Journal of Cancer Therapy
Vol.3 No.5(2012), Article ID:23448,9 pages DOI:10.4236/jct.2012.35071
Rejection of Experimental Hodgkins Lymphoma by T-Cells Engineered with a CD19 Chimeric Antigen Receptor
![]()
1The University of Edinburgh, Centre for Infectious Diseases, Edinburgh, UK; 2The University of Manchester, Clinical and Experimental Immunology Group, Manchester, UK; 3Targeted Therapy Group, Paterson Institute for Cancer Research, Christie Hospital, Manchester, UK; 4The University of Edinburgh, Division of Pathway Medicine, Chancellor’s Building, Edinburgh, UK; 5Laboratory Medicine (Virology), Royal Infirmary of Edinburgh, Edinburgh, UK.
Email: i.johannessen@ed.ac.uk
Received August 20th, 2012; revised September 21st, 2012; accepted October 2nd, 2012
Keywords: Hodgkin’s Lymphoma; CD19; Chimeric Antigen Receptor; Immunotherapy
ABSTRACT
T cells engineered to express chimeric antigen receptors (CARs) combining an external antibody binding domain with the CD3ζ T cell receptor (TCR) signaling domain for triggering cell activation are being used for immunotherapeutic targeting of tumor cells in a non-HLA restricted manner. In this study we transduced T cells with a CD19-CAR construct containing a truncated CD34 gene (tCD34) marker and used these to target the B cell antigen CD19 on the surface of a Hodgkin’s lymphoma (HL) cell line (L591) both in vitro and in vivo. Levels of tCD34 expression in transduced peripheral blood mononuclear cells (PBMCs) ranged from 6% - 20% and this was increased to 82% after selection for transduced tCD34+ cells. In vitro cytotoxicity testing on a CD19+ HL cell line (L591) showed specific cell lysis initiated by the CD19-CAR transduced PBMCs. Importantly, CD19-CAR T cells prevented the growth of L591 HL tumor cells when co-injected subcutaneously (sc) in 6/6 severe combined immunodeficient (SCID) mice. There was no evidence of anti-tumor activity when CD19-CAR T cells were infused intravenously (iv) at the same time as L591 HL tumor cells were injected sc. However, 3/6 SCID mice showed tumor rejection within 83 days after iv infusion of CD19-CAR T cells 3 - 9 days after establishment of L591 HL tumors, while all control animals succumbed to tumors within 60 days. Interestingly, immuno-histochemical analysis of L591 HL tumors demonstrated that CD19-CAR T cells were detected not earlier than 11 days after infusion within the tumor mass. These results suggest that CD19 is a potentially attractive target for the immunotherapy of HL.
1. Introduction
Hodgkin’s lymphoma (HL) is the most common lymphoma in young people in industrialised countries, with around 1500 new cases diagnosed per year in the UK. It is unusual histologically in that less than 2% of the tumor is made up of the malignant B cells; mononuclear Hodgkin or multinuclear Reed-Sternberg cells (collectively referred to as HRS cells). The remaining tumour bulk consists of a non-neoplastic infiltrate of leucocytes including lymphocytes and prominent eosinophils. In western countries, up to 50% of HL is associated with Epstein-Barr virus (EBV), a ubiquitous gamma-herpesvirus which persists for life in B lymphocytes of the host after primary infection [1]. In EBV-positive HL, HRS cells express a limited number of latent EB viral genes including the viral oncogene latent membrane protein
(LMP)-1. HL is one of the most treatable adult cancers with over 80% of patients achieving complete remission following chemotherapy and/or radiotherapy. However, a proportion of patients have resistant primary disease and up to a third of those achieving remission later relapse. Alternative therapies are required to treat these patients.
Immunotherapy of EBV-associated malignancies using adoptive transfer of autologous or allogeneic virus-specific cytotoxic T lymphocytes (CTLs) has proven successful, especially in the treatment of post-transplant lymphoproliferative disease (PTLD) [2-4]. Disappointingly, despite being well tolerated, this form of T cell therapy has proven less effective in the treatment of EBV positive HL [5]. This poor response is possibly because HRS cells lack expression of EBV nuclear antigens 3a, b, c, the immunodominant antigens for CD8 T cells, which are expressed in most PTLDs. Additionally, the nonneoplastic leucocyte infiltrate characteristic of HL tumors contains Th2 and T regulatory cells and is generally devoid of Th1 cells, CD8+ T cells and NK cells [6] which might mount an anti-tumor immune response. HRS cells appear to maintain this suppressive milieu by the secretion of Th2-type chemokines such as the thymus and activation regulated chemokine (TARC) [7] and cytokines like IL10 that inhibit Th1 responses [8].
One strategy which has been developed to surmount this tumor immune evasion is the use of engineered T cells that express chimeric antigen receptors (CARs), otherwise known as T-bodies [9]. These molecules generally contain an antibody-based external domain such as the single chain variable fragment (scFv), and a TCRderived internal domain for signal transduction and T cell activation [10]. They are expressed on the surface of T cells as a single gene-encoded homodimer. Advantages of this strategy include the ability of therapeutic cells to traffic to tumor sites, and to expand and persist in response to target recognition. Also, the MHC-independent recognition of targets circumvents the necessity for donor/recipient HLA matching and the propensity of tumor cells to down-regulate antigen presentation. Cells engineered with CARs directed against a range of tumor associated antigens have been shown to be capable of specific tumor cell lysis in vitro, including those targeted at CD19, CD30, HER2/neu and prostate-specific membrane antigen [11-14]. T-cells bearing first generation CARs (CD3z only signaling domain) are highly efficacious in mouse models of B-cell lymphoma [15,16] but, to date, there are no published studies investigating the potency of CAR T-cells targeting CD19 to control the growth of HL cells.
CD19 is a signaling molecule expressed at all stages of B cell development until terminal differentiation into plasma cells [17], and is constitutively expressed in B-non Hodgkin’s lymphomas (NHL), and on the majority of chronic lymphocytic leukaemias (CLL) and acute lymphoblastic leukaemias (ALL) [18]. CD19-specific CAR (CD19-CAR) engineered T cells have been shown to specifically lyse the CD19+ ve Raji (Burkitt’s lymphoma) cell line, CLL and ALL cells, as well as NHL lymph node tumor biopsy cells [11,19,20]. Expression of CD19 is not consistently observed in HL, however a variable percentage of classical HL biopsies (0 to 75%) and cell lines (20% to 30%) express the marker [21-24], and thus there is potential for the use of CD19-CAR T cells in the therapy of HL.
Here we show how a CAR construct incorporating a CD19-specific scFv linked to a CD3ζ signaling domain was used to engineer CD19-specificity in T cells. These cells were able to target and kill HL-derived cells via CD19 in both an in vitro cytotoxicity assay and a SCID mouse model of HL.
2. Materials and Methods
2.1. Blood Donors
Anonymised buffy coat donations were provided by the Scottish National Blood Transfusion Service (SNBTS) from registered blood donors with informed consent. Heparin (1000 IU into 50 mL; CP Pharmaceuticals, Wrexham, UK) was added to buffy coat samples, and peripheral blood mononuclear cells (PBMCs) were separated using Histopaque-1077 (Sigma, Irvine, UK). PBMCs were washed in RPMI 1640 (Invitrogen), and then cryopreserved in 10% dimethyl sulphoxide (Sigma) in fetal bovine serum (FBS).
