Journal of Cancer Therapy
Vol.2 No.5(2011), Article ID:16613,8 pages DOI:10.4236/jct.2011.25090
Molecular Markers for the Prediction of Anti-EGFR Monoclonal Antibody Treatment Efficacy in Metastatic Colorectal Cancer
![]()
1The First Department of Oncology, Shengjing Hospital of the China Medical University, Shenyang, China; 2College of Basic Science, China Medical University, Shenyang, China.
E-mail: *hancb@126.com
Received October 20th, 2011; revised November 21st, 2011; accepted December 5th, 2011.
Keywords: molecular marker, epidermal growth factor receptor, Kras, EGFR monoclonal antibody, metastatic colorectal cancer
ABSTRACT
The implementation of individualized targeted therapy for metastatic colorectal cancer (mCRC), in addition to standard chemotherapeutic regimens, currently is a topic under debate. Approximately 35% - 45% of mCRC patients exhibit mutated KRAS, which is considered to be an independent predictor of poor response to treatment with epidermal growth factor receptor (EGFR) monoclonal antibody. However, only about 50% of patients with wild-type KRAS respond to anti-EGFR therapy. Two major EGFR-dependent signaling pathways, RAS-RAF-MAPK and PI3K-PTENAKT, may be involved in the poor response to anti-EGFR. Increased EGFR gene copy number as detected by fluorescence in situ hybridization, but not increased EGFR protein expression, correlates with efficacy of anti-EGFR treatment. The identification of mutations in BRAF and PIK3CA (exon 20) and deletions in PTEN also may help clinicians screen for anti-EGFR resistance in mCRC patients with wild-type KRAS. To guide health professionals through the realm of individualized targeted therapies for mCRC, we review recent progress on identifying negative predictors and prognostic markers of anti-EGFR treatment efficacy.
1. Introduction
Combination chemotherapy with irinotecan or oxaliplatin and fluorouracil or its derivatives is the primary treatment for recurrent or metastatic colorectal cancer (mCRC). Despite an improved efficacy compared to chemotherapy alone, the median survival time under this regimen is still less than 2 years [1,2]. Molecularly targeted drug therapy has demonstrated encouraging results and is now regarded as a standard treatment for mCRC. Epidermal growth factor receptor (EGFR, HER1, and ErbB1) is one of the most commonly targeted components in mCRC therapy. Clinical application of anti-EGFR monoclonal antibodies (anti-EGFR) such as cetuximab and panitumumab extends the median survival time for mCRC patients beyond 2 years [3,4]. However, several studies have shown that the efficacy of cetuximab and panitumumab monotherapy for mCRC patients who had been previously treated with chemotherapy is only around 10%. The efficacy is marginally improved by 8% - 10% in combination with first-line chemotherapy (FOLFOX/ FOLFIRI regimen), when compared to conventional chemotherapy [5,6]. The current challenge is the individualization of targeted anti-EGFR therapy for mCRC. Mutations in the EGFR gene are poor predictors for its clinical efficacy. In contrast, genetic variations in the two EGFR-dependent signaling pathways (RAS-RAF-MAPK and PI3K-PTEN-AKT) may reveal more information for predicting the clinical efficacy of anti-EGFR therapies [7,8].
2. EGFR
EGFR (HER1/ErbB1) is a transmembrane glycoprotein in the HER/ErbB receptor tyrosine kinase family, which consists of four members. EGFR is activated by ligand binding and then forms homologous or heterologous dimers with other monomers in the HER/ErbB family. Upon dimerization, EGFR undergoes a conformational change, activates its intrinsic tyrosine kinase activity, and autophosphorylates the tyrosine residue at its C-terminus. The phosphorylated tyrosine becomes a binding site for SH2 or PTB domain-containing signaling molecules such as growth factor receptor-bound protein (GRB2) and phosphatidylinositol-3,4,5-triphosphate (PI3K), thereby initiationg a cascade of intracellular signal transduction pathways [9]. Among the activated pathways, the two major EGFR-dependent signaling pathways (RAS-RAFMAPK and PI3K-PTEN-AKT), and interactions between these two, play important roles in tumor growth, invasion, and metastasis. Therefore, the RAS-RAF-MAPK and PI3K-PTEN-AKT signaling pathways are regarded as potential molecular targets for anti-tumor therapies [10] (Figure 1).
