in tumor enhancement by dynamic contrast-enhanced magnetic resonance imaging and reductions in circulating VEGF-A and stromal-derived factor-1 levels [11].
Bevacizumab has also been studied in conjunction with cytotoxic chemotherapy, gemcitabine and oxaliplatin (GEMOX) for the treatment of unresectable HCC. In a phase II study of 33 patients with advanced metastatic HCC, treatment with combination regimen of bevacizumab and GEMOX resulted in a 20% response rate (RR) with a median OS of 9.6 months. While there were no patients with a complete response (CR), 27% had stable disease [12].
2.1.2. Vatalanib (PTK787)
Vatalanib (PTK787) an oral angiogenesis inhibitor, targets VEGFR-1/flt-1, VEGFR-2/KDR, and VEGFR-3/Flt- 4, all VEGFR tyrosine kinases and PDGFR, and the c-kit [13,14]. This drug interferes with the ATP binding sites of VEGF receptors. In a phase I study vatalanib was well tolerated and showed clinical activity in a variety of solid tumors. Administered once daily, at a dose of 750 mg to 1250 mg in patients with unresectable HCC, no CR or PR was observed. Nine patients had stable disease, and nine had progressive disease [15].
2.1.3. Cediranib (AZD2171)
Cediranib (AZD2171) is a potent inhibitor of both VEGFR-1 and VEGFR-2. It also has activity against c-kit, PDGFR-β, and FLT4 at Nano molar concentrations [16]. Cediranib has been shown to inhibit VEGF signaling. Cediranib was well tolerated up to 45 mg per day in patients with a broad range of solid tumors. The most common toxicities included diarrhea, dysphonia, and hypertension. In a phase II study of cediranib in 28 patients with advanced HCC, 19 patients were evaluable for toxicity [17]. The main adverse events were fatigue, hypertension and anorexia. The primary objective of this phase II study was to assess 6-month survival. Secondary objectives were to assess tumor response, time to progression (TTP), and toxicity. Twelve patients (42.9%) survived 6 months, 15 (53.6%) died within 6 months, and 1 (3.6%) was lost to follow-up before 6 months. The median OS was 5.8 months. No patients experienced a confirmed response. The median time to progression (mTTP) was 2.8 months [18].
2.1.4. Brivanib
Brivanib alaninate is an investigational, oral, anti-tumor agent that inhibits vascular endothelial growth factor receptor (VEGFR) and fibroblast growth factor receptors (FGFR). It has shown tumor growth inhibition in mouse HCC xenograft models. A phase II study evaluated the efficacy and safety of brivanib in patients with unresectable, locally advanced, or metastatic HCC who had received either no prior systemic therapy or one prior regimen of angiogenesis inhibitor [19]. At 800 mg oral daily dose brivanib provided a median OS of 10 months and TTP of 2.8 months with 5% PR and 47% disease control rate in previously untreated HCC patients. Most frequently observed grade 3 - 4 adverse events included fatigue (16%), high levels of AST (19%), and hyponatremia (41%). In a randomized phase III trial, BRISK-FL, brivanib versus sorafenib were evaluated in patients with advanced HCC who had not received prior systemic treatment. The study did not meet its primary overall survival objective based upon a non-inferiority statistical design.
2.1.5. Sunitinib
Sunitinib, an oral multikinase inhibitor targeting VEGFR- 1, VEGFR-2, also has inhibitory activities for other receptor tyrosine kinases including PDGFR-a/b, c-KIT, FLT3, and RET kinases [20]. In a study of 34 patients with advanced HCC, sunitinib was given 37.5 mg orally once daily on a 4-weeks-on, 2-weeks-off schedule (6 weeks per cycle). There was one patient with a PR of 20 months, and an additional 10 patients (38.5%) of stable disease for 12 weeks. The PFS was 3.9 months and OS was 9.8 months [21]. In a European/Asian phase II study, sunitinib was dosed at 50 mg daily for 4 weeks every 6 weeks in 37 unresectable HCC patients. Results reported one patient with a PR (2.7%), and 13 patients with stable disease (35%). The median OS was 8 months and PFS was 3.7 months [22]. In terms of toxicity, the studies that used the lower dose (37.5 mg) reported acceptable safety profiles. The most common adverse events included hematologic toxicities, fatigue, and an increase in transaminase levels. Grade 3 or 4 adverse events occurred in no more than 20% of the patients in any category. At the higher dose of 50 mg daily, sunitinib treatment led to more pronounced grade 3 - 4 toxicities and a higher death rate of 10% in this patient population [21,22]. Although the lower dose at 37.5 mg seems to be more tolerable, it remains uncertain whether the continuous or intermittent schedule is better. A randomized phase III study comparing sunitinib at 37.5 mg continuous daily dosing versus sorafenib at 400 mg twice daily in advanced HCC (clinical trial identifier: NCT00699374) was terminated on April 22th, 2010, based on a higher incidence of serious adverse events in the sunitinib arm compared to the sorafenib arm, and the fact that sunitinib did not meet the criteria to demonstrate that it was neither superior or non-inferior to sorafenib in the survival of patients with advanced hepatocellular cancer.
2.1.6. Pazopanib (GW786034)
Pazopanib is an oral angiogenesis inhibitor targeting VEGFR, PDGFR, and c-Kit. A phase I dose-escalating study of pazopanib in 28 Asian patients utilized oral pazopanib (200 - 800 mg) once daily on 21-day cycles. Results revealed dose-limiting toxicities (grade 3 aspartate aminotransferase (AST)/alanine aminotransferase (ALT) elevations) and grade 3 malaise at the 800 mg dose level. The maximum tolerated dose (MTD) in patients with HCC (Child-Pugh class A) was 600 mg daily [23]. Diarrhea, skin hypopigmentation, and AST elevation were the most commonly reported adverse events at the MTD. PR was observed in two patients (7%; one at 800 mg, one at 600 mg) and stable disease of 4 months in 11 patients (41%). Median TTP at the MTD was 137.5 days (range, 4 - 280 days). Changes in tumor dynamic contrast-enhanced magnetic resonance imaging parameters were seen after repeated dose pazopanib administration [24]. Pazopanib has a manageable safety profile in patients with advanced HCC, and 600 mg was chosen for further development of pazopanib in advanced HCC.
2.1.7. Linifanib (ABT869)
Linifanib (ABT869), a novel potent and selective inhibitor of the VEGF and PDGF platelet-derived growth factor families of receptor tytosine kinases, is designed to inhibit angiogenesis, tumor growth, and metastasis. An open-label, multicenter trial of oral linifanib dosed at 0.25 mg/kg daily in patients with a Child-Pugh A (C-PA) or dosed every other day in patients with a Child-Pugh B (C-PB) evaluated a total of 44 patients. Most common linifanib-related adverse events were fatigue (55%) and diarrhea (48%). Most common linifanib-related grade 3 or 4 toxicity were hypertension (18%) and fatigue (14%). Sixty-eight percent of patients had dose interruptions due to adverse events; PFS at 16 weeks were 31.8%, overall RR 6.8%, TTP 3.7 months and OS was 9.7 months [25]. An open-label, randomized phase 3 study of the efficacy and tolerability of linifanib (ABT869) versus sorafenib in advanced HCC (clinical trials identifier: NCT01009593) was discontinued in July 2012 by the independent data monitoring committee. The primary objective of this trial was to assess the overall survival of oral linifanib given as monotherapy once daily as compared to sorafenib given twice daily per standard of care. So far no publications available.
2.2. Epidermal Growth Factor and Receptor (EGF/EGFR) Pathway
The expression of several EGF family members, specifically EGF, TGF-a, and heparin binding epidermal growth factor, as well as EGFR, has been described in several HCC cell lines and tissues. The EGFR (also known as ErbB1 and Her1) belongs to the ERB family of receptor tyrosine kinases, which includes ErbB2 (also known as Her2), ErbB3 (also known as Her3) and ErbB4 (also known as Her4). All members (except Erb3) have tyrosine kinase activity and they all share a common structure with an extracellular ligand-binding domain, a transmembrane domain and an intracellular domain where the tyrosine kinase activity resides. EGFR forms homo or heterodimers upon ligand binding. Dimerization results in auto-phosphorylation of EGFR with the subsequent activation of a number of downstream signaling pathways, including the PI3K/Akt/mTOR and the Ras/Raf/ MEK/ERK pathways. ErbB2 has no known ligand, but the rest of the members can bind to a family of growth factors including EGF, TGF-α, epigenin (EPG), amphiregulin (AREG), heparin-binding-EGF (HB-EGF), epirugulin (EREG) and β-cellulin (BTC) and the last three ligands are also able to bind to ErbB4/Her4. The neuregulin (NRG) ligands NRG-1 and NGR-2 bind to both ErbB3/Her3 and ErbB4/Her4, whereas NGR-3 and NGR-4 only recognize ErbB4/Her4 [26-31]. The receptor most studied in HCC is EGFR/ErbB1. The EGF/EGFR pathway has been targeted mainly by two approaches; using either a single agent monoclonal antibody directed against EGFR (cetuximab) and a combination of cetuximab with cytotoxic chemotherapy or by small-molecule tyrosine kinase inhibitors [26-30] (Table 1).