2.2. Cell Lines
Cell lines used in the study included the HL cell line L591 [25]. Lymphoblastoid cell lines (LCLs) were derived by in vitro EBV infection of PBMCs from healthy donors. The Epstein Barr Virus (EBV)-specific CTL-A was produced as described [26] using PBMCs from a donor (HLA type: A1, A3, B35, B57). This CTL was chosen as it matched the HLA type of L591 cells (A1, A33, B8, B35) at two loci.
2.3. Cell Culture
Culture media was supplied by Invitrogen (Paisley, UK) unless otherwise stated. Cells were cultured in RPMI 1640 supplemented with 2 mM L-glutamine, 100 U/mL penicillin, 100 U/mL streptomycin and FBS (Perbio) at 10% v/v (LCL) or 20% v/v (transduced PBMCs, L591, PG13, CTL-A).
2.4. scFv Construct and Retroviral Production
The chimeric receptor construct was composed of a CD19 specific scFv isolated from the HD37 hybridoma, a flexible hinge region, and the transmembrane and intracellular signaling domains of CD3ζ [11]. A truncated CD34 (tCD34) gene was included as a marker of transduction. The construct was codon optimised and cloned into the pMP71 retroviral vector [27]. An empty vector with the expression cassette replaced by green fluorescent protein (GFP) was used as a virus control. Virus was collected from the PG13 packaging cell line stably expressing the construct as previously described [27].
2.5. T Cell Transduction
PBMCs were thawed and washed twice by centrifugation in RPMI with 20% FBS, first at 800 g for 8 min, then 400 g for 7 min. Cells were activated with 1 µg/mL antiCD3 (Becton Dickinson) and 1 µg/mL anti-CD28 (Becton Dickinson) in complete RPMI for 72 hours. Activated PBMCs were resuspended in undiluted virus supernatant (MOI of approximately 1.5) with 6 µg/mL polybrene (Sigma) and centrifuged at 1200 g and 4˚C for 3 hours. Transduced cells were resuspended at approximately 5 × 105 cells/mL in medium supplemented with 100 IU/mL IL-2 (UDG) and cultured overnight. Transduction was repeated the following day. Transduced cells were cultured for up to 2 weeks, with IL-2 (100 IU/mL) added to the culture three times a week.
2.6. Selection Protocols
EasySep systems (StemCell Technologies) were used to separate cell populations on the basis of cell surface antigen expression, following the manufacturer’s instructtions. Briefly, cells were washed in cell separation buffer (CSB) at 180 g for 7 minutes and resuspended at 1 × 108 cells/mL in CSB. EasySep positive selection cocktail was added at a concentration of 100 µL/mL cells for CD56 selection and 200 µL/mL cells for CD34 selection, and the mixture incubated for 15 minutes at room temperature. Nanoparticles were added at a concentration of 50 µL/mL, and the mixture incubated for 10 minutes at room temperature. Total volume was brought to 2.5 mL with CBS and the tube placed in an EasySep magnet for 5 minutes. The supernatant was poured off, the tube removed from the magnet, 2.5 mL CSB added to the remaining cells, and the tube placed back in the magnet. The separations were repeated a total of 4 times for CD34 and 3 times for CD56 selection.
2.7. Flow Cytometry
Cells to be analysed were washed twice in PBSA (1% w/v BSA, 0.1% w/v sodium azide, 0.2% w/v EDTA) by centrifugation at 120 g for 5 minutes. The supernatant was decanted and cells resuspended in residual liquid. 10 µL of the appropriate antibody was added and the tubes incubated for 20 minutes at 4˚C in the dark. The antibodies used were anti-CD4 (clone RPA-T4), anti CD8 (clone RPA-T8), anti-CD19 (clone HIB19), and CD34 (clone 582) and anti CD56 (clone B159), all supplied by Becton Dickinson. Cells were washed twice in PBSA then resuspended in 1 × CellFix (Becton Dickinson) and evaluated on either a FACScan (Becton Dickinson) or FACSCalibur (Becton Dickinson) flow cytometer. At least 10,000 events were acquired for each sample.
2.8. Cytotoxicity Assay
Cytotoxic activity in vitro was measured using a standard chromium (Cr) release assay [28]. Cells from each target cell line were labelled with 51Cr for 1 hour at 37˚C before plating with effector cells at an effector: target ratio of 20:1, 10:1 and 5:1. After incubation at 37˚C for 4 hours the release of 51Cr from lysed cells was measured on a gamma counter. Spontaneous release was calculated by incubating target cells without effector cells, and maximum release calculated by lysing target cells with 1% Triton-X (Sigma).
2.9. Mouse Model
CB17 scid/scid mice were supplied and maintained by the University of Edinburgh Biological Services Unit. All animal work was carried out in full compliance with the Animals (Scientific Procedures) Act 1986. Tumor cells were inoculated subcutaneously (sc) in the flank. Engineered cells were delivered sc or intravenously (iv) by tail vein injection. Mice were observed daily for signs of illness and sc tumor formation. Using callipers, macroscopic sc tumors were measured in 2 planes 90˚ to each other; tumor volume (mm3) calculated using the formula ab2/2 (a: length; b: width). Animals were observed for up to 100 days.
2.10. Immunohistochemistry
Formalin-fixed tissue samples were embedded in paraffin wax and cut into 4 mm sections. Antigen retrieval was performed on hydrated sections by immersing in 1 L of pH 6 10 mM citric acid and microwaving at full power (800 W) for 25 minutes, followed by 15 minutes cooling. Endogenous peroxidase was blocked using 0.03% hydrogen peroxide for 10 minutes, followed by a non specific blocking step using 10% goat serum (GS) for 20 mins prior to antibody incubation. The CD34 antibody (Dako M7165) was used at a dilution of 1 mg/mL with the Animal Research Kit (Dako K3954), as per instructtions. Incubation with the biotinylated CD34 antibody for 15 minutes is followed by detection with streptavidin peroxidase and DAB. Nuclei were counterstained with Gill 1 haematoxylin (Thermo-Electron, Runcorn, UK).
2.11. Statistics
Data are presented as mean ± standard deviation (SD). The non-parametric Mann Whitney t test was used to compare levels of specific lysis in chromium release assays. Fisher’s exact test for comparison of proportions, and Wilcoxon rank sum test for comparison of two groups, was used to analyse immunotherapy data. In all cases, p < 0.05 was accepted as indicating a significant difference. Tests were carried out using Prism 4.0 for Windows (GraphPad).
3. Results
3.1. T Cell Transduction
Stored donor PBMCs were thawed and transduced with the retroviral vector encoding the chimeric anti-CD19 receptor. Transduced cells were cultured for up to two weeks in IL-2 containing media. In six experiments cell expansion over the two week period averaged 84-fold (range 50- to 120-fold). Transduction levels were determined using flow cytometery and staining for the tCD34 molecule included in the vector. The average level of transduction from experiments on three different PBMC donors was 13% ± 6% (range 6% - 20%). At the end of the culture period, cells were predominantly CD8+ ve (57% ± 6%), with minority CD4+ ve (39% ± 4%) and CD56+ ve (14% ± 4%).