2.1. EGFR Gene Copy Number
Studies have shown that increased EGFR gene copy number (GCN) was positively associated with efficacy of colon cancer therapies [11-14]. Moroni et al. [15] reported that in patients responding well to anti-EGFR treatment, tumor growth was mainly driven by the EGFR signaling pathway, which could be evoked by increased EGFR GCN. In patients with high EGFR GCN but poor responses to anti-EGFR treatment, it is possible that downstream signaling pathways had been constitutively activated, for instance, via mutations in KRAS/BRAF/ PIK3CA oncogenes or deletions in the tumor suppressor gene, PTEN. More than 70% of cases involving increased EGFR GCN are accompanied by wild-type KRAS, and in the KRAS wild-type subgroup, the EGFR fluorescence in situ hybridization (FISH) or automated silver in situ hybridization (SISH)-positive phenotype was associated with a higher response rate (>70%) than in tumors with normal EGFR GCN [16,17]. Scartozzi et al. [14] found that in mCRC patients with wild-type KRAS and increased increased EGFR GCN who have not responded to irinotecan treatment, continuous combination therapy with cetuximab could achieve an efficacy of 60%. However, the efficacy in patients without increased EGFR GCN was only 9%, suggesting that EGFR GCN can be used to predict the most optimal treatments for mCRC patients with wild-type KRAS. In addition, most studies trying to detect EGFR GCN by quantitative PCR yielded negative results. This may result from dilution of the tumor DNA by normal tissue DNA in the process of DNA extraction. To avoid this, the use of FISH or chromogenic in situ hybridization (CISH) to detect EGFR GCN is recommended.
2.2. EGFR Protein Expression and Gene Mutation
From 25% to 80% of CRC patients concurrently exhibit EGFR overexpression, which appears to be unrelated to gene amplification [7,18,19]. EGFR and phospho-EGFR expression levels are associated with tumor cell hyperproliferation. Phospho-EGFR was not prognostic, whereas increased EGFR immunohistochemistry intensity was independently associated with poor disease-free survival [20]. Although anti-EGFR acts on EGFR located on the outer cell membrane, the expression level of EGFR does not correlate with the efficacy of anti-EGFR therapy, which was only about 10% in monotherapy of cetuximab [21,22]. In addition, many patients with low or even undetectable expression of EGFR respond well to antiEGFR therapy [23]. Chung et al. [24] reported that after failed irinotecan treatment, anti-EGFR therapy was effective in 25% (4/16 cases) of EGFR-negative mCRC patients. Thus, EGFR overexpression may not be a reliable predictor of anti-EGFR treatment efficacy.
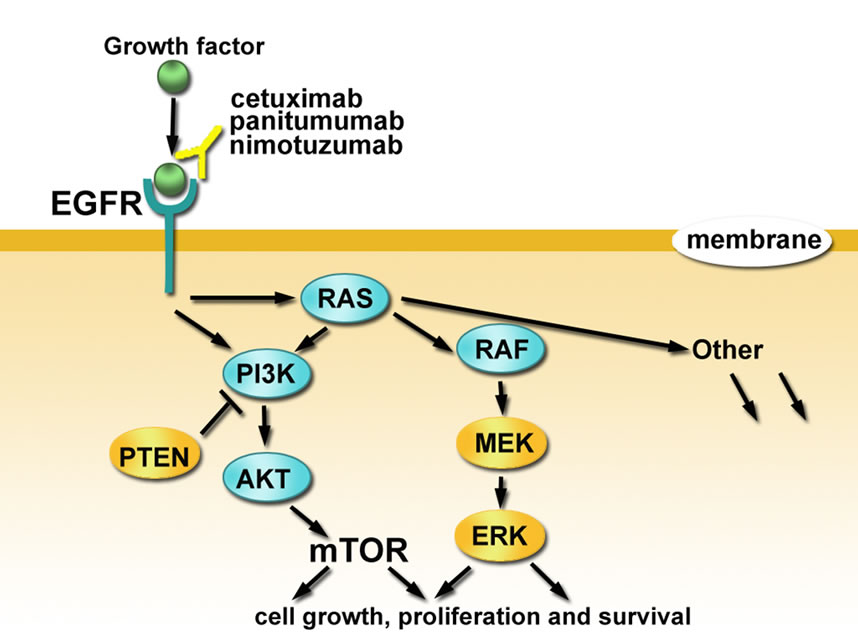
Figure 1. Schematic representation of molecular markers for predicting anti-EGFR treatment efficacies in mCRC patients.
