Journal of Cancer Therapy
Vol.3 No.4(2012), Article ID:21594,9 pages DOI:10.4236/jct.2012.34040
Current Trends and Future Directions in Clinical Trials for Malignant Melanoma Treatment Using Anti-Angiogenic Strategies
![]()
1Department of Burns & Plastic Surgery, Whiston Hospital, Prescott, UK; 2Department of Plastic Surgery, Whiston Hospital, Prescott, UK; 3Mersey Academic Plastic Surgery Group, Liverpool Cancer Research UK Centre, Department of Molecular and Clinical Cancer Medicine, The Duncan Building, Liverpool, UK; 4Department Plastic Surgery, The Christie NHS Foundation Trust, Manchester, UK.
Email: *daemon.dewing@gmail.com
Received June 12th, 2012; revised July 15th, 2012; accepted July 28th, 2012
Keywords: Angiogenesis; Malignant Melanoma; Drug Trials; Vemurafinib; BRAF Mutation
ABSTRACT
Melanoma is the most lethal skin cancer with a high propensity to metastasis and conventionally is poorly responsive to non-surgical treatments including chemotherapy and radiotherapy. Considerable advances have been made recently targeting BRAF mutations and immune regulation and, for the first time, credible options exist for patients with metastatic disease. Angiogenesis, the growth of new blood vessels, is an absolute prerequisite for tumour growth beyond a few millimetres in size. Melanoma neovascularisation is correlated with poor prognosis, reduced overall survival, ulceration and increased rate of relapse. Melanoma cells secrete several pro-angiogenic cytokines including Vascular Endothelial Growth Factor VEGF-A and raised levels of expression are associated with the switch from indolent radial, to invasive vertical and then metastatic growth phases. Understanding the processes underlying angiogenesis and how it relates to tumour growth broadly and to melanoma specifically is instrumental in the current drive to develop new treatments that target a range of tumour cell receptors and intracellular processes from receptor antagonism to monoclonal antibodies aimed at the disruption of the process of tumour angiogenesis. We discuss recent and current trials for metastatic melanoma therapy, and discuss potential directions of future treatment scheduling considering different treatment scheduling approaches beyond the parameters of standard drug trials.
1. Introduction
Inhibition of angiogenesis may make a tumour further susceptible to chemo and radiotherapies as has been demonstrated in pre-clinical trials in mouse models with the anti-angiogenic drug TL-118 (www.tiltanpharma.com). Angiogenesis in the adult is restricted to wound healing and female menstrual cyclein normal physiology, making inhibition of angiogenesis attractive, with potentially manageable side effects. Therapies can target different aspects of angiogenesis, including growth factors and their receptors, extra-cellular matrix (ECM) receptors, or target specific components of the ECM.
Malignant Melanoma (MM) is a highly angiogenic tumour, which is refractory to treatment after metastasis. It is demonstrated experimentally that vascular endothelial growth factor (VEGF), when over-expressed, transforms non-aggressive melanoma cell lines into vascularised and highly metastatic phenotypes. Literature in excess of 20,000 publications, evidences VEGF’s central role in angiogenesis. In attempting to translate experimental insights into clinical gains, VEGF is now being exploited as a potential future serum marker to act as a prognostic biomarker and monitor of treatment responses to chemotherapy, as well as a potential therapeutic target. Improved mean overall survival in patients with BRAF mutation, treated with Vemurafinib or Ipilimumab give hope to challenge the poor prognosis of metastatic MM; and phase III trials with Ipilimumab are now underway. This article reviews current trials and their approaches suggesting new developments likely to emerge in the future treatment of metastatic melanoma. We include ipilimumab on the basis that it is demonstrating a beneficial effect when used in combination with anti-angiogenic therapeutic agents.
2. Targeting Angiogenic Growth Factors and Their Receptors
2.1. Bevacizumab
Patients with Stage IV MM have a median survival of 6 - 9 months, depending on tumor bulk and location at time of recurrence. Treatment of Stage IV disease has not improved significantly in decades with the current chemotherapy regimens. Dacarbazine, the generally accepted standard, has response rates in Phase III trials of 9.8% - 12% [1]. Overall chemotherapy is disappointing. VEGF is the principle ligand targeted by antiangiogenic therapies. Bevacizumab (Avastin (Roche)), is a humanized monoclonal IgG antibody against VEGF. It has demonstrated anti-angiogenic effects in numerous tumours [2], recognizing all isoforms of VEGF. It is the first antiangiogenic agent to be FDA approved for use (in 2004), and 28 US trials are listed assessing bevacizumab, alone or in different combinations with chemotherapeutic agents for MMtherapy. Currently there are 7 completed clinical trials, with 9 recruiting, 7 in progress and 5 recorded as unknown (www.clinicaltrials.gov).
Initial evaluations of bevacizumab were conducted in 2007 in a Phase II trial as a monotherapeutic agent, or in combination with low dose IFN-a2b (inhibiting FGF). 25% of patients had increased disease stabilization ranging 24 - 146 weeks [3]. In 2009 a Phase II trial combining bevacizumab twice weekly at 10 mg/kg in 53 patients with a regime of paclitaxel/carboplatin in Stage IV unresectable MM, demonstrated disease stabilization in 57% of patients for 8 weeks or more, with median progresssion-free survival of 6 months and overall survival (OS) of 12 months [4]. The BEAM trial (NCT00434252), a randomized multi-center Phase II trial for MM treatment with around 200 patients, examined carboplatin/paclitaxel with or without bevacizumab in patients chemo-, bioor VEGF-naive. With results not statistically significant, bevacizumab still appeared beneficial, improving OS with chemotherapy alone by 21% [5]. See Table 1.