3.2. In Vitro Cytotoxicity
T cells transduced with the CD19-CAR were tested for cytotxic activity using a standard four hour chromium release assay in 5 repeat experiments (Figure 1). The targets used were the CD19-expressing HL cell line L591, and a CD19-positive LCL. Mock transduced PBMCs were used as a negative control, and the EBV specific CTL-A was used as a positive control. At effector:target ratios of 20:1, 10:1 and 5:1 specific lysis of both target cell lines by the CD19-CAR transduced cells and CTL-A was observed. Lysis by CD19-CAR transduced cells was significantly higher than that effected by mock transduced cells at all effector to target ratios. At 20:1, T cells expressing the CD19-CAR receptor lysed the L591 cell line at 24% ± 11% (range 12% - 35%), and the LCL
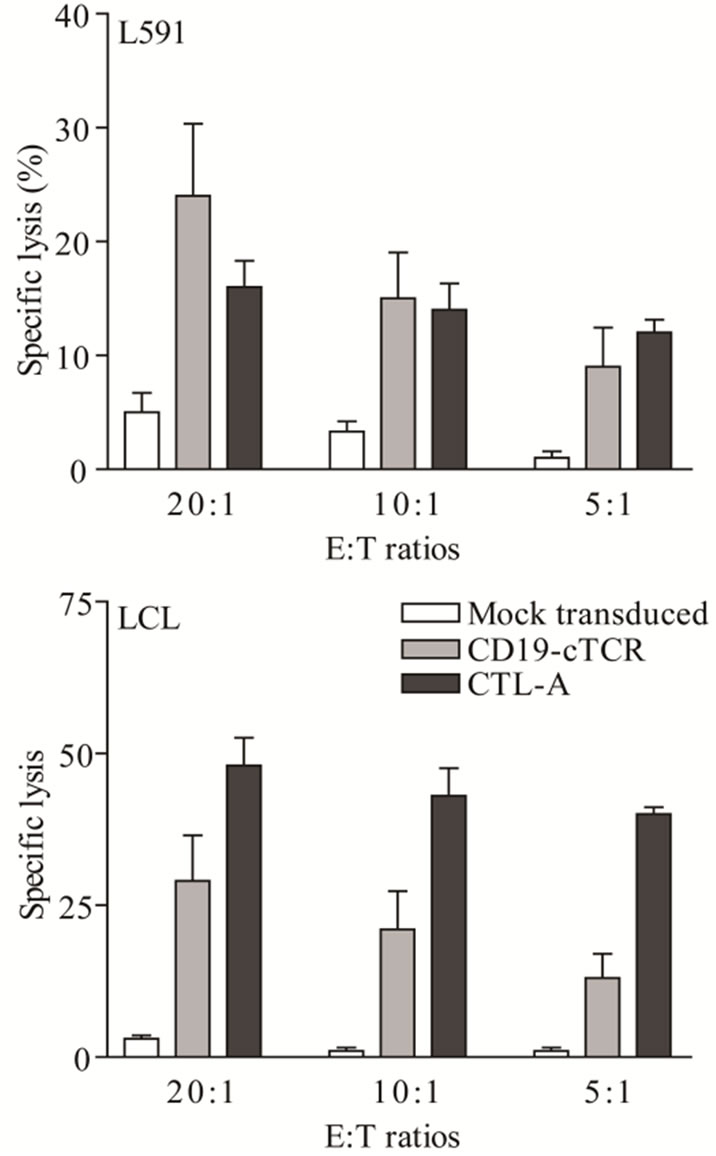
Figure 1. Specific lysis of the L591 and LCL target lines by CD19-CAR engineered T cells and CTL-A was significantly higher at all effector:target (E:T) ratios than that achieved by mock transduced cells in a 51Cr release assay. Bars show mean ± SD for five experiments.
at 29% ± 13% (range 12% - 44%), compared with mock transduced cells at 5% ± 3% (range 2% - 9%) and 3% ± 2% (range 1% - 6%).
In an attempt to increase the level of T cell transduction, a population of CD19-CAR transduced cells was enriched using positive selection for the tCD34 marker. Before selection, 8% of cells expressed tCD34, and following selection the depleted and enriched fractions contained 1% and 82% transduced cells respectively. Increasing the fraction of transduced cells in the total population improved levels of specific lysis (Figure 2). When the percentages of tCD34+ cells in CD19-CAR transduced cells, the tCD34-depleted fraction, and the tCD34-enriched fraction were 8%, 1% and 82%, respectively, specific lysis at an effector to target ratio of 20:1 of L591 cells was 12%, 10%, and 21%, respectively, while specific lysis of LCLs was 12%, 7%, and 41%, respectively.
3.3. In Vivo Tumor Prevention
T cells transduced with the CD19-CAR were assessed for their ability to prevent tumor formation in an in vivo HL model. Groups of 6 SCID mice received sc injection of 5
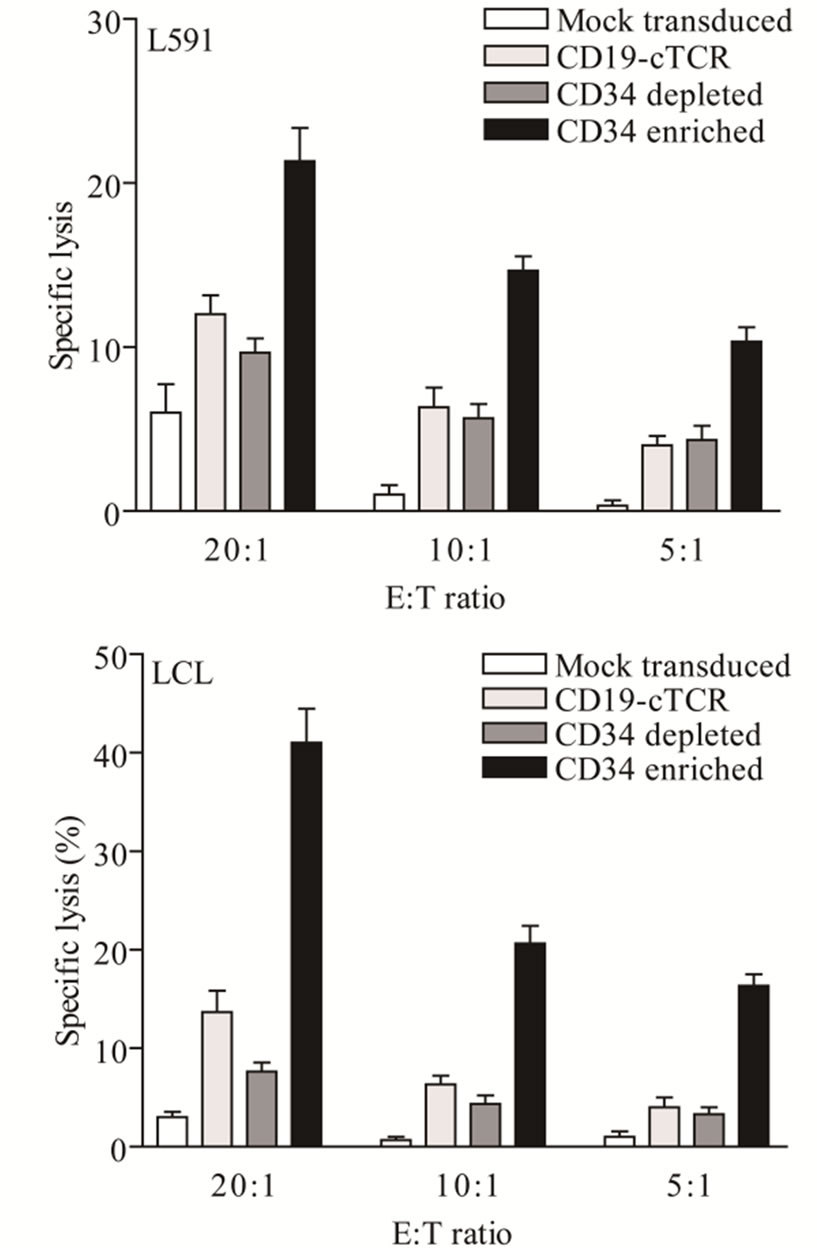
Figure 2. Effect of transduced cell numbers on in vitro cytotoxicity. Specific lysis of the L591 and LCL target lines by CD19-CAR engineered T cells was improved in a 51Cr release assay after the culture was enriched by selection of CD19-CAR cells using tCD34 as a marker of transduction.