The incidence of somatic mutations of EGFR in advanced colorectal cancer is less than 1%. These mutations are independent of K-Ras mutation [19], and not associated with cetuximab and panitumumab treatment responses [25,26]. Therefore, the validity of using EGFR gene status as a predictor of mCRC prognosis currently is debatable, and EGFR mutations may not be suitable indicators for survival and prognosis [20,27]. As an IgG1 antibody, cetuximab may exert its antitumor efficacy through both EGFR antagonism and antibody-dependent cell-mediated cytotoxicity (ADCC) [28]. ADCC effectiveness against tumors is dependent on antibody glycosylation, IgG1 isotype, and EGFR expression by tumor cells [29]. Thus, ADCC may play a significant role in targeted therapy.
3. RAS-RAF-MAPK Signaling Pathway
After autophosphorylation of the intracellular tyrosine kinase domain on EGFR, the signal is transduced to Ras, a small guanosine triphosphate (GTP) binding protein, via the adaptor protein GRB2 and the guanine nucleotide exchange factor SOS (son of sevenless). Ras then activates several key molecules of the RAS-RAF-MAPK signal cascade, such as RAF, MEK1/2, and Erk to control cell growth, differentiation, and apoptosis. Previous studies have demonstrated that the RAS-RAF-MAPK signaling pathway is a primary contributor to the antiEGFR treatment response in mCRC patients [7,10].
3.1. KRAS Mutation
KRAS executes signal transduction between several extracellular ligands and intracellular molecules. Approximately 35% - 45% of mCRC patients also exhibit KRAS mutations, and unlike correlations detected among nonsmall cell lung cancer patients, the mutation rate of the KRAS gene in mCRC is unassociated with race [3,30,31]. The KRAS activating mutations are considered to be an independent predictor of poor anti-EGFR treatment efficacy.
Currently, the efficacy of cetuximab and panitumumab monotherapy in chemotherapy-resistant patients with unknown KRAS gene status has been 8% - 10% compared with 0% for best supportive care [5,6]. The efficacy of panitumumab monotherapy for chemotherapy-resistant mCRC patients exhibiting the wild-type KRAS gene is 10% - 17% compared with 0% for patients with KRAS mutations [3,6]. The efficacy of cetuximab is 12.8% for patients with wild-type KRAS and 1.2% for patients with KRAS mutations [30]. Cetuximab combined with firstline chemotherapy achieves efficacies of 57% - 61% in mCRC patients with wild-type KRAS and 33% - 43% in patients with unknown KRAS status. In contrast, the efficacy for mCRC patients with mutations in KRAS was only 0% - 6% [4,32]. An update of the CRYSTAL and OPUS meta-analyses [4] reported that among mCRC patients with wild-type KRAS, the addition of cetuximab to standard first-line chemotherapy could significantly prolong the overall survival (OS) of patients (23.5 months vs. 19.5 months; hazard ratio [HR] = 0.81, P = 0.0062). Additionally, using highly sensitive methods for the detection of KRAS mutations significantly improves the identification of mCRC patients resistant to anti-EGFR therapy [33].