A more recently published multi-centre Phase II single arm trial with treatment of bevacizumab 15 mg/kg every 3 weeks, and fotemustine (100 mg/m2 by intravenous administration on days 1, 8, and 15, repeated after 4 weeks) showed average disease progression time to be 8 months and OS 20.5 months in 20 chemo-naive advanced MM patients. Serum VEGF-A levels were reduced post treatment as well as VEGF-C, VEGFR-1 and VEGFR-2 and overall all 16 measured pro-angiogenic serum markers were significantly reduced post treatment [6].
Fotemustine is of interest, because as a first-line chemotherapeutic, when compared to dacarbazine in a randomized trial in France in 2004 with 229 MM patients, fotemustine showed an overall response rate of 15.5 % vs 6.8% (P = 0.043), and remission time in the subgroup with brain metastases at inclusion of 22.7 months compared to 7.2 months (P = 0.059) [7].
Bevacizumab in a randomized Phase III trial with temozolamide or dacarbazine, yielded a mOS of 7.6 monthsand demonstrated an improved quality of life profile [8]. Results published this year combining temozolamide (150 mg/m2) and bevacizumab (10 mg/kg per 2 weeks) as combination therapy in a Phase II trial [9], demonstrated OS of 12 months vs 9.2 months (mOS 9.6), with mOS interestingly higher in BRAF mutation melanomas. There was a disease stabilization rate of 52%.
Temozolamide is interesting, as it crosses the blood brain barrier and may improve the palliative treatment of cerebral metastases, which drive mortality in stage IV disease [10]. Overall, temozolamide combined with bevacizumab improves the quality of life in end-stage of disease [8].
Given the long established focus on treating advanced metastatic disease which has so far yielded marginal improvements in survival; the UK Adjuvant Avastin Trial in High-Risk Melanoma (AVAST-M) trial has adopted a different approach, and focuses on prevention by inhibitingangiogenesis to disrupt early metastasis.
This trial is a Phase III randomized trial and offers adjuvant therapy to 1320 patients following resection of AJCC stage IIB (T3bN0M0 and T4aN0M0), IIC (T4bN0M0)
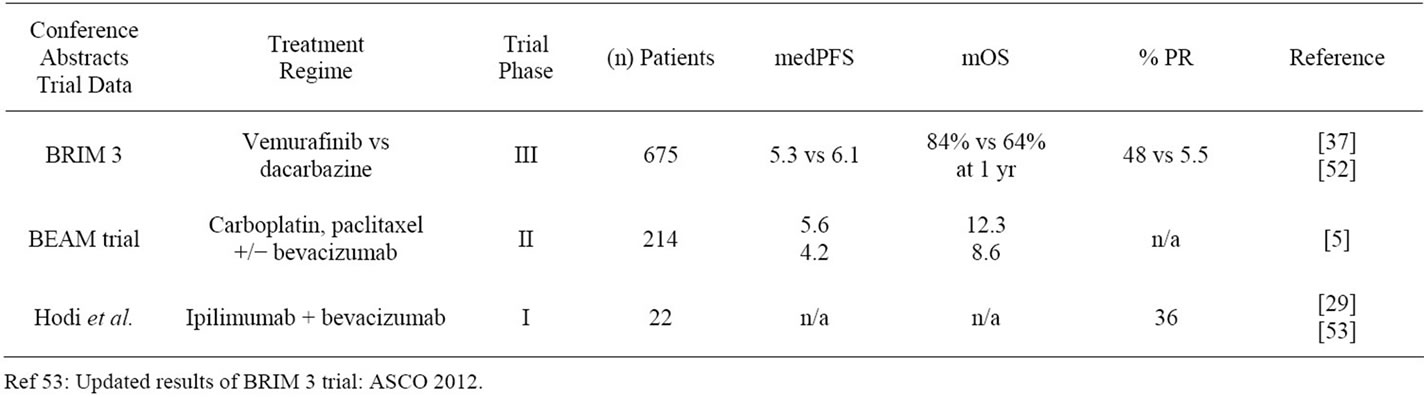
Table 1. American Society of Clinical Oncology (ASCO) conference abstracts.
and III (TxN1-3M0) cutaneous melanoma with either bevacizumab for 1 year or observation. This trial has recently been closed to further patient recruitment, and results are awaited.
In view of the observation that anti-angiogenic splice variants of VEGF appear to be expressed in primary melanomas of lower metastatic potential [11], the AVASTM trial could potentially yield equivocal clinical results. This will be anticipated with interest in view of bevacizumab’s indiscriminate inhibition of both proand antiangiogenic VEGF isoform expressing primary tumours. [12].
2.2. Targeting VEGFR Tyrosine Kinase Receptors
2.2.1. Sorafenib
Sorafenib tosylate (Nexavar, BAY 43-9006), is an orally active multikinase inhibitor, and is the first targeted drug approved for the treatment of advanced renal cell carcinoma (RCC) by the US Food and Drug Administration (FDA) but was first developed as a BRAF inhibitor. Later it was found to have anti-angiogenic properties inhibiting VEGFR in several xenograft models.
Sorafenib functions therapeutically by selective blockade of vascular endothelial growth factor receptors (VEGFR) 3 and 2, and platelet-derived growth factor recaptor β (PDGFR-β) and several other important receptors. [13].
As a monotherapy in BRAF mutant MM patients, sorafenib confers minor benefits [14-16] or no benefit, with a recent Phase II trial publishing a lack of correlation between BRAF mutational status and clinical activity, and minimal disease responses to sorafenib [16]. (see Table 2).