× 106 L591 cells concurrently with sc or iv inoculation of either 5 × 107 CD19-CAR engineered cells or CTL-A cells (an EBV-specific CTL, matching two MHC loci with L591). Seventeen out of 22 (77%) control animals that received no therapy developed sc tumors as a result of sc injection of 5 × 106 L591 cells. Time to tumors onset (Table 1) was monitored and compared between groups. Whilst mice given CTL-A cells sc developed tumors at a rate that was not statistically different to control mice [4/6 (67%) vs 17/22 (77%)], none of the mice that received CD19-CAR sc developed tumors [0/6 (0%); Fisher’s exact test: p = 0.001]. Conversely, administration of CD19-CAR-transduced cells via the iv route did not significantly affect tumor development, with 4 out of 6 (67%) iv CD19-CAR treated mice developing tumors (Fisher’s exact test: p = 0.6219). Interestingly, median time to tumor in untreated mice was 20 days compared with 29 days in iv administered CD19-CAR treated mice although this was not a significant difference (Wilcoxon rank sum test: p = 0.0625). Of the four mice treated with iv administered CD19-CAR cells that developed sc tumors, three showed sustained tumor growth which did not differ from control animals whereas one mouse had completely cleared its tumor burden by day 64 post injection. Of the 17 control animals that developed sc tumors, none showed spontaneous tumor regression over an observation period of 100 days.
3.4. In Vivo Tumor Therapy
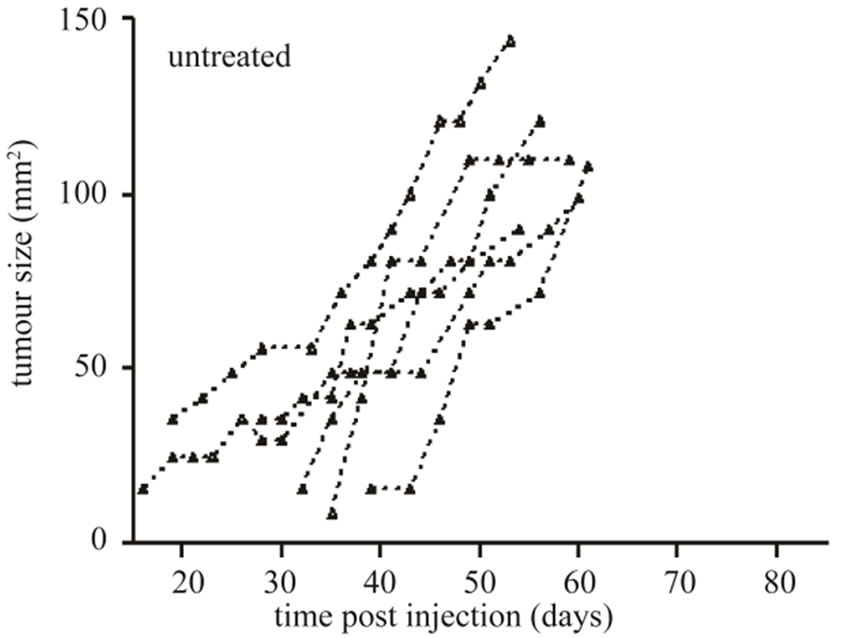
The ability of CD19-CAR engineered T cells to treat established sc L591 tumors was also tested. Six tumor-bearing SCID mice were injected iv with 5 × 107 engineered cells between three and nine days after first signs of visible sc tumor formation, which was 19 to 25 days following sc injection of L591 tumor cells, and subsequent tumor growth observed (Figure 3). Three mice were culled prior to the conclusion of the experiment due to skin necrosis at the tumor site, their lesions displaying sustained tumor growth up to time of culling. The remaining three mice had completely cleared their tumor burden by 65, 69 and 83 days post L591 injection. This was a significant effect when compared with the 17
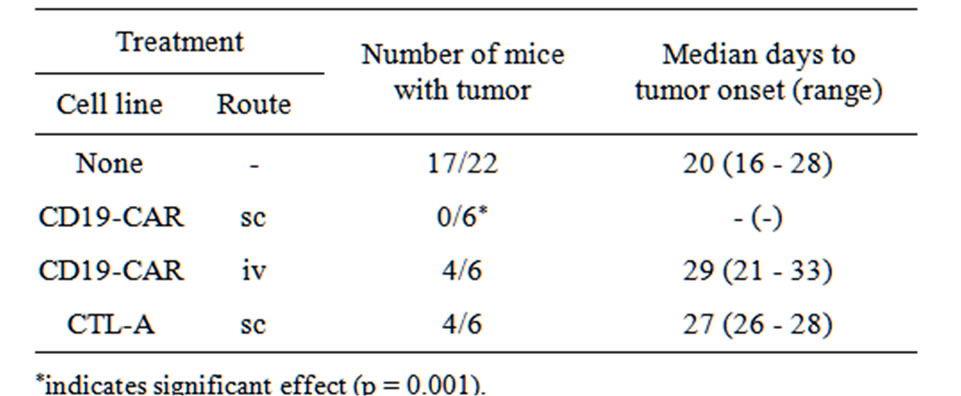
Table 1. Tumor incidence in HL SCID mice given prophylactic immunotherapy.