It is not clear whether mutated KRAS is a reliable prognostic marker. A retrospective study by Andreyev et al. [34] analyzed 2721 cases of colorectal cancer and observed an association between KRAS mutations and an increased risk of death from colorectal cancer. However, Karapetis et al. [30] compared cetuximab to the best supportive care for the treatment of mCRC in a prospective phase III clinical trial and found that KRAS gene mutation did not increase the risk of death in patients treated with best supportive care, suggesting that KRAS mutation may not be a reliable indicator of poor mCRC prognosis.
The incidence of NRAS oncogene mutation is less than 5%. De Roock et al. [35] reported that in patients with wild-type and mutant NRAS, the response rates after cetuximab therapy were 7.7% and 38.1%, respectively (OR = 0.14, P = 0.013). Since NRAS is almost always mutually exclusive with KRAS and BRAF mutations, addition of NRAS may help to further screen antiEGFR-resistant mCRC patients with wild-type KRAS and BRAF.
3.2. BRAF Mutation
Approximately 6% - 10% of mCRC patients exhibit BRAF gene mutations; in several studies, these are associated with cetuximab resistance. Therefore, BRAF mutation may be an independent predictor of cetuximab efficacy [36-39]. Di Nicolantonio et al. [37] reported that 9.7% (11/113) of mCRC patients exhibited wild-type KRAS and mutant-type BRAF (KRAS WT/BRAF MT), whereas none of these 11 patients responded effectively to targeted therapy. In another study, Di Nicolantonio et al. found that 93.75% (15/16) of patients with BRAF mutations were resistant to cetuximab treatment. In the CAIRO2 [39] trial, 559 mCRC patients were screened for mutated BRAF. The results suggested that regardless of cetuximab application, the progression-free survival (PFS) and OS of KRAS WT/BRAF WT patients was significantly improved as compared with the KRAS WT /BRAF MT patients. This indicates that BRAF mutation may be a useful prognostic factor.
Studies have found that mutations in KRAS and BRAF were mutually exclusive [40,41], so 45% - 55% of mCRC patients might avoid unnecessary anti-EGFR therapy through mutation screening in these two genes. The 2010 version of the national cancer comprehensive network (NCCN) guidelines recommends further testing for BRAF mutation in patients with wild-type KRAS. In addition, the 2010 ASCO (American society of clinical oncology) update of the CRYSTAL trial [4,42] showed that cetuximab combined with chemotherapy did not significantly improve the treatment efficacy of mCRC patients who were KRAS WT/BRAF MT. The HRs for OS and PFS were 0.62 (95% CI 0.36 - 1.06) and 0.67 (95% CI 0.34 - 1.29), respectively, indicating only a marginal improvement. Furthermore, in the group receiving chemotherapy alone, the efficacy among KRAS WT/BRAF MT patients was significantly lower than among KRAS WT/BRAF MT patients. Specifically, KRAS WT/BRAF MT patients had an OS of 9.9 months, versus 21.1 months among KRAS WT/BRAF WT patients. The PFS of KRAS WT/ BRAF MT patients was 3.7 months, compared to 7.7 months for KRAS WT/BRAF WT patients. These results suggest that BRAF mutations are not only predictors for anti-EGFR therapy, but may also be an important indicator for prognosis. Given the paucity of KRAS WT/ BRAF MT cases in the above study, the value of BRAF mutations for the prediction of cetuximab efficacy and prognosis still needs to be independently confirmed.
4. PI3K-PTEN-AKT Signaling Pathway
Neither genetic variations in EGFR amplification nor mutations in KRAS or BRAF can explain the 50% rate of poor response to anti-EGFR treatment in patients with wild-type KRAS. Instead, other EGFR-dependent signaling pathways—such as PIK3CA mutation, PTEN deletion, or pAKT overexpression—may be involved in treatment resistance. It is well known that phosphorylation and dephosphorylation play key roles in effecting basic cellular activities. PI3K activates the PI3K/AKT pathway by phosphorylating phosphatidylinositol-3,4-bisphosphate (PIP2), whereas PTEN negatively modulates the PI3K/ AKT signaling pathway by dephosphorylation, resulting in the inhibition of cell proliferation and the activation of apoptosis. PIK3CA mutations or PTEN deletions lead to PIP3 accumulation in the cytosol and sustained activation of AKT. The result is inhibition of apoptosis and stimulation of continuous cell growth. Recent studies have shown that the PI3K-PTEN-AKT signaling pathway is a key factor determining the response to anti-EGFR treatment in mCRC [8,30,41-44].