2.2.1. Monoclonal Antibodies against EGFR
Cetuximab/Cetuximab plus Cytotoxic Chemotherapy Cetuximab is a chimeric monoclonal antibody against EGFR. In a phase II study of 32 patients with advanced HCC, cetuximab was given at 400 mg/m2 intravenously on day 1 followed by weekly intravenous infusions at 250 mg/m2. There were no responses seen and median TTP for all patients was 8.0 weeks [31]. In another phase II trial evaluated 30 patients with advanced HCC, again with no responses seen. Five patients had stable disease (median time, 4.2 months; range, 2.8 - 4.2 months) with an OS of 9.6 months and the median PFS of 1.4 months [32].
The combination of cetuximab with cytotoxic chemotherapy has been evaluated in Capecitabine plus Oxaliplatin (CAPOX) and Gemcitabine plus Oxaliplatin (GEMOX) regimens. In CAPOX/Cetuximab trial capecitabine was given at 850 mg/m2 twice daily for 14 days and oxaliplatin on day 1 at 130 mg/m2 intravenously, along with cetuximab at 400 mg/m2 on day 1 followed by 250 mg/m2 weekly on a 21-day cycle. RR was 10%, and TTP was 4.3 months in 20 evaluable patients. While most patients tolerated the treatment well, diarrhea and electrolyte abnormalities including hypomagnesemia and hypocalcemia were more pronounced [33]. In GEMOX/ cetuximab trial 45 patients were given cetuximab at an initial dose of 400 mg/m2 followed by 250 mg/m2 weekly, gemcitabine 1000 mg/m2 on day 1, and oxaliplatin at 100 mg/m2 on day 2, repeated every 14 days until disease progression or dose limiting toxicity. The RR was 20% with a disease stabilization rate of 40%. The median PFS and OS were 4.7 months and 9.5 months, respectively [34]. Due to the lack of activity of cetuximab as single agent and to reported antitumor activity of GEMOX and CAPOX in prior phase II studies in HCC, the relative contribution of cetuximab to these regimens remains.
2.2.2. Tyrosine Kinase Inhibitors against EGFR
2.2.2.1. Erlotinib In a phase II trial of 40 patients with unresectable HCC with no prior systemic therapy, erlotinib 150 mg orally per day was evaluated. There were no CR or PR; however, 17 of 40 patients achieved stable disease at 16 weeks of continuous therapy. The PFS at 16 weeks was 43%, and the median OS was 43 weeks (10.75 months). No patients required dose reductions of erlotinib. In this study no correlation between EGFR expression and outcome was found [35]. Combination of erlotinib with cytotoxic and biologics has also been studied. In a phase II trial combination of erlotinib with docetaxel in 14 patients with advanced HCC showed no objective responses but 6 patients (46%) had stable disease with a median duration of 17.6 weeks. The 16-week PFS was 38%, median OS was 6.7 months [36]. Another phase II trial evaluated erlotinib in combination with bevacizumab. Fourty patients who had advanced HCC that was not amenable to surgical or regional therapies, with up to one prior systemic treatment were given bevacizumab 10 mg/kg every 14 days and erlotinib 150 mg orally daily for 28-day cycles. The primary end point of PFS at 16 weeks was 62.5%. Ten patients achieved a PR for a confirmed overall RR of 25%. The median PFS was 39 weeks, and the median overall survival was 68 weeks. Grades 3 to 4 drug-related toxicity included fatigue 20%, hypertension 15%, diarrhea 10%, and elevated transaminases 10% [37].
2.2.2.2. Gefitinib Gefitinib is an oral selective inhibitor of EGFR that has shown growth inhibition in HCC cell lines. Up to 47% of HCCs express the epidermal growth factor receptor which predicts survival. Gefitinib was evaluated in a single arm phase II Eastern Cooperative Oncology Group (ECOG) study E1203, in advanced HCC. Daily oral Gefitinib 250 mg was given for 21-day cycles. Tumor assessments were performed every 6 weeks. Thirty-one patients were accrued with a median follow-up duration of 13.2 months. PFS was 2.8 months, OS 6.5 months. One PR and 7 stable disease were observed. There was only one grade 4 toxicity (neutropenia). The criterion for second-stage accrual was not met. Gefitinib as a single agent was not found to be active in advanced HCC [38].
2.2.2.3. Lapatinib Lapatinib is an inhibitor of epidermal growth factor receptor and HER2/NEU both implicated in hepatocarcinogenesis. In a phase II trial 25 patients with advanced unresectable HCC were given lapatinib, 1500 mg per day orally in 28-day cycles. Tumor and blood specimens were analyzed for expression of HER2/NEU and status of downstream signal pathway proteins. Twenty-six patients were enrolled. No objective responses were observed. Ten (40%) patients had stable disease as their best response including 6 (23%) with stable disease lasting more than 20 days. Median PFS was 1.9 months and median OS was 12.6 months. Patients who developed a rash had a borderline statistically significant longer survival. Tissue and blood specimens were available for more than 90% of patients. No somatic mutations in EGFR (exons 18 - 21) and no evidence of HER2/NEU somatic mutations were found. PTEN, P-AKT, and P70S6K expression did not correlate with survival. Lapatinib was well-tolerated but seemed to benefit only a subgroup of patients for whom predictive molecular or clinical characteristics are not yet fully defined [39].
2.3. Insulin-Like Growth Factor (IGFR) Pathway
Obesity and diabetes have been shown to be associated with an increased risk of cancer in humans [40,41]. IGF signaling system has attracted a lot of research attention in cancer. The essential role of IGF axis in HCC has been illustrated in cell lines and in animal xenograft models. Preclinical evidence provides ample indication that all four components of IGF axis are crucial in the carcinogenic and metastatic potential of HCC. Several strategies targeting this system including monoclonal antibodies against the IGF-1 receptor (IGF-1R) and small molecule inhibitors of the tyrosine kinase function of IGF-1R are under active investigation. Constitutive activation of the IGF-signaling axis has been well known in HCC. This pathway consists of circulating ligands, IGF-I and IGF-II, interacting with a membrane receptor, IGF-1R. The IGF- 1R is a heterotetramer consisting of two extracellular ligand-binding α subunits and two β subunits with transmembrane and tyrosine kinase domains. Upon ligand binding IGF-1R undergoes conformational changes and phosphorylation, leading to the recruitment of insulin-receptor substrates and/or Src homology-2 domaincontaining proteins, with the consequential activation of pathways common to EGFR, the PI3K/Akt/ mTOR-axis and the Ras/MEK/ERK-pathway [42,43]. The overexpression of IGF-II, IGF-1R, and IRS contributes to cell proliferation and the inhibition of apoptosis, as well as increased invasive behavior [44]. Overexpression of IGFII has been observed in 16% 40% of patients, and around 30% of HCC cases has been shown to overexpress IGF-1R [45,46]. Small molecules (such as OSI- 906, BMS-554417) and monoclonal antibodies (such as IMCA12 and AVE-1642) targeting IGF signaling are currently under evaluation in clinical trials of HCC patients (Table 1).
2.3.1. Small Molecule Inhibitors Targeting IGF-1R
2.3.1.1. OSI-906 OSI-906 (OSI) is a novel potent dual tyrosine kinase inhibitor of both IGF-1R and insulin receptor. The unique advantage of OSI-906 over previous class of anti-IGF drugs is its ability to minimize the activity of IGF-2 where IGF-1R inhibition alone will not be sufficient. In cancers such as adrenocortical carcinoma and HCC, where insulin receptor binds to IGF ligand with higher affinity, OSI-906 is able to inhibit both insulin receptor and IGF-1R to achieve maximum inhibition of the IGF axis [46-48]. A phase III study using OSI-906 in patients with adrenocortical carcinoma is currently ongoing. OSI-906 is considered one of the desirable drugs to be tested in patients with HCC. A randomized, placebocontrolled, double-blinded phase II study of second-line treatment involving patients with advanced HCC patients who failed first-line treatment with sorafenib (Clinical trial identifier: NCT01101906 ) was launched in 2011. The sponsor decided not to pursue the development of this drug and the study was terminated in December 2012.