 (a)
(a)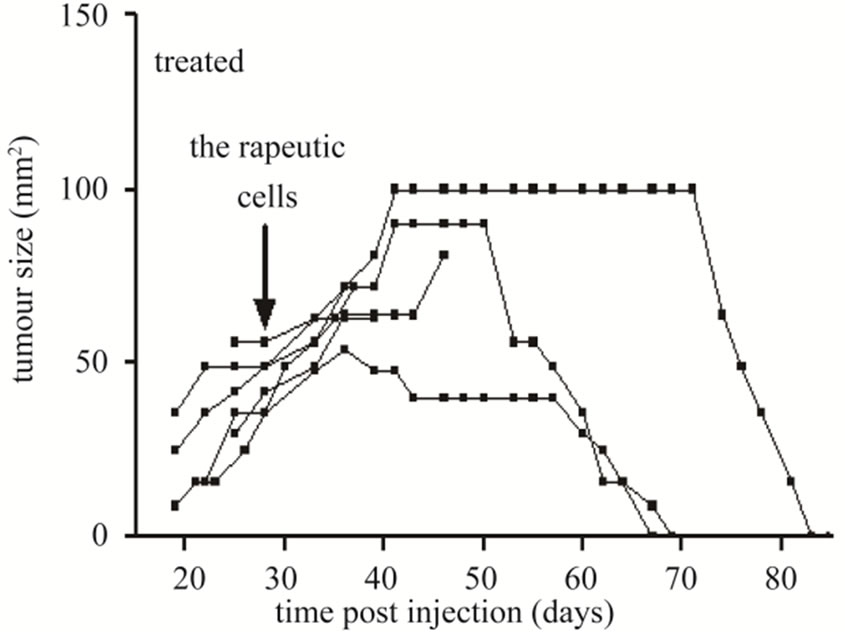 (b)
(b)
Figure 3. Tumor growth curves of SCID mice bearing sc tumors derived from HL L591 cells, either with no therapeutic intervention (a), or treated with CD19-CAR transduced T cells (b). Complete tumor regression was observed in three of six treated mice, which was not seen in any control mice. Each line represents an individual mouse.
untreated mice with established tumors derived from L591 cells which showed no tumor regression (Fisher’s exact, p = 0.011).
In a time course experiment to investigate trafficking of transduced CAR cells to the sc tumour site, five tumour-bearing SCID mice were injected iv with 5 × 107 CD19-CAR engineered cells. A single mouse was culled on days 1, 4, 7, 11, and 18 post treatment and immunohistochemistry performed on tumour sections. AntiCD34 antibodies were used to detect transduced human T cells infiltrating the tumours. tCD34+ cells were seen in tumour sections on day 11 and 18 post treatment, but not days 1, 4 or 7. Tumor tissue taken on days 11 and 18 showed 8% and 41% of the total cell population positive for tCD34 respectively (Figure 4).
4. Discussion
The results demonstrate that activated PBMCs transduced with a retroviral vector to express a CAR specific
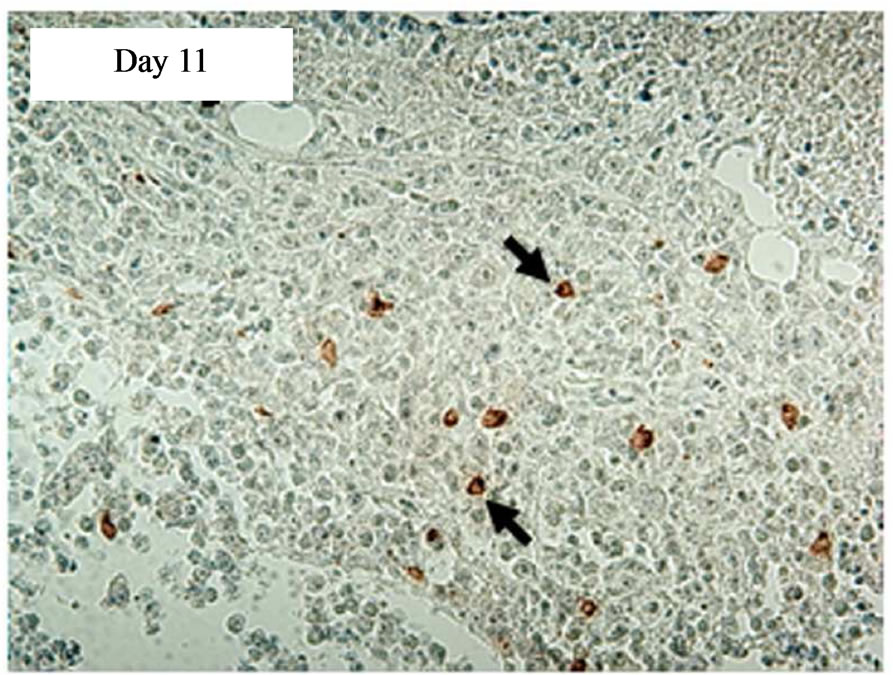
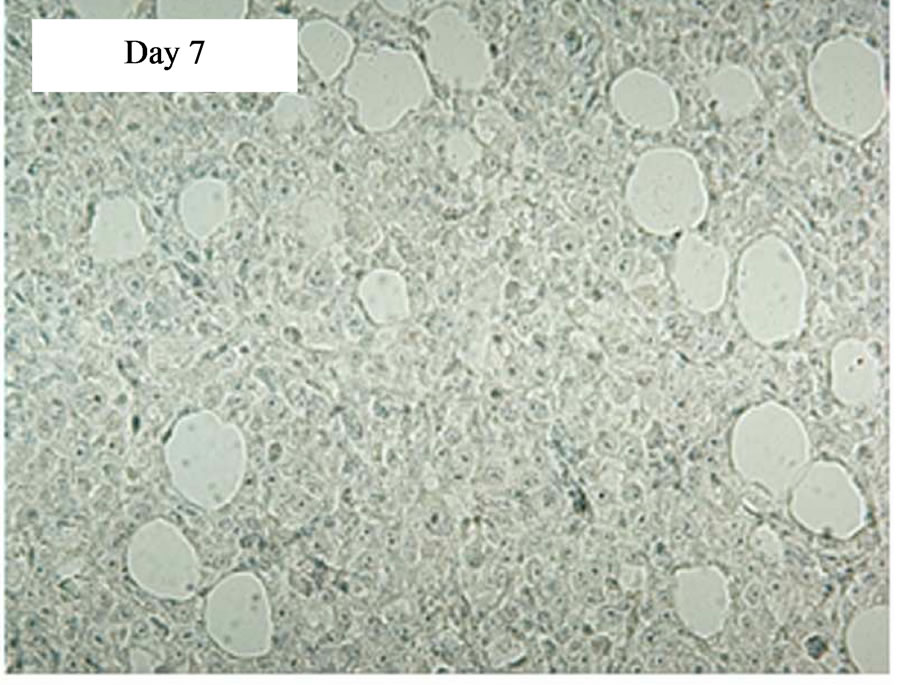
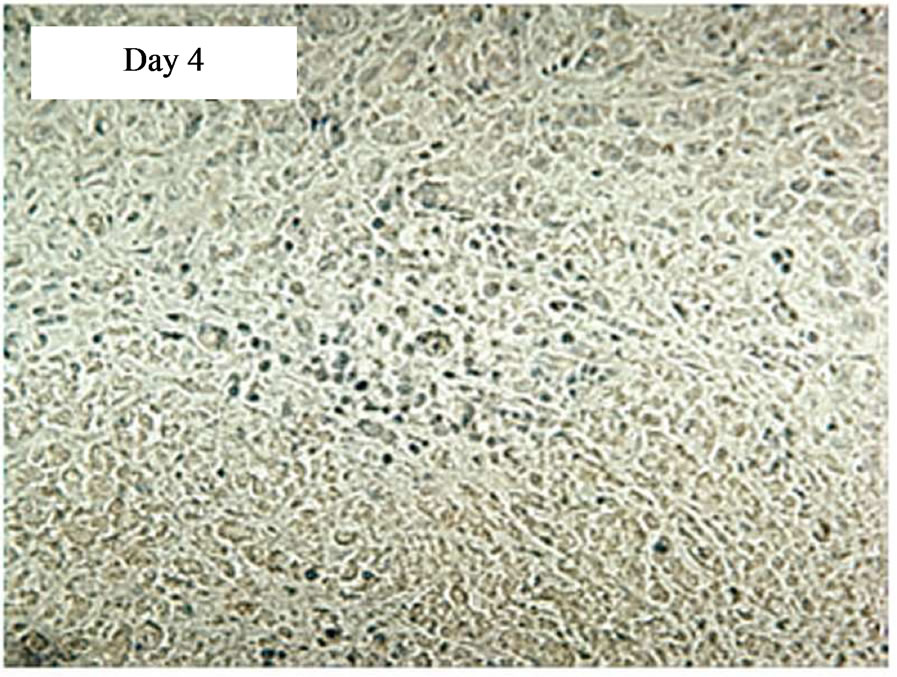
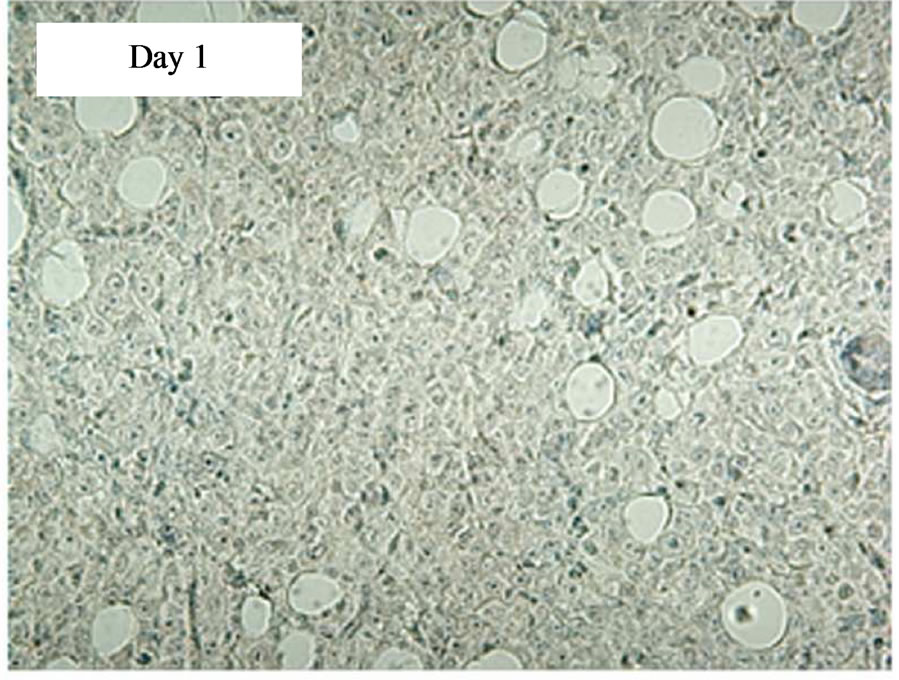
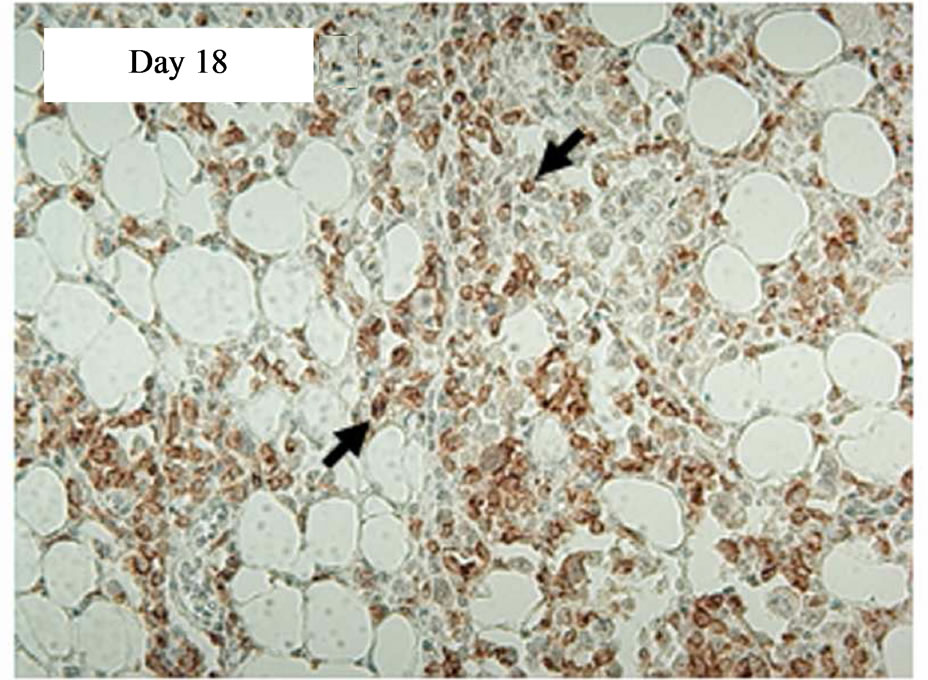
Figure 4. Immunostaining showing tCD34+ cells in L591 tumors treated with CD19-CAR transduced PBMCs at day 11 and 18, but not days 1, 4 or 7 post infusion. HRP detection system shows positive brown membrane staining. Arrows indicate individual positive cells. Sections were counterstained with haematoxylin.
for CD19 are capable of specific lysis of CD19+ ve HL target cells in an in vitro cytotoxicity assay and can mediate significant tumor regression in an in vivo model of HL.
Levels of retrovirus transduction varied between 6% - 20%, with a mean of 13% ± 6% cells expressing CD34 as a marker of transduction. Increasing the fraction of transduced cells in the total population by CD34 selection improved levels of specific lysis (Figure 2). However there was not a simple relationship between number of CD34+ cells and level of specific lysis. Others have also observed that levels of T cell transduction do not correlate with absolute levels of