Combination with established agents in Phase III trials has also failed to show benefits with a placebo-controlled trial of 270 patients treated with carboplatin, paclitaxel and sorafenib not improving median free progression survival (mPFS) [15]. A recent Phase II trial combining sorafenib and pegylated interferon-a2b treatment in 55 grade IV MM patients showed modest benefits with partial response (PR) in 3% of patients and mean progresssion free survival time of 2.47 months. Importantly, numerous hematological side-effects were encountered with 1 case of fatal bleeding [17] (see Table 2).
Currently sorafenib and bevacizumab are being assessed (NCT00387751) in a multicentre Phase II trial with 45 patients, with another ongoing study (NCT0053- 8005) combining a Phase I (bevacizumab)/Phase II dose escalation study (oxaliplatin/sorafenib). Temozolamide, which has recently shown improved disease stabilization combined with bevacizumab in a Phase III trial [9] (see Table 2), also showed benefits in combination therapy with sorafenib in a Phase II trial treating patients with advanced MM [18].
Sorafenib’s success in renal cell carcinoma as an antiangiogenic agent rather than BRAF inhibitor suggests there may be viable future roles for sorafenib as part of combination therapy to harness demonstrable anti-angiogenic properties [19].
2.2.2. Axitinib
Axinitib (AGO13736) is a small molecule tyrosine kinase inhibitor, and acts as an inhibitor of VEGFR-1, VEGFR-2 and VEGFR-3. It is being assessed under a Phase III trial for RCC (NCT00678392). An earlier Phase II trial with 32 MM patients demonstrated a median OS of 6.8 months and OR rate of 15.6% [20]. Currently a two-arm trial of axitinib and carboplatin/paclitaxel in MM is measuring the primary outcome Objective

Table 2. Recently published trials data.
Response Rate (ORR) which is measured by radiographic response per 21 day cycles of treatment. One arm is being followed up with PET-CT, CT or MRI scanning and the second arm with additional FLT PET scanning (NCT01174238). This involves the use of theimaging agent 3’-deoxy-3’-[F-18] fluorothymidine, (18-F FLT) acombined radionuclide and analog of thymidine. It is worth noting FLT PET scanning is reported in the literature as poorly discriminatory between non-metastatic reactive nodes and histologically proven nodes yielding a high false positive rate [21]. A 60-patient trial (NCT- 01321437) is projected to start recruiting shortly, looking at axitinib monotherapy in Stage III MM.
2.3. PDGFR TKIs
Imatinib targets platelet derived growth factor receptor (PDGFR), which is identified in playing a role in pericyte recruitment and modulation of autocrine growth of tumour.
Pericytes are required for normal microvascular stability and function; deficiency, as seen in mice lacking PDGF-B and its cognate receptor PDGF-Rβ, promotes a range of microvascular changes, such as endothelial hyperplasia, vessel dilation, tortuosity, leakage, and rupture. This leads to widespread and lethal microhaemorrhaging and oedema at late gestation. Genetic ablation of PDGFB or PDGF-Rβ leads to the formation of microvessels with many of the typical hallmarks of tumor vessels. [22].
Imatinib also targets c-kit. This mutation was observed to occur in 28% of MM arising from chronically sundamaged skin in an array comparative genomic hybridization study [23] and this finding may offer a potential future role for imatinib which as yet, has overall shown little difference in improving survival outcomes when compared to other therapy options. Recently in a Phase II trial with 16 patients, an OS of 3.9 months was achieved with imatinibmonotherapy. Another study is looking at temozolamide with imatinib (NCT00667953) with no published results available.
2.4. Ipilimumab
Recently for the first time in more than two decades of clinical trials of chemotherapy for the treatment of Stage IV metastatic melanoma, two agents, the immunotherapeutic ipilimumab and the kinase inhibitor vemurafinib [24], show improvements in overall survival that represent significant improvements over previous trial regimes. Whilst ipilimumab is not anti-angiogenic in its action, it is of interest having demonstrated synergistic benefit when used as combination therapy with anti-angiogenic drugs.
Ipilimumab demonstrates a survival advantage in advanced MM, achieved by induction of an immune mediated tumor vasculopathy. It is the first Phase III trialed drug that demonstrates improved OS, and median OS of 10 months with 676 enrolled MM patients treated with ipilimumab with or without gp100 protein. Survival analyses showed 1 and 2-year survival rates of 45.6% and 23.5%, respectively [25] (see Table 2) and compared well to recent, randomized, Phase III trials involving patients with unresectable Stage III or IV melanoma who had received previous treatment. 1-year survival rates ranged 22% to 38% with treatment regimens including lenalidomide [26] or sorafenib in combination [15]. The median OS in these studies ranged from 5.9 to 9.7 months. In 2011 a double blinded randomized trial with ipilimumab and dacarbazine with 250 patients in each treatment arm demonstrated an OS of 47.3% at 1 year and 20.3% at 3 years with ipilimumab monotherapy [27]. Further evidence of the benefit of ipilimumab comes from a meta-analysis of 38 trials last year, with ipilimumab demonstrating a superior OS than alternative therapies in group IV MM patients [28]. Recently published data shows ipilimumab and bevacizumab combination therapy in a Phase I trial demonstrates a synergistic effect more beneficial than either drug used alone, with 8 partial responders (8/22) and 6/22 with stabilization of disease [29]. As of 2012, a randomized Phase III trial with Ipilimumab month erapy is underway NCT01515189, with overall survival (OS) and progression free survival (PFS) as primary and secondary endpoints respectively. See Table 1.