4.1. PIK3CA Mutation
The incidence of PIK3CA mutations and PTEN deletions is approximately 15% - 20%, of which about 8% may coexist with KRAS and BRAF mutations [8,35,43-46]. About 70% of PIK3CA mutations are accompanied by KRAS mutations. Of these, almost all mutations in exon 20 are associated with mutations in KRAS, whereas only 50% of mutations in exon 9 are associated with KRAS mutations [40]. Most studies support that activation of the PIK3CA/PTEN signaling pathway predicts resistance to anti-EGFR treatment [43-46], but some discrepancies exist [47]. De Roock et al. [35] retrospectively analyzed the PIK3CA genotype in 743 mCRC tumor samples using mass spectrometry. These researchers observed that the overall frequency of PIK3CA mutations was 14.5% (108/743 cases). Of these, 68.5% (74/108 cases) occurred in exon 9, and 20.4% (22/108 cases) occurred in exon 20. Compared with wild-type PIK3CA, mutations in exon 9 do not affect treatment efficacy, but mutations in exon 20 are suggestive of poor prognoses. The response rates for patients with mutant and wide-type exon 20 were 0 and 36.8%, respectively (OR = 0.00, P = 0.029). These response rates were associated with median PFS times of 11.5 weeks and 24 weeks, respectively (HR = 2.52, P = 0.013), and median OS times of 34 weeks and 51 weeks, respectively (HR = 3.29, P = 0.0057). Ogino et al. [48] analyzed the correlation between PIK3CA mutations and tumor-related deaths in early resectable CRC and found that the incidence of PIK3CA mutation was 18% (82/450 cases) and was correlated with tumor-related deaths in patients with wild-type but not mutant KRAS, implying that PIK3CA may be of value for prognosis.
4.2. AKT Expression
Overexpression of the PI3K downstream factor phosphor-AKT (pAKT) has been linked to tumor proliferation, invasion, and angiogenesis [49]. Therefore, it is conceivable that pAKT overexpression may be associated with poor CRC prognosis because activation of oncogenes or inactivation of tumor suppressor genes may correlate with highly aggressive tumors. However, a retrospective study (n = 717 cases) [50] reported that pAKT overexpression correlated with a lower grade leading to a good prognosis for CRC patients. That is, pAKT overexpression is an indicator of good prognosis and thus is a promising new target for cancer therapy. Researchers believe that CRC patients without pAKT overexpression acquire worse prognostic factors and maintain malignant tumor behaviors [50].
4.3. PTEN Deletion
PTEN plays an important role in the pathogenesis and development of colorectal cancers, and PTEN inactivation is not only a predictor of nonresponse to anti-EGFR therapy [45-47] but also is a marker for poor prognosis [50]. Studies suggest that PTEN deletions in metastases may predict resistance to cetuximab and irinotecan combination therapy [51]. Specifically, the efficacy of cetuximab was significantly higher for mCRC patients expressing PTEN-positive metastases (PTEN+) compared with those expressing PTEN-negative metastases, with objecttive response rates (ORR) of 36% versus 5% (P = 0.007), respectively, and PFS of 4.7 months versus 3.3 months (HR = 0.49, P = 0.005), respectively. PTEN+/KRAS WT patients had significantly longer PFS times than other patients (5.5 months vs 3.8 months; HR = 0.42, P = 0.001), indicating that detection of PTEN deletions and KRAS mutations together may help in screening efforts for anti-EGFR reactivity among mCRC patients. However, Tol et al. [38,39] reported that in wild-type KRAS patients, only BRAF mutations (not PTEN deletions or PIK3CA mutations) independently predicted cetuximab efficacy. Of course, standardization of detection methods for PTEN gene deletions will facilitate multi-center trials and improve comparability of results.