2.3.1.2. BMS554417 Several small molecule tyrosine kinase inhibitors of IGF- 1R such as BMS554417 (Bristol-Myers-Squibb) are under development [49]. There have been encouraging in vitro and in vivo data in broad range of cancers with activated IGF axis. Current phase I data on drug tolerability will provide more information regarding the feasibility of such medications in the future potential treatment of advanced HCC.
2.3.2. Monoclonal Antibodies Targeting IGFR-1R
2.3.2.1. IMC-A12 Pre-clinical evidence obtained in HCC cells showed that IMC-A12, a human monoclonal antibody that blocks IGF-1R, both in vitro and in vivo decreases cell viability and proliferation and blocks ligand-induced IGF-1R activation. In vivo IMC-A12 delayed tumor growth and prolonged survival, reducing proliferation rates and inducing apoptosis [50]. IMC-A12 effectively blocked IGF signaling, providing the rationale for testing this therapy in clinical trials. A phase I study of IMC-A12 (cituxumumab) yielded a partial response in HCC [51]; however, a subsequent phase II study in patients with advanced HCC showed that IMC-A12 is inactive as monotherapy [52].
2.3.2.2. AVE1642 AVE1642 is a humanized monoclonal antibody that specifically blocks IGF-1R signaling. A phase I study showed that AVE1642 can be safely combined with active doses of sorafenib, and the pharmacokinetics of both AVE1642 and sorafenib were not modified at the concentrations tested. Interestingly, long-lasting disease stabilizations were observed in most patients with progressive disease [53]). Although IMC-A12 lacks single agent activity in HCC, its combination with sorafenib could potentially yield synergy. It is currently undergoing phase I study in combination with sorafenib in patients with HCC. The result of this clinical trial may help understand the clinical benefits of combining IGFR-1R monoclonal antibodies and sorafenib in this disease.
2.3.2.3. BIIB022 BIIB022 showed inhibition of tumor growth in HCC cell line HepG2, and this inhibitory effect was enhanced by addition of sorafenib [54]. A planned phase I/II study comparing sorafenib with or without BIIB022 in patients with advanced HCC was terminated due to a business decision of the sponsor company.
2.4. Ras/Raf/MEK/ERK Pathways
Also known as, the mitogen-activated protein kinase (MAPK) pathway, Ras/Raf/MEK/ERK pathway, consists of a kinase cascade that is regulated by phosphorylation and de-phosphorylation by specific kinases, phosphatases, and GTP/GDP exchange proteins. Ras is activated by cellular stimuli to a GTP-bound state, leading to the recruitment of Raf from the cytosol into the cell membrane where it becomes activated [55]. Activated Raf leads to the activation and phosphorylation of MEK1 and MEK2, which then activates ERK1 and ERK2. When ERK is activated, it translocates into the nucleus and phosphorylates transcription factors that can promote cellular growth and prevent apoptosis [56]. Dysregulation of the Ras/Raf/MEK/ERK pathway is positively associated with HCC. A recent study showed that 100% of HCC specimens analyzed contained Ras pathway activation [57]. Because of the cross talk at the cellular level, upregulation of IGF [58], aberrant upstream EGFR signaling and other receptor signaling (i.e. VEGFR and PDGFR) may cause activation of Ras/Raf/MEK/ERK signaling in HCC [59]. Thus, an effective blockade of the Ras/Raf/ MEK/ERK pathway can be achieved using small molecules, such as sorafenib, selumetinib (AZD6244), and regorafenib (Table 1).
2.4.1. Sorafenib
Sorafenib, is a multikinase inhibitor, which in addition to targeting Raf kinases also inhibits VEGFR-2/-3, PDGFR- β, Flt-3 and c-Kit. Based on the findings of the pivotal SHARP trial sorafenib has been approved by the United States Food and Drug Administration for the treatment of patients with advanced HCC [60]. Sorafenib increased OS from 7.9 months in the placebo group to 10.7 months in the treatment group. Time to progression was 5.5 months in the sorafenib group and 2.8 months in the placebo group.
2.4.2. Selumetinib (AZD6244)
Selumetinib is an oral non-ATP-competitive small-molecule inhibitor of the mitogen-activated protein kinase MEK1/2. The recommended dose found in early dose escalation studies were established as 100 mg twice per day [61]. In an open-label, phase II trial, researchers evaluated the efficacy of selumetinib in patients with confirmed advanced or metastatic HCC. Patients were given 100 mg of selumetinib twice per day, fasting for a minimum of 2 hours before their dose. A cycle of therapy was 21 days. PK samples were taken 48 hours after initiating treatment for monitoring. Of the 19 patients enrolled, 2 failed to complete a full cycle of therapy due to elevated transaminases and voluntary withdrawal. Among the 17 remaining patients, there was no PR or CR. The study was terminated early due to a lack of radiographic response and short PFS, reflecting the lack of adequate antitumor activity of selumetinib as monotherapy [62].
2.4.3. Regorafenib
Regorafenib is an oral multikinase inhibitor of MAPK, Raf, FGFR, RET, KIT, VEGFR, and PDGFR. The safety and tolerability of regorafenib was evaluated in 36 patients with hepatocellular carcinoma (HCC) who have progressed on first-line sorafenib. Treatment consisted of regorafenib 160 mg once daily on a 3-weeks-on/ 1-weekoff schedule. The dosing regimen was based on a European dose-finding study. The median duration of treatment was 15.5 weeks. Common treatment-related adverse events included hand-foot skin reaction in 50% of patients, diarrhea in 50%, fatigue in 47%, and hypothyroidism. The median time to disease progression was 4.1 months, and nearly three-quarters of patients achieved disease control. Disease progression led to treatment withdrawal in 6 patients (17%), and 15 of the 36 patients remained on treatment at the data cut-off. The mechanism by which regorafenib might overcome resistance to sorafenib needs to be examined in future studies [63].
2.5. PI3K/Akt/mTOR Pathways
The PI3K/Akt/mTOR pathway is crucial in carcinogenesis [64]. This pathway is activated when growth factors bind to membrane receptors, such as EGFR and IGF-R1, which engage and activate PI3K. When PI3K is activated, a cascade of downstream effectors such as Akt and mTOR are produced. Once activated, Akt leaves the cell membrane to phosphorylate intracellular substrates. Phosphorylation can lead to the promotion of cell survival as well as positively modulating mTOR function. The involvement of Akt in HCC is still being evaluated; however, early observations have found tumors with Akt41 expression have worse prognosis [65]. As many as 50% of patients with HCC, has a dysfunction in the components of the PI3K/Akt/mTOR pathway [66]. Acting as the primary regulator of cell growth, survival, and proliferation, mTOR is an important mediator of the PI3K-Akt pathway. Other downstream effectors of mTOR, including 4E-BP1 and S6K1, have been associated with higher tumor severity [67]. Negative regulation of PI3K is predominantly accomplished by the PTEN tumor suppressor protein. PTEN dephosphorylates, and thus inhibits the PI3K/Akt pathway. Mutations to the PTEN protein can lead to improper regulation and promotion of PI3K/ Akt/mTOR signaling [68]. Although some preclinical studies have demonstrated that PI3K inhibitors such as perifosine, LY29004 and wortmannin have anti-HCC activity, no studies have been conducted so far at the clinical level. However several novel agents are being developed to target different components of the PI3K/ AKt/mTOR pathway in HCC (Table 1).
2.5.1. MK2206
MK2206 is a novel selective non-ATP-competitive allosteric inhibitor of Akt and is being investigated in sorafenib resistant HCC. In a phase II study, data showed that combination treatment of sorafenib and MK-2206 overcame the resistance to sorafenib resistant HCC and increased the percentage of apoptotic cells. MK2206 also down-regulated Akt and induced cell death. Experimenters found that using MK2206 alone did not show significant effects on apoptosis in sorafenib resistant HCC cells, determining co-administration of sorafenib and MK2206 to be the most effective and novel strategy in promoting apoptosis [69]. A phase II study of MK2206 (a novel oral potent allosteric Akt inhibitor) in advanced HCC patients who have not responded or are intolerant to one previous line of anti-angiogenic therapy is currently recruiting patients (Clinical trial identifier: NCT01239355).
2.5.2. AZD8055
AZD8055 is a novel ATP-inhibitor of mTOR kinase undergoing Phase I/II studies in Asia in patients with HCC. Early dose finding studies in Japan concluded that AZD8055 at 90 mg twice a day to be the MTD. Researchers also found mean Akt levels to be reduced in the treatment cohorts [70]. Further studies are needed to determine the role of this novel agent.