target cell lysis [11], suggesting that it may not be necessary to achieve high levels of expression of the CAR to induce a therapeutic response. Indeed the relationship between transduction rates and specific lysis in in vitro assays and the in vivo effect of engineered cells is unclear. Whilst we observed significant target cell lysis by a CTL line (CTL-A) in vitro, this didn’t correspond to tumor prevention in vivo (Table 1 and Figure 1). In a clinical trial using EBVspecific CTLs to treat PTLD, no correlation was found between levels of specific lysis as measured by 51Cr release assays and tumor response [3]. It remains to be seen whether this is also the case when the therapeutic cells are directed against a single epitope via a CAR rather than multiple CTL specificities.
The aim of this cellular immunotherapy is to produce engineered T cells that home to primary and metastatic tumor sites to give an immediate effect on tumor burden as well as persisting and proliferating to protect against disease relapse. In our experiments the SCID mouse tumors continued to increase in size for up to 10 days after infusion of the engineered T cells before showing signs of regression (Figure 3), and tCD34+ T cells were not detectable in tumors until day 11 post infusion (Figure 4). It is possible that the transduced cells required a period of expansion before migrating to the tumor site in sufficient numbers to effect a clinical response. This may also explain why no significant effect was seen in mice that received iv engineered T cells on the same day as tumor cells but were effective in mice which received T cells after tumor formation (it is possible that a minimum tumor volume is required before effective migration and stimulation of transduced T-cells can be achieved). By day 18 post infusion, the level of transduced cells in the tumor (41%) was high and these cells were able to effect complete tumor regression in 50% of SCID mice carrying an established HL tumor with tumor regression beginning at the time of high T-cell infiltration. In addition several animals were culled due to necrosis of the skin at the tumor site rather than tumor progression perse. This unusual response may have been caused by an inflamematory response initiated by the T cell themselves, and it is possible that a higher dose of cells would have produced a more rapid treatment effect and avoided this complication.