5. Evaluation of RAS-RAF-MAPK and PI3K-PTEN-AKT Signaling Pathways for Individualized Treatment
Regardless of whether the EGFR gene is activated or blocked by drugs, mutations in KRAS, BRAF, or PIK3CA genes lead to sustained activation of the RAS-RAFMAPK and PI3K-PTEN-AKT signaling pathways [52]. Comprehensive detection of these genetic variations may facilitate the identification of more mCRC patients predicted to be resistant to anti-EGFR therapy [35,39,46]. Most of the molecular markers reported here were evaluated alone as indicators for the efficacy of anti-EGFR treatment. However, some overlapping features of these molecules may further increase the complexity of applying these genes as efficacy predictors. Some prognostic and predictive molecular markers of anti-EGFR for CRC are listed in Table 1.
Using laser microdissection and sequencing analyses, Baldus et al. [8] found that 60% of mCRC patients have at least one mutation in KRAS, BRAF, or PIK3CA. Additional detection of BRAF and PIK3CA mutations can identify 15% more anti-EGFR-resistant patients than detecting the KRAS mutation alone. Similarly, SartoreBianchi et al. [53] observed that 70% (74/106) of mCRC patients exhibited variations in at least one of these genes (KRAS, BRAF, PIK3CA mutations; PTEN deletions). The clinical efficacies for patients according to gene variations were as follows: 51% for those without gene mutation, 4% for those with one gene mutation, and 0% for those with ≥2 gene mutations.
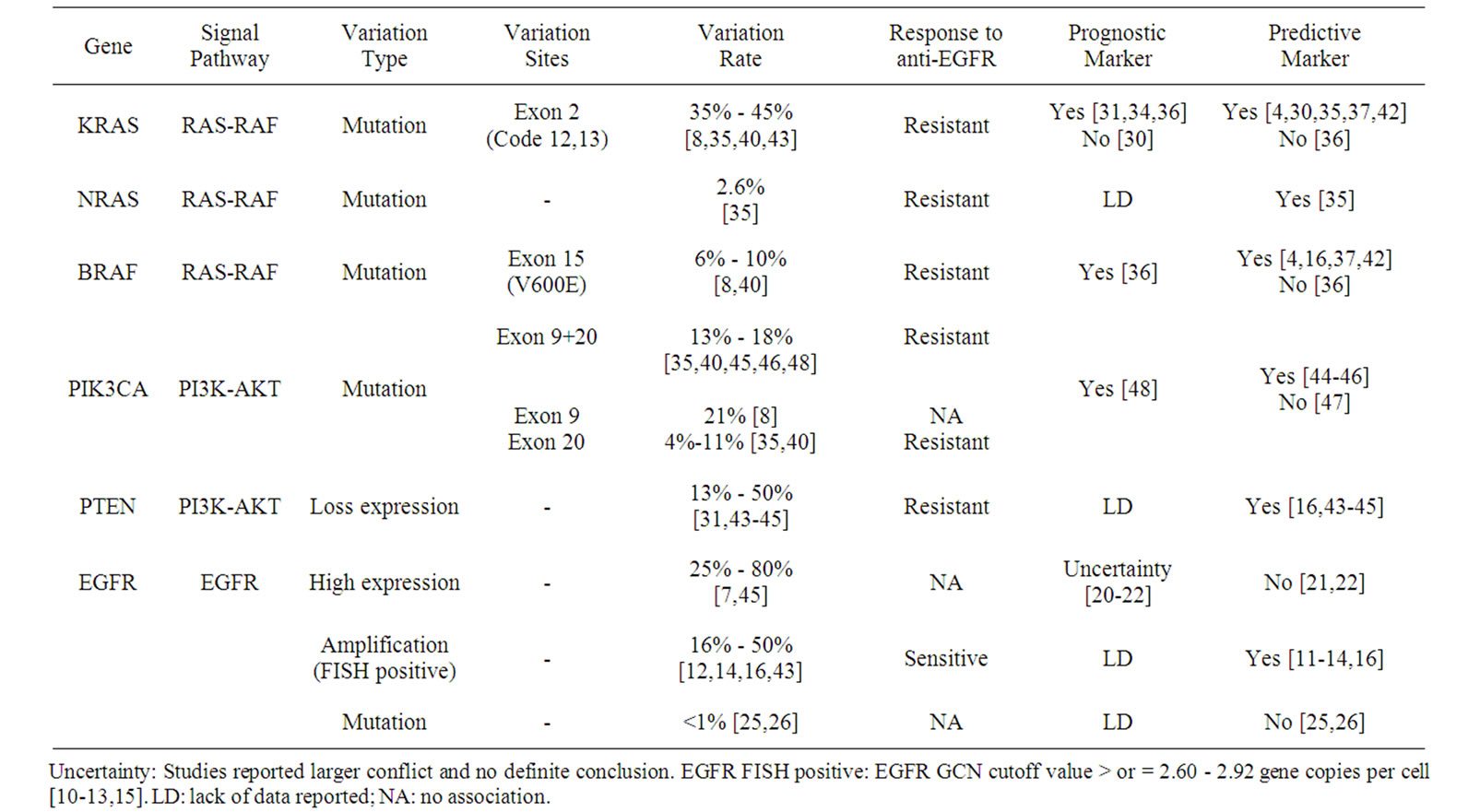
Table 1. Promising prognostic and predictive molecular markers for the response to EGFR-targeted monoclonal antibody treatment for advanced colorectal cancer.
De Roock et al. [35] used mass spectrometry to determine genotypes of KRAS, BRAF, NRAS, and PIK3CA in 747 mCRC tumor samples and correlated the results with efficacies of cetuximab. In KRAS wild-type and mutant patients, the treatment efficacies of cetuximab were 35.8% and 6.7%, respectively (OR = 0.13, P < 0.0001). The median PFS times were 24 weeks and 12 weeks (HR = 1.98, P < 0.0001), respectively, and the median OS times were 50 weeks and 32 weeks (HR = 1.75, P < 