2.5.3. Everolimus
Everolimus is a macrolide immunosuppressant and mTOR inhibitor with antiproliferative and anti-angiogenic properties. Its current place in therapy is for treatment of breast and kidney cancer, neuroendocrine tumors, and certain lymphomas. Its benefit for HCC is currently being evaluated. In a phase I/II study, in patients with histologically confirmed, measurable, and advanced HCC received everolimus orally at 5 mg/day or 10 mg/day. The phase I endpoint was to determine a safe dosage regimen, while the phase II endpoint meant to assess PFS at 24 weeks. Results from thus study found everolimus dosed at 10 mg/day was well tolerated and used this dose for the phase II stage of the study. Although the study did not proceed to the second stage, preliminary observations found antitumor activity with everolimus in patients with advanced HCC, most of whom had tried other prior systemic treatments [71].
2.5.4. Sirolimus
Sirolimus has been used as an immunosuppressant agent and works by inhibiting T-lymphocyte activation and proliferation in response to antigenic and cytokine stimulation and inhibits antibody production. Differing from other immunosuppressants, sirolimus binds to the intracellular protein, FKBP-12, to form an immunosuppressive complex which inhibits mTOR. A phase II study had been performed to examine the efficacy of sirolimus as an antitumor agent in HCC. In this study, patients with advanced HCC and underlying cirrhosis received 20 mg/week of sirolimus for 1 month and were then increased to 30 mg/week thereafter. Tumor response was assessed every 8 weeks as the primary endpoint. In this small study, 25 patients received sirolimus for a median of 20.6 weeks. Two patients had an objective response including one CR and 8 patients had stable disease. Grade 5 toxicities were observed in 2 patients (infections) and grade 3 toxicities were seen in 5 patients. Median time to radiological progression and OS were 15.3 weeks and 26.4 weeks respectively. The study concluded sirolimus did show some antitumor activity in patients with advanced HCC; however, larger studies are needed to determine its use [72]. In a separate phase II study, the effects of sirolimus were investigated in 21 patients with advanced HCC. Sirolimus was dosed daily by mouth and dosages were adjusted to a serum trough level between 4 and 15 µg/ml. One patient had partial remission, and 5 patients had stable disease at 3 months [73].
2.5.5. Temsirolimus
Temsirolimus, an mTOR inhibitor is approved for treatment of advanced renal cell carcinoma. Its efficacy and potential utility for HCC is currently being studied. In vitro studies showed that effective inhibition of mTOR signaling with temsirolimus alone was able to suppress HCC cell growth in a dose-dependent manner. Different cell lines were tested with varying temsirolimus resistance. In cells that had high temsirolimus resistance, investigators co-targeted mTOR with temsirolimus and vinblastine resulting in growth inhibition; demonstrating potent antitumor activity of this novel combination. In vivo studies showed that temsirolimus treatment alone for one week was able to inhibit the growth of susceptible xenografts. The temsirolimus/vinblastine combination induced a significant and sustained antitumor activity (up to 27 days post-treatment), with effective reduction of tumor vessel density in sensitive and resistant models [74]. These early findings may provide additional treatment options for inhibition of the mTOR kinase. Further studies are needed to fully understand its place in therapy.
2.6. Wnt-Beta-Catenin Pathway
Wnt-β-catenin pathway controls biochemical processes related to cell growth and differentiation. Alterations of this pathway have been associated with tumors such as HCC or hepatoblastoma. In normal cell homeostasis, Wnt proteins are absent allowing β-catenin, the key player in the Wnt pathway, to be phosphorylated and targeted for ubiquination and protein degradation by the proteosome. When Wnt signaling events are initiated, a series of events leads to the absence of β-catenin phosphorylation releasing it from degradation. β-catenin then accumulates in the cytoplasm and translocates into the nucleus where it binds to lymphoid enhancer factor (LEF) or T-cell factor (TCF) displacing the transcriptional inhibitor Groucho, and in complex with LEF/TCF activates the expression of different genes which regulate cell proliferation and apoptosis. Hepatocytes with nuclear translocation of β-catenin have abnormal cellular proliferation, and expresses membrane proteins involved in HCC, metastatic behavior, and cancer stem cells [75].
Activating mutations in β-catenin (CTNNB1) were found in about 20% - 40% of human HCC lines [76]. Also, HCC occurring in patients with HCV were shown to have high incidence of β-catenin mutations, at nearly 40%. HCC occurring in HBV patients showed β-catenin activation induced in a mutation-dependent manner by the expression of HBx protein [77,78]. Future experiments are needed to understand the role β-catenin in HCC treatment using molecular targeting agents (Table 1).
PKF118-310, PKF115-584, CGP049090
Pharmacologic agents targeting Wnt-β-catenin are being developed. Currently researchers are evaluating the activity of small molecules that antagonize the TCF/β-catenin complex on HCC cell lines in vitro and in vivo. Researchers discovered that after injecting mice with β- catenin expressing HCC tumors, the small molecule antagonists of the TCF/β-catenin complex exhibited antitumor activity by inducing apoptosis in the G1/S phase and reduced the expression of downstream target gene cyclin D1. The small molecules also led to dose-dependent reductions in the expression of other TCF/β-catenin downstream oncoproteins such as c-Myc and survivin [79]. Preliminary studies targeting the Wnt-β-catenin pathway demonstrates a potential place for new novel therapies to treat HCC.
2.7. Hedgehog Pathway
The role of the hedgehog pathway in human cancers has been established through studies of basal cell nevus syndrome, prostate cancer and gastrointestinal cancers. The hedgehog pathway is essential for embryonic development, tissue polarity and cell differentiation. The hedgehog signaling pathway has emerged as a critical mediator in the development of various diseases, including cancer, when aberrantly activated [80]. Some early studies have shown activation of the Hedgehog pathway in liver carcinogenesis suggesting a potential therapeutic target [81, 82]. Furthermore, it has been shown that this pathway can cross-talk with the Wnt-β-catenin signaling pathway, a well-known oncogenic pathway implicated in HCC development [83,84]. Currently no hedgehog inhibitor drugs in clinical trials exist for HCC; however, a recent preclinical study has revealed profound tumor growth and metastasis inhibitory effect of a polymeric nanoparticle-encapsulated hedgehog pathway inhibitor, HPI-1 (NanoHHI), in an orthotopic model of human HCC [85]. (Table 1)
2.8. Other Pathways and Therapeutic Targets
Various other pathways in the pathogenesis of HCC are being discovered. Some of these pathways and targeted therapies for them are already making their way into clinical trials. Transforming growth factor (TGF)-β [86], hepatocyte growth factor (HGF)/c-MET [87,88], fibroblast growth factor (FGF), Hyppo [89], and Notch [90,91] signaling patways are just a few to mention. Clinical data targeting at least two of these pathways, HGF/c-MET and FGF, are available (Table 1).
2.8.1. Tivantinib
Tivantinib is a selective oral tyrosine kinase inhibitor of the hepatocyte growth factor receptor known as c-MET. In a Phase II multicenter clinical trial of 107 patients with unresectable HCC who either experienced disease progression after first-line therapy or were unable to tolerate first-line treatment, patients were randomly assigned (2:1) to receive tivantinib 360 mg or 240 mg twice-daily, or placebo; the initial 360 mg dose was lowered to 240 mg after the first set of patients developed low white blood cell count. In an intent-to-treat analysis, the median TTP was 1.7 months for the tivantinib arm compared with 1.5 months for the placebo arm. Benefits were greater for the subset of participants with high c-MET expression with TTP of 2.7 months with the tivantinib arm versus 1.4 months in placebo arm. Overall survival durations were 7.2 versus 3.8 months, respectively. Adverse events included fatigue, weakness, loss of appetite, and blood cell deficiencies [92].
2.8.2. Lenalidomide
Lenalidomide inhibits fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF). FGF has recently been reported to be an important growth factor in HCC. Patients with advanced HCC who progressed or were intolerant to sorafenib were eligible. Forty patients were enrolled. They were treated with lenalidomide 25 mg orally days 1 - 21 of a 28 day cycle until disease progression or unacceptable toxicity. Thirty-two patients experienced elevated baseline alpha-feto protein (AFP), nine had more than 50% reduction in tumor size (28%). Six of the first 37 patients (16%) had a radiographic PR while two patients achieved a CR and have not progressed at 36 and 32 months, respectively. One patient had grade 4 neutropenia. Grade 3 toxicities included fatigue, rash, arthritis, and low blood counts [93].