In summary, this study demonstrates that CD19-CAR T cell adoptive therapy may be a suitable approach to target Hodgkin lymphoma. Recent studies demonstrating spectacular anti-tumor responses in three patients with advanced chronic lymphocytic leukaemia (CLL) treated with CD19-CAR T cells highlights the strong potential for this form of therapy [29,30]. In this study, a second generation CAR consisting of the CD137 (4-1BB) fused to the CD3z signalling chain was used together with lentiviral gene transfer and advanced ex vivo T cell culture systems. Other studies using retroviral vectors encoding CAR’s employing CD28-CD3z second generation CAR’s are also underway to challenge CLL [31,32]. Exploring these technological developments to deliver an optimised CAR-T cell therapy for Hodgkins Lymphoma clearly warrants further examination.
5. Acknowledgements
This work was supported by grant CZB/4/521 from the Chief Scientist Office (CSO), Scotland. We would like to acknowledge Garry Ashton and Caron Abbey at the Paterson Institute for Cancer Research histology lab for help with immunohistochemistry.
REFERENCES
- A. B. Rickinson and E. Kieff, “Epstein-Barr Virus,” In: D. Knipe and P. Howley, Eds., Fields Virology, Lippincott Williams and Wilkins, Philadelphia, Vol. 2. 2007, pp. 2655-2700.
- C. M. Bollard, C. M. Rooney and H. E. Heslop, “T-Cell Therapy in the Treatment of Post-Transplant Lymphoproliferative Disease,” Nature Reviews Clinical Oncology, 2012 (in press).
- T. Haque, G. M. Wilkie, M. M. Jones, C. D. Higgins, G. Urquhart, P. Wingate, D. Burns, K. McAulay, M. Turner, C. Bellamy, P. L. Amlot, D. Kelly, A. Macgilchrist, M. K. Gandhi, A. J. Swerdlow and D. H. Crawford, “Allogeneic Cytotoxic T Cell Therapy for EBV-Positive Post Transplant Lymphoproliferative Disease: Results of a Phase II Multicentre Clinical Trial,” Blood, Vol. 110, No. 4, 2007, pp. 1123-1131. doi:10.1182/blood-2006-12-063008
- C. M. Rooney, C. A. Smith, C. Y. Ng, S. Loftin, C. Li, R. A. Krance, M. K. Brenner and H. E. Heslop, “Use of Gene-Modified Virus-Specific T Lymphocytes to Control Epstein-Barr-Virus-Related Lymphoproliferation,” Lancet, Vol. 345, No. 8941, 1995, pp. 9-13. doi:10.1016/S0140-6736(95)91150-2
- C. M. Bollard, L. Aguilar, K. C. Straathof, B. Gahn, M. H. Huls, A. Rousseau, J. Sixbey, M. V. Gresik, G. Carrum, M. Hudson, D. Dilloo, A. Gee, M. K. Brenner, C. M. Rooney and H. E. Heslop, “Cytotoxic T Lymphocyte Therapy for Epstein-Barr Virus+ Hodgkin’s Disease,” Journal of Experimental Medicine, Vol. 200, No. 12, 2004, pp. 1623-1633. doi:10.1084/jem.20040890
- S. Poppema, “Immunobiology and Pathophysiology of Hodgkin Lymphomas,” Hematology American Society of Hematology Education Program Book, Vol. 2005, No. 1, 2005, pp. 231-238. doi:10.1182/asheducation-2005.1.231
- A. van den Berg, L. Visser and S. Poppema, “High Expression of the CC Chemokine TARC in Reed-Sternberg Cells. A Possible Explanation for the Characteristic T-Cell Infiltratein Hodgkin’s Lymphoma,” The American Journal of Pathology, Vol. 154, No. 6, 1999, pp. 1685- 1691. doi:10.1016/S0002-9440(10)65424-7
- H. Herbst, H. D. Foss, J. Samol, I. Araujo, H. Klotzbach, H. Krause, A. Agathanggelou, G. Niedobitek and H. Stein, “Frequent Expression of Interleukin-10 by Epstein-Barr Virus-Harboring Tumor Cells of Hodgkin’s Disease,” Blood, Vol. 87, No. 7, 1996, pp. 2918-2829.
- Z. Eshhar, T. Waks, G. Gross and D. G. Schindler, “Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-Cell Receptors,” Proceedings of the National Academy of Sciences, Vol. 90, No. 2, 1993, pp. 720-724. doi:10.1073/pnas.90.2.720
- Z. Eshhar, T. Waks, A. Bendavid and D. G. Schindler, “Functional Expression of Chimeric Receptor Genes in Human T Cells,” Journal of Immunological Methods, Vol. 248, No. , 2001, pp. 67-76. doi:10.1016/S0022-1759(00)00343-4
- E. J. Cheadle, D. E. Gilham, F. C. Thistlethwaite, J. A. Radford and R. E. Hawkins, “Killing of Non-Hodgkin Lymphoma Cells by Autologous CD19 Engineered T Cells,” British Journal of Haematology, Vol. 129, No. 3, 2005, pp. 322-332. doi:10.1111/j.1365-2141.2005.05456.x
- M. C. Gong, J. B. Latouche, A. Krause, W. D. Heston, N. H. Bander and M. Sadelain, “Cancer Patient T Cells Genetically Targeted to Prostate-Specific Membrane Antigen Specifically Lyse Prostate Cancer Cells and Release Cytokines in Response to Prostate-Specific Membrane Antigen,” Neoplasia, Vol. 1, No. 2, 1999, pp. 123-127. doi:10.1038/sj.neo.7900018
- N. M. Haynes, J. A. Trapani, M. W. Teng, J. T. Jackson, L. Cerruti, S. M. Jane, M. H. Kershaw, M. J. Smyth and P. K. Darcy, “Single-Chain Antigen Recognition Receptors That Costimulate Potent Rejection of Established Experimental Tumors,” Blood, Vol. 100, No. 10, 2002, pp. 3155-3163. doi:10.1182/blood-2002-04-10411
- B. Savoldo, C. M. Rooney, A. Di Stasi, H. Abken, A. Hombach, A. E. Foster, L. Zhang, H. E. Heslop, M. K. Brenner and G. Dotti, “Epstein Barr Virus-Specific Cytotoxic T Lymphocytes Expressing the Anti-CD30{Zeta} Artificial Chimeric T-Cell Receptor for Immunotherapy of Hodgkin’s Disease,” Blood, Vol. 110, No. 7, 2007, pp. 2620-2630. doi:10.1182/blood-2006-11-059139
- E. J. Cheadle, R. E. Hawkins, H. Batha, D. G. Rothwell, G. Ashton and D. E. Gilham, “Eradication of Established B-Cell Lymphoma by CD19-Specific Murine T Cells Is Dependent on Host Lymphopenic Environment and Can Be Mediated by CD4+ and CD8+ T Cells,” Journal of Immunotherapy, Vol. 32, No. 3, 2009, PP. 207-218. doi:10.1097/CJI.0b013e318194a921
- E. J. Cheadle, R. E. Hawkins, H. Batha, A. L. O’Neill, S. J. Dovedi and D. E. Gilham, “Natural Expression of the CD19 Antigen Impacts the Long-Term Engraftment but Not Antitumor Activity of CD19-Specific Engineered T Cells,” Journal of Immunotherapy, Vol. 184, No. 4, 2010, pp. 1885-1896. doi:10.4049/jimmunol.0901440
- L. M. Nadler, K. C. Anderson, G. Marti, M. Bates, E. Park, J. F. Daley and S. F. Schlossman, “B4, a Human B Lymphocyte-Associated Antigen Expressed on Normal, Mitogen-Activated, and Malignant B Lymphocytes,” Journal of Immunotherapy, Vol. 131, No. 1, 1983, pp. 244-250.