0.0001), respectively. Among patients exhibiting wild-type KRAS, the response rates of patients with BRAF and NRAS mutations were significantly lower than those of wild-type BRAF/NRAS patients. In this study, the overall response rate of randomly selected patients was 24.4%, whereas that of the KRAS wild-type patients was 36.3%. The ORR of patients with wild-type KRAS, BRAF, NRAS, and PIK3CA (exon 20) genes was 41.2%. Recent results of the CAIRO2 clinical trial (n = 559) [39] suggest that, except for KRAS and BRAF gene mutations, other genetic variants alone or in combination cannot be used as predictors for treatment efficacy and prognosis of mCRC.
6. Conclusions
Further analyses of BRAF and PIK3CA mutations and of PTEN deletions in mCRC patients with wild-type KRAS may lend additional insight into their predictive values for anti-EGFR therapy. Approximately 60% - 70% of mCRC patients can be identified as nonresponders for anti-EGFR therapy using these genetic characteristics. In the remaining 30% - 40% of mCRC patients, the effectiveness of anti-EGFR combined with first-line chemotherapy was up to 60%. Inclusion of anti-EGFR therapy can achieve an efficacy of close to 40%, even in chemotherapy-resistant mCRC patients. NRAS mutations and EGFR GCN also seem to be relevant for treatment efficacy and necessitate further study. At present, prospective data from comprehensive mutation analyses for both the KRAS-BRAF and the PIK3CA-PTEN signaling pathways are needed to assess their predictive power for anti-EGFR treatment response in mCRC patients. Considering the high cost of treatment and low cost of detection, it seems both economical and reasonable to perform routine tests for BRAF and PIK3CA mutations and for PTEN deletions in patients exhibiting wild-type KRAS. Additionally, a set of standardized and effective detection methods needs to be established to unify the results of current clinical trials, and to objectively judge the predictive and prognostic value of these molecular markers.
7. Acknowledgements
This study was supported by a grant from the National Natural Science Foundation of China (No.30700979) to Dr. Cheng-Bo Han.