2.5. Targeting Multiple Pathways
The BRAF mutation occurs in 50% - 60% of metastatic primary tumours, and arises in the kinase domain resulting in a glutamate substitution for valine at the position 600 (V600E). This activates BRAF, and results in overactivity of the RAS/RAF (MAPK) cascade. This cascade is integral to angiogenesis regulating endothelial cell (EC) survival and proliferation [30] and engages a pathway independent of the hypoxia responsive HIF-a pathways [31]. BRAF mutations demonstrate an inverse relationship between BRAF mutation rate and age and evidence suggests that BRAF mutation genotype varies signifycantly with age. V600E predominates in the young <40 years (80% - 92%), and V600K in older populations (21%). BRAF wild-type melanoma may be also associated with higher BMI in patients <40 yr [32]. Recently a Phase I/II trial commenced using RAF265 whichcombines RAF and VEGFR-kinase inhibition in solid tumours which have a confirmed BRAFV600E positive status (NCT01352273). RAF265 is currently being trialed in melanoma patients with AJCC IIIB locally advanced disease to Stage IV MM in a Phase I/II trial that is ongoing (NCT00304525). First results this year of RAF- 265 from a Phase I study define maximum treatment dose as 48 mg daily with higher doses limited by hematological side-effects [33].
2.6. Vemurafinib
Vemurafenib has been FDA approved for the treatment of unresectable or metastatic BRAFV600E mutated melanoma since August 2011 based on results of the BRIM 3 trial.
Vemurafinib (PLX4032) is an inhibitor of the BRAFV600E mutation. In a Phase I study, high doses gave response rates greater than 50%, providing proof of concept for specifically targeting the BRAFV600E mutation [34]. In a multicenter, phaseI, dose escalation trial with extension phase involving maximum dose, no adverse effects were reported. Patients received vemurafinib twice daily until disease progression. 55 patients (49 MM) were enrolled in the dose-escalation phase, and 32 additional BRAF MM patients with the V600E mutation enrolled in the extension phase. In the dose-escalation cohort, of 16 MM patients with V600E BRAF mutation receiving 240 mg or more of vemurafinib twice daily, 10 partially responded and 1 completely. In the extension cohort, 24/32 had a partial response and 2 had a complete response with an estimated median progression-free survival among all patients of >7 months. Treatment of MM with PLX4032 carrying the V600E BRAF mutation resulted in complete or partial tumor regression in the majority of patients [35]. The same group recently conducted the BRIM 2 trial, a multi-centre single-arm Phase II trial treating 90 Stage IV previously treated BRAFV600E patients with 960 mg bd vemurafinib. With a median progression free survival of 6.7 months the overall response rate (ORR) was 53% with an OS at 12 months of 58%, and with an acceptable toxicity profile at maximum dose reversible with dose modification [36].
BRIM3 is a Phase III multi-centre randomized trial comparing vemurafinib (960 mg bd) with dacarbazine (1 gm/m2) in 675 pre-screened BRAFV600E mutation positive patients with previously untreated Grade IV disease. It is also the first Phase III trial with an alternative single therapy to standard chemotherapy to demonstrate objectively verified improvements in OS (84% at 6 months vs 64%, mPFS (5.3 months vs 1.6 months) and ORR (48.4% vs 5.5%) [37]. Data presented this year has shown improved OS, building on vemurafinibs early encouraging data of 2011 which prompted FDA approval for vemurafinib as a therapy for MM treatment. See Table 1.
Despite vemurafinib’s impressive response rates few patients enter full remission with most demonstrating later disease progression indicating tumor resistance in the longer-term. Recent in vitro investigations established from cell lines cultured from treatment resistant patient MM cells, a reactivation of RAS/RAF pathway with an activating mutation identified in the KRAS gene. Future targets anticipated would combine vemurafinib therapy with MEK and AKT inhibitors as a potential strategy for overcoming treatment resistance [38]. MEK being downstream of BRAF presents another target.
The inhibitor AZD6244 (ARRY-142886) is under investigation in a Phase II double-blind randomized trial as a selective inhibitor in MEK1/2 in 91 BRAF positive MM patients treated AZD6244 and dacarbazine or dacarbazine alone (NCT00936221).
A more focused targeting of therapy using predictive biomarkers for tumour characterization to identify patients able to benefit therapy, is demonstrated by recent findings with dasatinib another multi-target tyrosine kinase inhibitor, with anti-proliferative and anti-invasive effects in vitro with melanoma cell lines. ANXA1, CAV- 1 and EphA2 are biomarkers identified by the research group, from a previously identified 6 gene panel in a panel of 8 melanoma cell lines that have been shown to correlate with a positive therapeutic response to dasatinib. Immunohistochemical analysis in 12% of melanomas (13/112) showed high levels of staining for these three markers and it is suggested that tumours expressing high levels of these biomarkers may be responsive to dasatinibtreatment [39].
Importantly it illustrates the benefits of predictive biomarkers which are likely to become a routine part of clinical strategy decision making as the move towards “personalised medicine” increasingly occurs.
2.7. Targeting the HIF Pathway
Given the importance of the HIF axis in hypoxic induced VEGF synthesis, mTOR-containing complexes, present another potential target for therapeutic exploitation, being important for HIF synthesis. The rapamycin analogue RAD001 (everolimus) is an mTOR inhibitor that has undergone Phase II trials in MM with modest benefits demonstrating a PFS of 3 months and a decrease in VEGF serum levels [40]. mTOR still remains a subject of interest in future MM treatment regimes, as it is clear that the HIF axis is important as part of a tumours adaptive response to intratumoural hypoxia.