3. Conclusion
Despite extensive efforts by many investigators, systemic therapy with many different classes of agents for HCC has been ineffective. Since the emergence of sorafenib as the new standard for the systemic treatment of advanced HCC, extensive research efforts have focused on several key molecular pathways implicated in the pathogenesis of HCC. These efforts are leading to a revolutionary change and progress in the treatment of this devastating disease. Several promising novel anticancer agents are currently under investigation. While many molecularly targeted agents are at different stages of clinical development, a strategy of combining these different targeted agents should be considered as a key approach for improving the effectiveness and usefulness of these agents. Future research will continue to unravel the mechanism of hepatocarcinogenesis and identify key relevant molecular targets for therapeutic intervention. During this process, focus on identifying and validating surrogate and predictive biomarkers to predict clinical efficacy, toxicity, and resistance to these agents would be indispensable. The field of molecularly targeted therapy in cancer has already come a long way and HCC model will continue to inspire many researchers for the development of novel therapies for other types of cancers.
REFERENCES
- F. X. Bosch, J. Ribes, R. Cleries and M. Diaz, “Epidemiology of Hepatocellular Carcinoma,” Clinical Liver Disease, Vol. 9, No. 2, 2005, pp. 191-211. doi:10.1016/j.cld.2004.12.009
- P. Pisani, F. Bray and D. M. Parkin, “Estimates of the World-Wide Prevalence of Cancer for 25 Sites in the Adult Population,” International Journal of Cancer, Vol. 97, No. 1, 2002, pp. 72-81. doi:10.1002/ijc.1571
- H. B. El-Serag, “Epidemiology of Hepatocellular Carcinoma in USA,” Hepatology Research, Vol. 37, Suppl. 2, 2007, pp. S88-S94. doi:10.1111/j.1872-034X.2007.00168.x
- Z. F. Yang and R. T. Poon, “Vascular Changes in Hepatocellular Carcinoma,” The Anatomical Record, Vol. 291, No. 6, 2008, pp. 721-734. doi:10.1002/ar.20668
- W. S. Moon, K. H. Rhyu, M. J. Kang, D. G. Lee, H. C. Yu, J. H. Yeum, et al., “Overexpression of VEGF and Angiopoietin 2: A Key to High Vascularity of Hepatocellular Carcinoma?” Modern Pathology, Vol. 16, No. 6, 2003, pp. 552-557. doi:10.1097/01.MP.0000071841.17900.69
- H. Yoshiji, S. Kuriyama, J. Yoshii, M. Yamazaki, M. Kikukawa, H. Tsujinoue, et al., “Vascular Endothelial Growth Factor Tightly Regulates in Vivo Development of Murine Hepatocellular Carcinoma Cells,” Hepatology, Vol. 28, No. 6, 1998, pp. 1489-1496. doi:10.1002/hep.510280607
- Y. Chao, C. P. Li, G. Y. Chau, et al., “Prognostic Significance of Vascular Endothelial Growth Factor, Basic Fibroblast Growth Factor, and Angiogenin in Patients with Resectable Hepatocellular Carcinoma after Surgery,” Annals of Surgical Oncology, Vol. 10, No. 4, 2003, pp. 355-362. doi:10.1245/ASO.2003.10.002
- K. S. Jeng, I. S. Sheen, Y. C. Wang, et al., “Prognostic Significance of Preoperative Circulating Vascular Endothelial Growth Factor Messenger RNA Expression in Resectable Hepatocellular Carcinoma: A Prospective Study,” World Journal of Gastroenterology, Vol. 10, No. 5, 2004, pp. 643-648.
- R. T. Poon, J. W. Ho, C. S. Tong, et al., “Prognostic Significance of Serum Vascular Endothelial Growth Factor and Endostatin in Patients with Hepatocellular Carcinoma,” British Journal of Surgery, Vol. 91, No. 10, 2004, pp. 1354-1360. doi:10.1002/bjs.4594
- J. M. Llovet, S. Ricci, V. Mazzaferro, et al., “Sorafenib in Advanced Hepatocellular Carcinoma,” The New England Journal of Medicine, Vol. 359, 2008, pp. 378-390. doi:10.1056/NEJMoa0708857
- A. B. Siegel, E. I. Cohen, A. Ocean, D. Lehrer, A. Goldenberg, J. J. Knox, et al., “Phase II Trial Evaluating the Clinical and Biologic Effects of Bevacizumab in Unresectable Hepatocellular Carcinoma,” Journal of Clinical Oncology, Vol. 26, No. 18, 2008, pp. 2992-2998. doi:10.1200/JCO.2007.15.9947
- A. X. Zhu, L. S. Blaszkowsky, D. P. Ryan, J. W. Clark, A. Muzikansky, K. Horgan, et al., “Phase II Study of Gemcitabine and Oxaliplatin in Combination with Bevacizumab in Patients with Advanced Hepatocellular Carcinoma,” Journal of Clinical Oncology, Vol. 24, No. 12, 2006, pp. 1898-1903. doi:10.1200/JCO.2005.04.9130
- J. M. Wood, “Inhibition of vascular Endothelial Growth Factor (VEGF) as a Novel Approach for Cancer Therapy,” Medicina (B Aires), Vol. 60, Suppl. 2, 2000, pp. 41- 47.
- J. Drevs, R. Muller-Driver, C. Wittig, et al., “PTK787/ZK 222584, a Specific Vascular Endothelial Growth Factor-Receptor Tyrosine Kinase Inhibitor, Affects the Anatomy of the Tumor Vascular Bed and the Functional Vascular Properties as Detected by Dynamic Enhanced Magnetic Resonance Imaging,” Cancer Research, Vol. 62, No. 14, 2002, pp. 4015-4022.
- I. Koch, A. Baron, S. Roberts, et al., “Influence of Hepatic Dysfunction on Safety, Tolerability, and Pharmacokinetics (PK) of PTK787/ZK 222584 in Patients (pts) with Unresectable Hepatocellular Carcinoma (HCC),” Journal of Clinical Oncology, Vol. 23, Suppl. 4134, 2007.
- S. R. Wedge, J. Kendrew, L. F. Hennequin, P. J. Valentine, S. T. Barry, S. R. Brave, N. R. Smith, N. H. James, M. Dukes, J. O. Curwen, et al., “AZD2171: A Highly Potent, Orally Bioavailable, Vascular Endothelial Growth Factor Receptor-2 Tyrosine Kinase Inhibitor for the Treatment of Cancer,” Cancer Research, Vol. 65, No. 10, 2005, pp. 4389-4400. doi:10.1158/0008-5472.CAN-04-4409
- J. Drevs, P. Siegert, M. Medinger, K. Mross, R. Strecker, U. Zirrgiebel, J. Harder, H. Blum, J. Robertson, J. M. Jurgensmeier, et al., “Phase I Clinical Study of AZD2171, an Oral Vascular Endothelial Growth Factor Signaling Inhibitor, in Patients with Advanced Solid Tumors,” Journal of Clinical Oncology, Vol. 25, No. 21, 2007, pp. 3045-3054. doi:10.1200/JCO.2006.07.2066
- S. R. Alberts, T. R. Fitch, G. P. Kim, B. W. Morlan, S. R. Dakhil, H. M. Gross and S. Nair, “Cediranib (AZD2171) in Patients with Advanced Hepatocellular Carcinoma: A Phase II North Central Cancer Treatment Group Clinical Trial,” American Journal of Clinical Oncology, Vol. 35, No. 4, 2012, pp. 329-333. doi:10.1097/COC.0b013e3182118cdf
- J. L. Raoul, R. S. Finn, Y. K. Kang, et al., “An OpenLabel Phase II Study of Firstand Second-Line Treatment with Brivanib in Patients with Hepatocellular Carcinoma (HCC),” Journal of Clinical Oncology, Vol. 27, Suppl. 4577, 2009.
- D. B. Mendel, A. D. Laird, X. Xin, et al., “In Vivo Antitumor Activity of SU11248, a Novel Tyrosine Kinase Inhibitor Targeting Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor Receptors: Determination of a pharmacokinetic/Pharmacodynamic Relationship,” Clinical Cancer Research, Vol. 9, No. 1, 2003, pp. 327- 337.