- F. M. Uckun, W. Jaszcz, J. L. Ambrus, A. S. Fauci, K. Gajl-Peczalska, C. W. Song, M. R. Wick, D. E. Myers, K. Waddick and J. A. Ledbetter, “Detailed Studies on Expression and Function of CD19 Surface Determinant by Using B43 Monoclonal Antibody and the Clinical Potential of Anti-CD19 Immunotoxins,” Blood, Vol. 71, No. 1, 1988, pp. 13-29.
- R. J. Brentjens, J. B. Latouche, E. Santos, F. Marti, M. C. Gong, C. Lyddane, P. D. King, S. Larson, M. Weiss, I. Riviere and M. Sadelain, “Eradication of Systemic B-Cell Tumors by Genetically Targeted Human T Lymphocytes Co-Stimulated by CD80 and Interleukin-15,” Nature Medicine, Vol. 9, No. 3, 2003, pp. 279-286. doi:10.1038/nm827
- L. J. Cooper, M. S. Topp, L. M. Serrano, S. Gonzalez, W. C. Chang, A. Naranjo, C. Wright, L. Popplewell, A. Raubitschek, S. J. Forman and M. C. Jensen, “T-Cell Clones Can Be Rendered Specific for CD19: Toward the Selective Augmentation of the Graft-versus-B-Lineage Leukemia Effect,” Blood, Vol. 101, No. 4, 2003, pp. 1637-1644. doi:10.1182/blood-2002-07-1989
- H. G. Drexler, “Recent Results on the Biology of Hodgkin and Reed-Sternberg Cells. I. Biopsy Material,” Leukemia & Lymphoma, Vol. 8, No. 4-5, 1992, pp. 283-313. doi:10.3109/10428199209051008
- H. G. Drexler, “Recent Results on the Biology of Hodgkin and Reed-Sternberg Cells. II. Continuous Cell Lines,” Leukemia & Lymphoma, Vol. 9, No. 1-2, 1993, pp. 1-25. doi:10.3109/10428199309148499
- H. Herbst, G. Tippelmann, I. Anagnostopoulos, J. Gerdes, R. Schwarting, T. Boehm, S. Pileri, D. B. Jones and H. Stein, “Immunoglobulin and T-Cell Receptor Gene Rearrangements in Hodgkin’s Disease and Ki-1-Positive Anaplastic Large Cell Lymphoma: Dissociation between Phenotype and Genotype,” Leukemia Research, Vol. 13, No. 2, 1989, pp. 103-116. doi:10.1016/0145-2126(89)90134-3
- C. Schmid, L. Pan, T. Diss and P. G. Isaacson, “Expression of B-Cell Antigens by Hodgkin’s and Reed-Sternberg Cells,” The American Journal of Pathology, Vol. 139, No. 4, 1991, pp. 701-707.
- V. Diehl, H. H. Kirchner, H. Burrichter, H. Stein, C. Fonatsch, J. Gerdes, M. Schaadt, W. Heit, B. UchanskaZiegler, A. Ziegler, F. Heintz and K. Sueno, “Characteristics of Hodgkin’s Disease-Derived Cell Lines,” Cancer Treatment Reports, Vol. 66, No. 4, 1982, pp. 615-632.
- G. M. Wilkie, C. Taylor, M. M. Jones, D. M. Burns, M. Turner, D. Kilpatrick, P. L. Amlot, D. H. Crawford and T. Haque, “Establishment and Characterization of a Bank of Cytotoxic T Lymphocytes for Immunotherapy of EpsteinBarr Virus-Associated Diseases,” Journal of Immunotherapy, Vol. 27, No. 4, 2004, pp. 309-316. doi:10.1097/00002371-200407000-00007
- E. J. Cheadle, D. E. Gilham and R. E. Hawkins, “The Combination of Cyclophosphamide and Human T Cells Genetically Engineered to Target CD19 Can Eradicate Established B-Cell Lymphoma,” British Journal of Haematology, Vol. 142, No. 1, 2008, pp. 65-68. doi:10.1111/j.1365-2141.2008.07145.x
- T. Haque, P. L. Amlot, N. Helling, J. A. Thomas, P. Sweny, K. Rolles, A. K. Burroughs, H. G. Prentice and D. H. Crawford, “Reconstitution of EBV-Specific T Cell Immunity in Solid Organ Transplant Recipients,” Journal of Immunology, Vol. 160, No. 12, 1998, pp. 6204-6209.
- M. Kalos, B. L. Levine, D. L. Porter, S. Katz, S. A. Grupp, A. Bagg and C. H. June, “T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia,” Science Translational Medicine Home, Vol. 3, No. 95, 2011, p. 95ra73. doi:10.1126/scitranslmed.3002842
- D. L. Porter, B. L. Levine, M. Kalos, A. Bagg and C. H. June, “Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia,” The New England Journal of Medicine, Vol. 365, No. 8, 2011, pp. 725-733. doi:10.1056/NEJMoa1103849
- R. J. Brentjens, I. Riviere, J. H. Park, M. L. Davila, X. Wang, J. Stefanski, C. Taylor, R. Yeh, S. Bartido, O. Borquez-Ojeda, M. Olszewska, Y. Bernal, H. Pegram, M. Przybylowski, D. Hollyman, Y. Usachenko, D. Pirraglia, J. Hosey, E. Santos, E. Halton, P. Maslak, D. Scheinberg, J. Jurcic, M. Heaney, G. Heller, M. Frattini and M. Sadelain, “Safety and Persistence of Adoptively Transferred Autologous CD19-Targeted T Cells in Patients with Relapsed or Chemotherapy Refractory B-Cell Leukemias,” Blood, Vol. 118, No. 18, 2011, pp. 4817-4828. doi:10.1182/blood-2011-04-348540
- J. N. Kochenderfer, M. E. Dudley, S. A. Feldman, W. H.Wilson, D. E. Spaner, I. Maric, M. Stetler-Stevenson, G. Q. Phan, M. S. Hughes, R. M. Sherry, J. C. Yang, U. S. Kammula, L. Devillier, R. Carpenter, D. A. Nathan, R. A. Morgan, C. Laurencot and S. A. Rosenberg, “B-Cell Depletion and Remissions of Malignancy along with Cytokine-Associated Toxicity in a Clinical Trial of Anti-CD19 Chimeric-Antigen-Receptor-Transduced T Cells,” Blood, Vol. 119, No. 12, 2012, pp. 2709-2720. doi:10.1182/blood-2011-10-384388