2.8. MMP Inhibitors
Several trials using MMP inhibitors such as marimastat or batimastat trialled in a range of metastatic tumours failed to translate encouraging preclinical trial data into clinically beneficial therapies, either as monoor combination therapy and demonstrated in Phase II and III trials, side-effects limiting their applicability [41]. These trials conducted during the late nineties were without the benefit of later knowledge concerning the more complex role played by MMPs. MMP-8 is a case in point. Its role as a tumour suppressor was later identified in melanoma. When wild type and mutated forms were compared, the wild-type forms demonstrated tumour suppressive effects in vitro and in vivo. This was absent in the mutant form identified in 23% of melanoma cell lines examined [42]. Currently no Phases II/III trials are utilizing MMP inhibitors, but future trials will likely clarify a more precise and targeted therapy in place of previous blanket therapy approaches.
2.9. Immune Modulation
Melanoma is a highly immunogenic tumour as evidenced by the small number of advanced cases of MM demonstrating complete remission with chemotherapy and primary melanomas can also undergo spontaneous regression in response to an immune mediated process. Previous investigations have not yet established a clear cutrole for interleukin (IL) treatment, although IL-2 and IL-8 have been extensively investigated as therapies for MM. Interferon is a case in point being immunomodulatory and anti-angiogenic, with effects against tumour cell biology (VEGF, B-FGF, IL-8 production) and endothelial cell behaviour (motility) [43], and is demonstrated experimentally to inhibit tumour growth in in vivo mouse models [44]. It is reasonable to suggest that interferon therapy may in a future role constitute a component of combination therapy as part of a multi-pronged anti-tumour strategy targeting multiple tumour angiogenic pathways simultaneously.
3. Discussion
Advanced MM has long had a poor prognosis as evidenced by the failure of any therapy in the last 20 years to demonstrate successful results in Phase II/III trials extending clinical disease progress markers such as OS well beyond a year, nor does the cure rate give hope for metastatic melanoma being treatable but by palliation.
Many aspects of tumour angiogenesis have demonstrated a responsiveness to anti-angiogenic therapies in a wide range of in vivo and in vitro experiments, and several pathways of angiogenic activity and cell-environment angiogenic interactions have been elucidated, with candidate VEGF receptors offering promising targets. However this has largely failed in respect of MM therapy to translate into more than modest gains in clinical trials. Clearly, tumour angiogenesis has a variety of pathways that equip a tumour with adaptability to counter antiangiogenic therapy, and this may explain the failure of therapies targeting single axes to make long-term gains.
Standard MM therapy has largely concentrated on single chemotherapies with dacarbazine the mainstay of therapy, against which new agents have been typically trialed to assess their monotherapeutic efficacy. In Phase I trials the purpose is to establish maximal tolerated dose (MTD), and in Phase II the highest tolerated dose and this sequential approach is well established historically. However itis a blunt tool for addressing the multifactorial complexities of the angiogenic process. Furthermorethese processes are poor at identifying the potentially synergistic benefit of a given drug as stringent criteria prevent many agents from ever making it beyond Phase II trials to be able to be considered for polytherapy. Licensing is further restrictive, being only given to drugs that demonstrate single agent safety and efficacy in Phase I trials for a specific clinical indication, thereby restricting the range of applications the drug may be used for.Many potentially useful drugs are therefore eliminated at the Phase I stage, with the consequence that potentialuse in combination therapy is lost. The typical goal of a randomized Phase II trial comparing regimes is intended to identify potentially successful agents for a Phase III investigation, rather than providing definitive information on efficacy per se and therefore it is clear that many potentially invaluable agents face serious obstacles to ever reaching Phase III trials.
Another issue of trials is the timing and duration of treatment, the benefits of which are gradually lost by disease progression in MM. Different strategies are being explored with continuous low dose (metronomic) chemotherapy as one possible strategy for maintaining suppression of tumour angiogenesis [45] and the concept of ongoing suppressive anti-angiogenic treatment may be an achievable alternative to the aim of total disease eradication. Another approach to treating MM could involve dose scheduling, with changes to treatment regime timed for maximal efficacy [46].
We may look to a strategy whereby therapies are intelligently escalated at time points where chemo-resistance is known to likely occur, thus staying one step ahead of progressive disease. Here then, a role for reliable markers of tumour angiogenic bioactivity and response to treatment should improve the decision making process of drug therapy making possible the concept of a treatment adapted and evolving to keep up with, and treat, an evolving disease process. In other words, trials of the future may be heading towards a personalised therapeutic strategy. As a picture of angiogenesis emerges revealing a complex interaction of melanoma cell, environment and immune system regulation/dysregulation, future therapies will likely develop an anti-tumour strategy that also counters the pro-angiogenic environment surrounding a tumour, and the pro-angiogenic mediators of inflammation such as macrophages [47-49], which will involve combination therapies focused at disrupting tumor and metastases, tumor environment and tumor interaction with the haemopoietic system simultaneously [50]. A multi-pronged approach tailoring therapy to target specific mutations such as BRAF heralds the advent of more targeted and personalized medicine, but also provides impetus for a paradigm shift in thinking to the idea of MM as a disease with diverse aetiology and underlying molecular mechanisms driving the process.
Angiogenesis is clearly a “hallmark of cancer” [51], but as mono-therapies have shown therapeutic limitations, so too focusing on a single “hallmark” of cancer may also be too limited. Combination therapies incorporating anti-angiogenic, cytotoxics and immunogenic strategies likely represent the therapeutic future. Anti-angiogenic therapy however, is likely to be an established part of future therapies for metastatic melanoma being shown by many trials to be safe with readily manageable side effects.
REFERENCES
- M. R. Middleton, et al., “Randomized Phase III Study of Temozolomide versus Dacarbazine in the Treatment of Patients with Advanced Metastatic Malignant Melanoma,” Journal of Clinical Oncology, Vol. 18, No. 1, 2000, pp. 158-166.