- A. X. Zhu, D. V. Sahani, D. G. Duda, et al., “Efficacy, Safety, and Potential Biomarkers of Sunitinib Monotherapy in Advanced Hepatocellular Carcinoma: A Phase II Study,” Journal of Clinical Oncology, Vol. 27, No. 18, 2009, pp. 3027-3035. doi:10.1200/JCO.2008.20.9908
- S. Faivre, E. Raymond, E. Boucher, et al., “Safety and Efficacy of Sunitinib in Patients with Advanced Hepatocellular Carcinoma: An Open-Label, Multicentre, Phase II Study,” The Lancet Oncology, Vol. 10, No. 8, 2009, pp. 794-800. doi:10.1016/S1470-2045(09)70171-8
- T. Yau, P. J. Chen, P. Chan, et al., “Phase I Dose-Finding Study of Pazopanib in Hepatocellular Carcinoma: Evaluation of Early Efficacy, Pharmacokinetics, and Pharmacodynamics,” Clinical Cancer Research, Vol. 17, No. 21, 2011, pp. 6914-6923. doi:10.1158/1078-0432.CCR-11-0793
- C. C. Yau, P. J. Chen, C. M. Curtis, et al., “A Phase I Study of Pazopanib in Patients with Advanced Hepatocellular Carcinoma,” Journal of Clinical Oncology, Vol. 27, Suppl. 3561, 2009 (Abstr 3561).
- H. Toh, P. Chen, I. Carr, et al., “Linifanib Phase II Trial in Patients with Advanced Hepatocellular Carcinoma (HCC),” Journal of Clinical Oncology, Vol. 28, Suppl. 15s, 2010 (Abstr 4038).
- C. R. Carlin, D. Simon, J. Mattison and B. B. Knowles, “Expression and Biosynthetic Variation of the Epidermal Growth Factor Receptor in Human Hepatocellular Carcinoma-Derived Cell Lines,” Molecular and Cellular Biology, Vol. 8, No. 1, 1998, pp. 25-34.
- K. Harada, G. Shiota and H. Kawasaki, “Transforming Growth Factor Alpha and Epidermal Growth Factor Receptor in Chronic Liver Disease and Hepatocellular Carcinoma,” Liver, Vol. 19, No. 4, 1999, pp. 318-325. doi:10.1111/j.1478-3231.1999.tb00056.x
- Y. Ito, T. Takeda, S. Higashiyama, et al., “Expression of Heparin Binding Epidermal Growth Factor-Like Growth Factor in Hepatocellular Carcinoma: An Immunohistochemical Study,” Oncology Reports, Vol. 8, No. 1, 2001, pp. 903-907.
- S. Kira, T. Nakanishi, S. Suemori, et al., “Expression of Transforming Growth Factor Alpha and Epidermal Growth Factor Receptor in Human Hepatocellular Carcinoma,” Liver, Vol. 17, No. 4, 1997, pp. 177-182. doi:10.1111/j.1600-0676.1997.tb00803.x
- A. Kiss, N. J. Wang, J. P. Xie and S. S. Thorgeirsson, “Analysis of Transforming Growth Factor (TGF)-Alpha/ Epidermal Growth Factor Receptor, Hepatocyte Growth Factor/c-Met, TGF-Beta Receptor Type II, and p53 Expression in Human Hepatocellular Carcinomas,” Clinical Cancer Research, Vol. 3, No. 7, 1997, pp. 1059-1066.
- V. Gruenwald, L. Wilkens, M. Gebel, et al., “A Phase II Open-Label Study of Cetuximab in Unresectable Hepatocellular Carcinoma: Final Results,” Journal of Clinical Oncology, Vol. 25, No. 18, 2007.
- A. X. Zhu, K. Stuart, L. S. Blaszkowsky, et al., “Phase 2 Study of Cetuximab in Patients with Advanced Hepatocellular Carcinoma,” Cancer, Vol. 110, No. 3, 2007, pp. 581-589. doi:10.1002/cncr.22829
- B. H. O’Neil, S. A. Bernard, R. M. Goldberg, et al., “Phase II Study of Oxaliplatin, Capecitabine, and Cetuximab in Advanced Hepatocellular Carcinoma,” Journal of Clinical Oncology, Vol. 26, Suppl. 4604, 2008 (Abstr 4604).
- S. Louafi, V. Boige, M. Ducreux, et al., “Gemcitabine plus Oxaliplatin (GEMOX) in Patients with Advanced Hepatocellular Carcinoma (HCC): Results of a Phase II Study,” Cancer, Vol. 109, 2007, pp. 1384-1390. doi:10.1002/cncr.22532
- M. B. Thomas, R. Chadha, K. Glover, et al., “Phase 2 Study of Erlotinib in Patients with Unresectable Hepatocellular Carcinoma,” Cancer, Vol. 110, No. 5, 2007, pp. 1059-1067. doi:10.1002/cncr.22886
- E. G. Chiorean, R. Ramasubbaiah, M. Yu, et al., “Phase II Trial of Erlotinib and Docetaxel in Advanced and Refractory Hepatocellular and Biliary Cancers: Hoosier Oncology Group GI06-101,” Oncologist, Vol. 17, No. 1, 2011, p. 13.
- M. B. Thomas, J. S. Morris and R. Chadha, “Phase II Trial of the Combination of Bevacizumab and Erlotinib in Patients Who Have Advanced Hepatocellular Carcinoma,” Journal of Clinical Oncology, Vol. 27, No. 6, 2009, pp. 843-850.
- P. J. O’Dwyer, B. J. Giantonio, D. E. Levy, et al., “Gefitinib in Advanced Unresectable Hepatocellular Carcinoma: Results from the Eastern Cooperative Oncology Group’s Study E1203,” Journal of Clinical Oncology, Vol. 24, No. 18S, 2006, p. 4143.
- T. Bekaii-Saab, J. Markowitz, N. Prescott, et al., “A Multi-Institutional Phase II Study of the Efficacy and Tolerability of Lapatinib in Patients with Advanced Hepatocellular Carcinomas,” Clinical Cancer Research, Vol. 15, No. 18, 2009, pp. 5895-5901.
- J. Bruix and J. M. Llovet, “Major Achievements in Hepatocellular Carcinoma,” Lancet, Vol. 373, No. 9664, 2009, pp. 614-616. doi:10.1016/S0140-6736(09)60381-0
- E. E. Calle, C. Rodriguez, K. Walker-Thurmond and M. J. Thun, “Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of US Adults,” The New England Journal of Medicine, Vol. 348, 2003, pp. 1625-1638. doi:10.1056/NEJMoa021423
- J. G. Scharf and T. Braulke, “The Role of the IGF Axis in Hepatocarcinogenesis,” Hormone and Metabolic Research, Vol. 35, No. 11-12, 2003, pp. 685-693. doi:10.1055/s-2004-814151
- T. Nussbaum, J. Samarin, V. Ehemann, M. Bissinger, E. Ryschich, A. Khamidjanov, X. Yu, N. Gretz, P. Schirmacher and K. Breuhahn, “Autocrine Insulin-Like Growth Factor-II Stimulation of Tumor Cell Migration Is a Progression Step in human Hepatocarcinogenesis,” Hepatology, Vol. 48, No. 1, 2008, pp. 146-156. doi:10.1002/hep.22297
- K. Breuhahn and P. Schirmacher, “Reactivation of the Insulin-Like Growth Factor-II Signaling Pathway in Human Hepatocellular Carcinoma,” World Journal of Gastroenterology, Vol. 14, No. 11, 2008, pp. 1690-1698. doi:10.3748/wjg.14.1690
- E. Cariani, D. Seurin, C. Lasserre, D. Franco, M. Binoux and C. Brechot, “Expression of Insulin-Like Growth Factor II (IGF-II) in Human Primary Liver Cancer: mRNA and Protein Analysis,” Journal of Hepatology, Vol. 11, No. 2, 1990, pp. 226-231. doi:10.1016/0168-8278(90)90118-B
- K. Breuhahn, S. Vreden, R. Haddad, S. Beckebaum, D. Stippel, P. Flemming, T. Nussbaum, W. H. Caselmann, B. B. Haab and P. Schirmacher, “Molecular Profiling of Human Hepatocellular Carcinoma Defines Mutually Exclusive Interferon Regulation and Insulin-Like Growth Factor II Overexpression,” Cancer Research, Vol. 64, No. 17, 2004, pp. 6058-6064. doi:10.1158/0008-5472.CAN-04-0292
- F. M. Barlaskar and G. D. Hammer, “The Molecular Genetics of Adrenocortical Carcinoma,” Reviews in Endocrine & Metabolic Disorders, Vol. 8, No. 4, 2007, pp. 343-348. doi:10.1007/s11154-007-9057-x
- Z. Wang, Y. B. Ruan, Y. Guan and S. H. Liu, “Expression of IGF-II in Early Experimental Hepatocellular Carcinomas and Its Significance in Early Diagnosis,” World Journal of Gastroenterology, Vol. 9, No. 2, 2003, pp. 267- 270.
- P. Haluska, J. M. Carboni, D. A. Loegering, et al., “In Vitro and in Vivo Antitumor Effects of the Dual Insulin-Like Growth Factor-I/Insulin Receptor Inhibitor, BMS- 554417,” Cancer Research, Vol. 66, No. 1, 2003, pp. 362-371. doi:10.1158/0008-5472.CAN-05-1107
- V. Tovar, C. Alsinet, A. Villanueva, et al., “IGF Activation in a Molecular Subclass of Hepatocellular Carcinoma and Pre-Clinical Efficacy of IGF-1R Blockage,” Journal of Hepatology, Vol. 52, No. 4, 2010, pp. 550-559. doi:10.1016/j.jhep.2010.01.015
- C. S. Higano, E. Y. Yu, S. H. Whiting, et al., “A Phase I, First in Man Study of Weekly IMC-A12, a Fully Human Insulin Like Growth Factor-I Receptor IgG1 Monoclonal Antibody, in Patients with Advanced Solid Tumors,” Journal of Clinical Oncology, Vol. 25, No. 18S, 2007, p. 3505.
- G. K. Abou-Alfa, B. Gansukh, J. F. Chou, et al., “Phase II Study of Cixutumumab (IMC-A12, NSC742460; C) in Hepatocellular Carcinoma (HCC),” Journal of Clinical Oncology, Vol. 29, 2011 (Abstract 4043).
- S. J. Faivre, L. Fartoux, M. Bouattour, F. et al., “A Phase I Study of AVE1642, a Human Monoclonal AntibodyBlocking Insulin-Like Growth Factor-1 Receptor (IGF- 1R), Given as a Single Agent and in Combination with Sorafenib as First-Line Therapy in Patients with Advanced Hepatocellular Carcinoma (HCC),” Journal of Clinical Oncology, Vol. 29, Suppl. 4, 2011 (Abstr 270).
- Biogen-Idec Inc., “Investigator’ Brochure of BIIB-022,” 2010.
- J. A. McCubrey, L. S. Steelman, S. L. Abrams, W. H. Chappell, S. Russo, R. Ove, M. Milella, A. Tafuri, P. Lunghi, A. Bonati, F. Stivala, F. Nicoletti, M. Libra, A. M. Martelli, G. Montalto and M. Cervello, “Emerging MEK Inhibitors,” Expert Opinion on Emerging Drugs, Vol. 15, No. 2, 2010, 203-223, pp. 633-648.
- J. Xing, D. D. Ginty and M. E. Greenberg, “Coupling of the RAS-MAPK Pathway to Gene Activation by RSK2, a Growth Factor-Regulated CREB Kinase,” Science, Vol. 273, No. 5277, 1996, pp. 959-963. doi:10.1126/science.273.5277.959
- D. F. Calvisi, S. Ladu, A. Gordon, M. Farina, E. A. Conner, J. S. Lee, V. M. Factor and S. S. Thorgeirsson, “Ubiquitous Activation of Ras and Jak/Stat Pathways in Human HCC,” Gastroenerology, Vol. 130, No. 4, 2006, pp. 1117-1128. doi:10.1053/j.gastro.2006.01.006
- H. Huynh, P. K. Chow, L. L. Ooi and K. C. Soo, “A Possible Role for Insulin-Like Growth Factor-Binding Protein-3 autocrine/Paracrine Loops in Controlling Hepatocellular Carcinoma Cell Proliferation,” Cell Growth Differ, Vol. 13, No. 3, 2002, pp. 115-122.
- R. Kannangai, F. Sahin and M. S. Torbenson, “EGFR Is Phosphorylated at Ty845 in Hepatocellular Carcinoma,” Modern Pathology, Vol. 19, No. 11, 2006, pp. 1456- 1461.
- [61] J. M. Llovet, S. Ricci, V. Mazzaferro, P. Hilgard, E. Gane, J. F. Blanc, A. C. de Oliveira, A. Santoro, J. L. Raoul, A. Forner, M. Schwartz, C. Porta, S. Zeuzem, L. Bolondi, T. F. Greten, P. R. Galle, et al., “SHARP Investigators Study Group. Sorafenib in Advanced Hepatocellular Carcinoma,” The New England Journal of Medicine, Vol. 359, 2008, pp. 378-390. doi:10.1056/NEJMoa0708857
- [62] B. R. Davies, A. Logie, J. S. McKay, et al., “AZD6244 (ARRY-142886), a Potent Inhibitor of Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Kinase 1/2 Kinases: Mechanism of Action in Vivo, Pharmacokinetic/Pharmacodynamic Relationship, and Potential for Combination in Preclinical Models,” Molecular Cancer Therapeutics, Vol. 6, No. 8, 2007, pp. 2209-2219. doi:10.1158/1535-7163.MCT-07-0231
- [63] A. A. Adjei, R. B. Cohen, W. Franklin, et al., “Phase I Pharmacokinetic and Pharmacodynamic Study of the Oral, Small-Molecule Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor AZD6244 (ARRY-142886) in Patients with Advanced Cancers,” Journal of Clinical Oncology, Vol. 26, No. 13, 2008, pp. 2139-2146. doi:10.1200/JCO.2007.14.4956
- [64] Bolondi, et al., “Phase II Safety Study of the Oral Multikinase Inhibitor Regorafenib (BAY 73-4506) as SecondLine Therapy in Patients with Hepatocellular Carcinoma September 26 at the 2011 European Multidisciplinary Cancer Congress,” Abstract 6576, 2011.
- [65] D. M. Sabatini, “mTOR and Cancer: Insights into a Complex Relationship,” Nature Reviews Cancer, Vol. 6, No. 9, 2006, pp. 729-734. doi:10.1038/nrc1974
- [66] K. J. Schmitz, J. Wohlschlaeger, H. Lang, G. C. Sotiropoulos, M. Malago, K. Steveling, H. Reis, et al., “Activation of the ERK and AKT Signalling Pathway Predicts Poor Prognosis in Hepatocellular Carcinoma and ERK Activation in Cancer Tissue Is Associated with Hepatitis C Virus Infection,” Journal of Hepatology, Vol. 48, No. 1, 2008, pp. 83-90. doi:10.1016/j.jhep.2007.08.018
- [67] A Villanueva, DY Chiang, P Newell, et al., “Pivotal Role of mTOR Signaling in Hepatocellular Carcinoma,” Gastroenterology, Vol. 135, No. 6, 2008, pp. 1972-1983, 1983.e1-e11.
- [68] L. D. Zhou, Y. Huang, J. D. Li and Z. M. Wang, “The mTOR Pathway Is Associated with the Poor Prognosis of Human Hepatocellular Carcinoma,” Medical Oncology, Vol. 27, No. 2, 2010, pp. 255-261. doi:10.1007/s12032-009-9201-4
- [69] T. H. Hu, C. C. Huang, P. R. Lin, H. W. Chang, L. P. Ger, Y. W. Lin, C. S. Changchien, C. M. Lee and M. H. Tai, “Expression and Prognostic Role of Tumor Suppressor gene PTEN/MMAC1 Mutations in Hepatocellular Carcinoma,” Cancer Investigation, Vol. 18, No. 2, 2000, pp. 123-129. doi:10.3109/07357900009038243
- [70] K. F. Chen, H. L. Chen, W. T. Tai, W. C. Feng, C. H. Hsu, P. J. Chen and A. L. Cheng, “Activation of Phosphatidylinositol 3-Kinase/Akt Signaling Pathway Mediates Acquired Resistance to Sorafenib in Hepatocellular Carcinoma Cells,” Journal of Pharmacology and Experimental Therapeutics, Vol. 337, No. 1, 2011, pp. 155- 161. doi:10.1124/jpet.110.175786
- [71] H. Asahina, H. Nokihara, N. Yamamoto, Y. Yamada, Y. Tamura, K. Honda, Y. Seki, Y. Tanabe, H. Shimada, X. Shi and T. Tamura, “Safety and Tolerability of AZD8055 in Japanese Patients with Advanced Solid Tumors; a Dose-Finding Phase I Study,” Investigational New Drugs, 2012.