- C. G. Willett, et al., “Direct Evidence That the VEGFSpecific Antibody Bevacizumab Has Antivascular Effects in Human Rectal Cancer,” Nature Medicine, Vol. 10, No. 2, 2004, pp. 145-147. doi:10.1038/nm988
- K. A. Varker, et al., “A Randomized Phase 2 Trial of Bevacizumab with or without Daily Low-Dose Interferon Alfa-2b in Metastatic Malignant Melanoma,” Annals of Surgical Oncology, Vol. 14, No. 8, 2007, pp. 2367-2376. doi:10.1245/s10434-007-9389-5
- D. G. Perez, et al., “Phase 2 Trial of Carboplatin, Weekly Paclitaxel, and Biweekly Bevacizumab in Patients with Unresectable Stage IV Melanoma: A North Central Cancer Treatment Group Study, N047A,” Cancer, Vol. 115, No. 1, 2009, pp. 119-127. doi:10.1002/cncr.23987
- S. J. O’Day, J. A. Sosman, et al., “BEAM: A Randomised Phase II Study Evaluating the Activity of Bevacizumab in Combination with Carboplatin plus Paclitaxel in Patients with Previously Untreated Advanced Melanoma., in ECCO-ESMO European Cancer Congress,” Cancer, Vol. 7, No. 3, 2009, p. 23.
- M. Del Vecchio, et al., “Bevacizumab plus Fotemustine as First-Line Treatment in Metastatic Melanoma Patients: Clinical Activity and Modulation of Angiogenesis and Lymphangiogenesis Factors,” Clinical Cancer Research, Vol. 16, No. 23, 2010, pp. 5862-5872. doi:10.1158/1078-0432.CCR-10-2363
- M. F. Avril, et al., “Fotemustine Compared with Dacarbazine in Patients with disseminated malignant melanoma: A Phase III Study,” Journal of Clinical Oncology, Vol. 22, No. 6, 2004, pp. 1118-1125. doi:10.1200/JCO.2004.04.165
- K. A. Varker, et al., “A Randomized Phase 2 Trial of bevacizumab with or without Daily Low-Dose Interferon Alfa-2b in Metastatic Malignant Melanoma,” Annals of Surgical Oncology, Vol. 14, No. 8, 2007, pp. 2367-2376. doi:10.1245/s10434-007-9389-5
- R. Von Moos, et al., “First-Line Temozolomide Combined with Bevacizumab in Metastatic Melanoma: A Multicentre Phase II Trial (SAKK 50/07),” Annals of Oncology, Vol. 23, No. 2, 2011, pp. 531-536.
- S. S. Agarwala, J. M. Kirkwood, M. Gore, B. Dreno, N. Thatcher, B. Czarnetski, M. Atkins, A. Buzaid, D. Skarlos and E. M. Rankin, “Temozolomide for the Treatment of Brain Metastases Associated with Metastatic Melanoma: A Phase II Study,” Journal of Clinical Oncology, Vol. 22, No. 11, 2004, pp. 2101-2107. doi:10.1200/JCO.2004.11.044
- R. O. Pritchard-Jones, et al., “Expression of VEGFxxxb, the Inhibitory Isoforms of VEGF, in Malignant Melanoma,” British Journal of Cancer, Vol. 97, No. 2, 2007, pp. 223-230. doi:10.1038/sj.bjc.6603839
- A. H. R. Varey, et al., “VEGF165b, an Antiangiogenic VEGF-A Isoform, Binds and Inhibits Bevacizumab Treatment in Experimental Colorectal Carcinoma: Balance of Proand Antiangiogenic VEGF-A Isoforms Has Implications for Therapy,” British Journal of Cancer, Vol. 98, No. 8, 2008, pp. 1366-1379. doi:10.1038/sj.bjc.6604308
- S. M. Wilhelm, C. Carter, L. Tang, et al., “BAY43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis,” Cancer Research, Vol. 64, 2004, pp. 7099- 7109. doi:10.1158/0008-5472.CAN-04-1443
- T. Eisen, et al., “Sorafenib in Advanced Melanoma: A Phase II Randomised Discontinuation Trial Analysis,” British Journal of Cancer, Vol. 95, No. 5, 2006, pp. 581- 586. doi:10.1038/sj.bjc.6603291
- A. Hauschild, et al., “Results of a Phase III, Randomized, Placebo-Controlled Study of Sorafenib in Combination with Carboplatin and Paclitaxel as Second-Line Treatment in Patients with Unresectable Stage III or Stage IV Melanoma,” Journal of Clinical Oncology, Vol. 27, No. 17, 2009, pp. 2823-2830. doi:10.1200/JCO.2007.15.7636
- P. A. Ott, et al., “A Phase II Trial of Sorafenib in Metastatic Melanoma with Tissue Correlates to Surgical Approach to Primary Cutaneous Melanoma Mucosal Melanomas: A Case-Based Review of the Literature A Phase II Trial of the Epothilone B Analog Ixabepilone (BMS- 247550) in Patients with Metastatic Melanoma,” PLoS One, Vol. 5, No. 12, 2010, p. 15588. doi:10.1371/journal.pone.0015588
- F. Egberts, et al., “Sorafenib and Pegylated Interferon- {alpha}2b in Advanced Metastatic Melanoma: A Multicenter Phase II DeCOG Trial,” Annals of Oncology, Vol. 10, 2011, p. 10.
- R. Amaravadi, D. F. Mc Dermott, et al., “Updated Results of a Randomised Phase II Study Comparing Two Schedules of Temozolamide in Combination with Sorafenib in Patients with Advanced Melanoma,” Journal of Clinical Oncology, Vol. 25, No. 18s, 2007, p. 8527.