- [72] A. X. Zhu, T. A. Abrams, R. Miksad, L. S. Blaszkowsky, J. A. Meyerhardt, H. Zheng, A. Muzikansky, J. W. Clark, E. L. Kwak, D. Schrag, K. R. Jors, C. S. Fuchs, A. J. Iafrate, D. R. Borger and D. P. Ryan, “Phase 1/2 Study of Everolimus in Advanced Hepatocellular Carcinoma,” Cancer, Vol. 117, No. 22, 2011, pp. 5094-5102. doi:10.1002/cncr.26165
- [73] T. Decaens, A. Luciani, E. Itti, A. Hulin, F. RoudotThoraval, A. Laurent, E. S. Zafrani, A. Mallat and C. Duvoux, “Phase II Study of Sirolimus in TreatmentNaive Patients with advanced Hepatocellular Carcinoma,” Digestive and Liver Disease, Vol. 44, No. 7, 2012, pp. 610-616. doi:10.1016/j.dld.2012.02.005
- [74] M. Rizell, M. Andersson, C. Cahlin, L. Hafström, M. Olausson and P. Lindnér, “Effects of the mTOR Inhibitor Sirolimus in Patients with Hepatocellular and Cholangiocellular Cancer,” International Journal of Clinical Oncology, Vol. 13, No. 1, 2008, pp. 66-70. doi:10.1007/s10147-007-0733-3
- [75] Q. Zhou, V. W. Lui, C. P. Lau, S. H. Cheng, M. H. Ng, Y. Cai, S. L. Chan and W. Yeo, “Sustained Antitumor Activity by Co-Targeting mTOR and the Microtubule with Temsirolimus/Vinblastine Combination in Hepatocellular Carcinoma,” Biochemical Pharmacology, Vol. 83, No. 9, 2012, pp. 1146-1158. doi:10.1016/j.bcp.2012.01.013
- [76] C. Herencia, J. M. Martínez-Moreno, C. Herrera, F. Corrales, R. Santiago-Mora, I. Espejo, M. Barco, Y. Almadén, M. de la Mata, A. Rodríguez-Ariza and J. R. MuñozCastañeda, “Nuclear Translocation of β-Catenin during Mesenchymal Stem Cells Differentiation into Hepatocytes Is Associated with a Tumoral Phenotype,” PLoS One, Vol. 7, No. 4, 2012, Article ID: e34656. doi:10.1371/journal.pone.0034656
- [77] H. Fujii, K. Moriya, Y. Shintani, T. Tsutsumi, T. Takayama, M. Makuuchi, S. Kimura and K. Koike, “Frequent Beta-Catenin Abberation in Human Hepatocellular Carcinomas,” Oncogene, Vol. 20, No. 1, 2007, pp. 39-51.
- [78] Y. Zhang, W. Wei, N. Cheng, K. Wang, B. Li, X. Jiang and S. Sun, “Hepatitis C Virus-Induced Up-Regulation of Microrna-155 Promotes Hepatocarcinogenesis by Activating Wnt Signaling,” Hepatology, Vol. 56, No. 5, 2012, pp. 1631-1640. doi:10.1002/hep.25849
- [79] R. Srisuttee, S. S. Koh, S. J. Kim, W. Malilas, W. Boonying, I. R. Cho, B. H. Jhun, M. Ito, Y. Horio, E. Seto, S. Oh and Y. H. Chung, “Hepatitis B Virus X (HBX) Protein Upregulates β-Catenin in a Human Hepatic Cell Line by Sequestering SIRT1 Deacetylase,” Oncology Reports, Vol. 28, No. 1, 2012, pp. 276-282.
- [80] W. Wei, M. Chua, S. Grepper and S. So, “Small Molecule Antagonists of Tcf4/b-Catenin Complex Inhibit the Growth of HCC Cells in Vitro and in Vivo,” International Journal of Cancer, Vol. 126, No. 10, 2010, pp. 2426-2436.
- [81] M. Pasca di Magliano and M. Hebrok, “Hedgehog Signalling in Cancer Formation and Maintenance,” Nature Reviews Cancer, Vol. 3, No. 12, 2003, pp. 903-911. doi:10.1038/nrc1229
- [82] M. A. Patil, J. Zhang, C. Ho, S. T. Fan and X. Chen, “Hedgehog Signaling in Human Hepatocellular Carcinoma,” Cancer Biology& Therapy, Vol. 5, No. 1, 2006, pp. 111-117.
- [83] J. K. Sicklick, “Dysregulation of the Hedgehog Pathway in human Hepatocarcinogenesis,” Carcinogenesis, Vol. 27, No. 4, 2006, pp. 748-757. doi:10.1093/carcin/bgi292
- [84] J. L. Mullor, N. Dahmane, T. Sun and A. A. Ruizi, “Wnt Signals Are Targets and Mediators of Gli Function,” Current Biology, Vol. 11, No. 10, 2001, pp. 769-773. doi:10.1016/S0960-9822(01)00229-9
- [85] C. Niemann, A. B. Unden, S. Lyle, Ch. C. Zouboulis, R. Toftgard and F. M. Watt, “Indian Hedgehog and β-Catenin Signaling: Role in the Sebaceous Lineage of Normal and Neoplastic Mammalian Epidermis,” Proceedings of the National Academy of Sciences of the United States, Vol. 100, Suppl. 1, 2003, pp. 11873-11880. doi:10.1073/pnas.1834202100
- [86] Y. Xu, V. Chenna, C. Hu, H. X. Sun, M. Khan, H. Bai, X. R. Yang, Q. F. Zhu, Y. F. Sun, A. Maitra, J. Fan and R. A. Anders, “Polymeric Nanoparticle-Encapsulated Hedgehog Pathway Inhibitor HPI-1 (NanoHHI) Inhibits Systemic Metastases in an Orthotopic Model of Human Hepatocellular Carcinoma,” Clinical Cancer Research, Vol. 18, No. 5, 2012, pp. 1291-1302. doi:10.1158/1078-0432.CCR-11-0950
- [87] G. Giannelli, A. Mazzocca, E. Fransvea, M. Lahn and S. Antonaci, “Inhibiting TGF-β Signaling in Hepatocellular Carcinoma,” Biochimica et Biophysica Acta, Vol. 1815, No. 2, 2011, pp. 214-223.
- [88] T. Ueki, J. Fujimoto, T. Suzuki, H. Yamamoto and E. Okamoto, “Expression of Hepatocyte Growth Factor and Its Receptor, the c-Met Proto-Oncogene, in Hepatocellular Carcinoma,” Hepatology, Vol. 25, No. 3, 1997, pp. 619- 623. doi:10.1002/hep.510250321
- [89] P. Kaposi-Novak, J. S. Lee, L. Gomez-Quiroz, C. Coulouarn, V. M. Factor and S. S. Thorgeirsson, “MetRegulated Expression Signature Defines a Subset of Human Hepatocellular Carcinomas with Poor Prognosis And Aggressive Phenotype,” Journal of Clinical Investigation, Vol. 116, No. 6, 2006, pp. 1582-1595. doi:10.1172/JCI27236
- [90] J. Avruch, D. Zhou, J. Fitamant and N. Bardeesy, “Mst1/2 Signalling to Yap: Gatekeeper for Liver Size and Tumour Development,” British Journal of Cancer, Vol. 104, No. 1, 2011, pp. 24-32. doi:10.1038/sj.bjc.6606011
- [91] L. Gramantieri, C. Giovannini, A. Lanzi, P. Chieco, M. Ravaioli, A. Venturi, G. L. Grazi and L. Bolondi, “Aberrant Notch3 and Notch4 Expression in Human Hepatocellular Carcinoma,” Liver International, Vol. 27, No. 7, 2007, pp. 997-1007. doi:10.1111/j.1478-3231.2007.01544.x
- [92] J. Gao, Z. Song, Y. Chen, L. Xia, J. Wang, R. Fan, R. Du, F. Zhang, L. Hong, J. Song, X. Zou, H. Xu, G. Zheng, J. Liu and D. Fan, “Deregulated Expression of Notch Receptors in Human Hepatocellular Carcinoma,” Digestive and Liver Disease, Vol. 40, No. 2, 2008, pp. 114-121. doi:10.1016/j.dld.2007.08.001
- [93] I. Borbath, C. Porta, L. Rimassa, et al., “Randomized Controlled Phase 2 Study (RCT) with Tivantinib in PreTreated Hepatocellular Carcinoma (HCC): Efficacy, Safety, and MET-Analysis,” 63rd Annual Meeting of the American Association for the Study of Liver Diseases (AASLD 2012), Abstract 114, Boston, 9-13 November 2012.
- [94] H. Safran, K. Charpentier, G. Dubel, et al., “Lenalidomide for Second-Line Treatment of Advanced Hepatocellular Cancer (HCC): A Brown University Oncology Group Phase II Study,” Journal of Clinical Oncology, Vol. 30, 2012 (Abstr 4098).
NOTES
*Corresponding author.