- B. Escudier, T. Eisen, W. M. Stadier, et al., “Sorafenib for Treatment of Renal Cell Carcinoma: Final Efficacy and Safety Results of the Phase III Treatment Approaches in Renal Cancer Global Evaluation Trial,” Journal of Clinical Oncology, Vol. 27, 2009, pp. 3312-3318. doi:10.1200/JCO.2008.19.5511
- J. P. Fruehauf, J. Lutzky and D. F. McDermott, “Axitinib (AG-013736) in Patients with Metastatic Melanoma: A Phase II Study,” Journal of Clinical Oncology, Vol. 26, 2008, p. 9006.
- E. G. Troost, et al., “18F-FLT PET Does not Discriminate between Reactive and Metastatic Lymph Nodes in Primary Head and Neck Cancer Patients,” Journal of Nuclear Medicine, Vol. 48, No. 5, 2007, pp. 726-735. doi:10.2967/jnumed.106.037473
- A. Abramsson, P. Lindblom and C. Betsholtz, “Endothelial and Nonendothelial Sources of PDGF-B Regulate Pericyte Recruitment and Influence Vascular Pattern Formation in Tumors,” Journal of Clinical Investigation, Vol. 112, No. 8, 2003, pp. 1142-1151.
- J. C. Becker, E. B. Bröcker, D. Schadendorf and S. Ugurel, “Imatinib in Melanoma: A Selective Treatment Option Based on KIT Mutation Status?” Journal of Clinical Oncology, Vol. 25, No. 7, 2007, p. 9.
- K. T. Flaherty, et al., “Inhibition of Mutated, Activated BRAF in Metastatic Melanoma,” The New England Journal of Medicine, Vol. 363, No. 9, 2010, pp. 809-819. doi:10.1056/NEJMoa1002011
- F. S. Hodi, et al., “Improved Survival with Ipilimumab in Patients with Metastatic Melanoma,” The New England Journal of Medicine, Vol. 363, No. 8, 2010, pp. 711-723. doi:10.1056/NEJMoa1003466
- T. Eisen, et al., “Results of a Multicenter, Randomized, Double-Blind Phase 2/3 Study of Lenalidomide in the Treatment of Pretreated Relapsed or Refractory Metastatic Malignant Melanoma,” Cancer, Vol. 116, No. 1, 2010, pp. 146-154.
- D. Wolchok, I. N. Bondarenko, S. O’Day, J. S. Weber, C. Garbe, S. Francis, R. A. Ibrahim, A. Hoos, C. Robert, “Phase III Randomized Study of Ipilimumab (IPI) plus Dacarbazine (DTIC) versus DTIC Alone as First-Line Treatment in Patients with Unresectable Stage III or IV Melanoma,” Journal of Clinical Oncology, Vol. 29, 2011.
- S. Kotapati, M. Ouwens, M. van Baardewijk, R. A. Ibrahim, S. Wagner and J. P. Jansen, “Overall Survival (OS) in the Management of Pretreated Patients with Unresectable Stage III/IV Melanoma: A systematic Literature Review and Meta-Analysis,” Journal of Clinical Oncology, Vol. 29, 2011.
- S. Hodi, M. B. Atkins, D. F. McDermott, D. P. Lawrence, N. Ibrahim, X. Wu, J. Zhou, A. Giobbie-Hurder, G. Murphy, T. Hollman, E. Velazquez, S. Russell, P. Dipiro, J. T. Yap and A. D. Van Den Abbeele, “A Phase I Trial of Ipilimumab plus bevacizumab in patients with Unresectable Stage III or Stage IV Melanoma,” Journal of Clinical Oncology, Vol. 29, 2011.
- H. Davies, et al., “Mutations of the BRAF Gene in Human Cancer,” Nature, Vol. 417, No. 6892, 2002, pp. 949- 954. doi:10.1038/nature00766
- Y. Mizukami, Y. Kohgo and D. C. Chung, “Hypoxia Inducible Factor-1 Independent Pathways in Tumor Angiogenesis,” Clinical Cancer Research, Vol. 13, No. 19, 2007, pp. 5670-5674. doi:10.1158/1078-0432.CCR-07-0111
- A. M. Menzies, M. D. Chatfield, M. S. Carlino, J. R. Howle, R. A. Scolyer, J. F. Thompson, R. F. Kefford and G. V. Long, “BRAF Mutation by Age-Decade and Body Mass Index in Metastatic Melanoma,” Journal of Clinical Oncology, Vol. 29, 2011.
- W. H. Sharfman, D. P. Lawrence, K. T. Flaherty, R. K. Amaravadi, K. B. Kim, R. Dummer, S. Gobbi, I. Puzanov, J. A. Sosman, K. Dohoney, L. P. Lam, S. Kakar, Z. Tang, O. Krieter and M. B. Atkins, “Results from the First-inHuman (FIH) Phase I Study of the Oral RAF inhibitor RAF265 Administered Daily to Patients with Advanced Cutaneous Melanoma,” Journal of Clinical Oncology, Vol. 29, 2011.
- K. T. Flaherty, H. Sosman, et al., “Phase I Study of PLX4032: Proof of Concept for V600E BRAF Mutation as a Therapeutic Target in Human Cancer,” Journal of Clinical Oncology, Vol. 27, No. 15S, 2009.
- P. B. Chapman, A. Hauschild, C. Robert, J. B. Haanen, P. Ascierto, J. Larkin, R. Dummer, C. Garbe, A. Testori, M. Maio, D. Hogg, P. Lorigan, C. Lebbe, T. Jouary , D. Schadendorf, A. Ribas, S. J. O’Day, J. A. Sosman, J. M. Kirkwood, A. M. Eggermont, B. Dreno, K. Nolop, J. Li, B. Nelson, J. Hou, R. J. Lee, K. T. Flaherty and A. G. McArthur, “Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation,” New England Journal of Medicine, Vol. 364, 2011, pp. 2507-2516. doi:10.1056/NEJMoa1103782
- A. Ribas, “An Open-Label, Multicentre Phase II Study of Vemurafenib (PLX4032, RG7204) in Previously Treated Patients with BRAFV600E Mutation Positive Metastatic Melanoma,” Journal of Clinical Oncology, Vol. 29, 2011.
- P. B. Chapman, et al., “Phase III Randomized, OpenLabel, Multicenter Trial (BRIM3) Comparing BRAF Inhibitor Vemurafenib with Dacarbazine in Patients with BRAFV600E-Mutated Melanoma,” Journal of Clinical Oncology, Vol. 29, 2011.
- F. Su, H. Y. B. Higgins, K. D. Kolinsky, K. Packman, M. Kim, B. J. Lestini, G. Bollag and D. C. Heimbrook, “Molecular Mechanisms Underlying Disease Relapse on Treatment with Selective BRAF Inhibitor Vemurafenib (PLX4032),” Journal of Clinical Oncology, Vol. 29, 2011.
- A. J. Eustace, S. K. A. Larkin, T. Mahgoub, D. Tryfonopoulos, L. O’Driscoll, M. Clynes, J. Crown and N. O’Donovan, “Identification of Predictive Biomarkers for Dasatinib Treatment Of Metastatic Melanoma,” Journal of Clinical Oncology, Vol. 29, 2011.
- R. D. Rao, J. B. Allred, et al., “Phase II Trial of the mTOR Inhibitor Everolimus (RAD-001) in Metastatic Melanoma,” Journal of Clinical Oncology, Vol. 24, No. 18S, 2006.
- I. Quirt, et al., “Phase II Study of Marimastat (BB-2516) in Malignant Melanoma: A Clinical and Tumor Biopsy Study of the National Cancer Institute of Canada Clinical Trials Group,” Investigational New Drugs, Vol. 20, No. 4, 2002, pp. 431-437. doi:10.1023/A:1020625423524
- L. H. Palavalli, et al., “Analysis of the Matrix Metalloproteinase Family Reveals that MMP8 Is Often Mutated in Melanoma,” Nature Genetics, Vol. 41, No. 5, 2009, pp. 518-520. doi:10.1038/ng.340
- D. Brouty-Boye and B. R. Zetter, “Inhibition of Cell Motility by Interferon,” Science, Vol. 208, No. 4443, 1980, pp. 516-518. doi:10.1126/science.6154315
- C. P. Dinney, et al., “Inhibition of Basic Fibroblast Growth Factor Expression, Angiogenesis, and Growth of Human Bladder Carcinoma in Mice by Systemic Interferon-Alpha Administration,” Cancer Research, Vol. 58, No. 4, 1998, pp. 808-814.
- J. Drevs, et al., “Antiangiogenic Potency of Various Chemotherapeutic Drugs for Metronomic Chemotherapy,” Anticancer Research, Vol. 24, No. 3a, 2004, pp. 1759- 1763.
- S. J. O’Day, M. B. Atkins, P. Boasberg, H. J. Wang, J. A. Thompson, C. M. Anderson, R. Gonzalez, J. Lutzky, T. Amatruda, E. M. Hersh and J. S Weber, “Phase II Multicenter Trial of Maintenance Biotherapy after Induction Concurrent Biochemotherapy for Patients with Metastatic Melanoma,” Journal of Clinical Oncology, Vol. 27, No. 36, 2009, pp. 6207-6212. doi:10.1200/JCO.2008.20.3075
- J. M. Pawelek and A. K. Chakraborty, “The Cancer CellLeukocyte Fusion Theory of Metastasis,” Advances in Cancer Research, Vol. 101, 2008, pp. 397-444. doi:10.1016/S0065-230X(08)00410-7
- J. Pawelek, et al., “Co-Opting Macrophage Traits in Cancer Progression: A Consequence of Tumor Cell Fusion?” Contrib Microbiol, 2006, Vol. 13, pp. 138-155. doi:10.1159/000092970
- J. M. Pawelek, “Tumour-Cell Fusion as a Source of Myeloid Traits in Cancer,” The Lancet Oncology, Vol. 6, No. 12, 2005, pp. 988-993. doi:10.1016/S1470-2045(05)70466-6
- J. M. Pawelek, “Viewing Malignant Melanoma Cells as Macrophage-Tumor Hybrids,” Cell Adhesion & Migration, Vol. 1, No. 1, 2007, pp. 2-6.
- D. Hanahan and R. A. Weinberg, “The Hallmarks of Cancer,” Cell, Vol. 100, No. 1, 2000, pp. 57-70. doi:10.1016/S0092-8674(00)81683-9
- J. Chapman, et al., “Updated Overall Survival (OS) Results for BRIM-3, a Phase III Randomized, Open-Label, Multicenter Trial Comparing BRAF Inhibitor Vemurafenib (vem) with Dacarbazine (DTIC) in Previously Untreated Patients with BRAFV600E-Mutated Melanoma,” Journal of Clinical Oncology, Vol. 30, 2012. doi:10.1056/NEJMoa1003466
- F. S. Hodi, et al., “Improved Survival with Ipilimumab in Patients with Metastatic Melanoma,” The New England Journal of Medicine, Vol. 363, No. 8, 2010, pp. 711-723. doi:10.1056/NEJMoa1003466
NOTES
*Corresponding author.