REFERENCES
- J. A. Meyerhardt and R. J. Mayer, “Systemic Therapy for Colorectal Cancer,” New England Journal of Medicine, Vol. 352, 2005, pp. 476-487. doi:10.1056/NEJMra040958
- C. Tournigand, T. André, E. Achille, G. Lledo, et al., “FOLFIRI Followed by FOLFOX6 or the Reverse Sequence in Advanced Colorectal Cancer: A Randomized GERCOR Study,” Journal of Clinical Oncology, Vol. 22, No. 2, 2004, pp. 229-237. doi:10.1200/JCO.2004.05.113
- R. G. Amado, M. Wolf, M. Peeters, E. Van Cutsem, et al., “Wildtype KRAS Is Required for Panitumumab Efficacy in Patients with Metastatic Colorectal Cancer,” Journal of Clinical Oncology, Vol. 26, No. 31, 2008, pp. 1626-1634. doi:10.1200/JCO.2007.14.7116
- C. Bokemeyer, C. Kohne, P. Rougier, C. Stroh, M. Schlichting and E. Van Cutsem, “Cetuximab with Chemotherapy (CT) as First-Line Treatment for Meta-static Colorectal Cancer (mCRC): Analysis of the CRYSTAL and OPUS Studies According to KRAS and BRAF Mutation Status,” Journal of Clinical Oncology, Vol. 28, 2010.
- D. J. Jonker, C. J. O’Callaghan, C. S. Karapetis, et al., “Cetuximab for the Treatment of Colorectal Cancer,” New England Journal of Medicine, Vol. 357, 2007, pp. 2040-2048. doi:10.1056/NEJMoa071834
- E. Van Cutsem, M. Peeters, S. Siena, et al., “Open-Label Phase III Trial of Panitumumab Plus Best Supportive Care Compared with Best Supportive Care Alone in Patients with Chemotherapy-Refractory Metastatic Colorectal Cancer,” Journal of Clinical Oncology, Vol. 25, No. 29, 2007, pp. 1658-1664. doi:10.1200/JCO.2006.08.1620
- M. Scaltriti and J. Baselga, “The Epidermal Growth Factor Receptor Pathway: A Model for Targeted Therapy,” Clinical Cancer Research, Vol. 12, 2006, pp. 5268-5272. doi:10.1158/1078-0432.CCR-05-1554
- S. E. Baldus, K. L. Schaefer, R. Engers, D. Hartleb, N. H. Stoecklein and H. E. Gabbert, “Prevalence and Heterogeneity of KRAS, BRAF, and PIK3CA Mutations in Primary Colorectal Adenocarcinomas and Their Corresponding Metastases,” Clinical Cancer Research, Vol. 16, 2010, pp. 790-799. doi:10.1158/1078-0432.CCR-09-2446
- Y. Yarden and M. X. Sliwkowski, “Untangling the ErbB Signalling Network,” Nature Reviews Molecular Cell Biology, Vol. 2, 2001, pp. 127-137. doi:10.1038/35052073
- L. N. Klapper, M. H. Kirschbaum, M. Sela and Y. Yarden, “Biochemical and Clinical Implications of the ErbB/HER Signaling Network of Growth Factor Receptors,” Advances in Cancer Research, Vol. 77, 2000, pp. 25-79. doi:10.1016/S0065-230X(08)60784-8
- A. Sartore-Bianchi, M. Moroni, S. Veronese, C. Carnaghi, et al., “Epidermal Growth Factor Receptor Gene Copy Number and Clinical Outcome of Metastatic Colorectal Cancer Treated with Panitumumab,” Journal of Clinical Oncology, Vol. 25, No. 22, 2007, pp. 3238-3245. doi:10.1200/JCO.2007.11.5956
- F. Cappuzzo, G. Finocchiaro, E. Rossi, et al., “EGFR FISH Assay Predicts for Response to Cetuximab in Chemotherapy Refractory Colorectal Cancer Patients,” Annals of Oncology, Vol. 19, No. 4, 2008, pp. 717-723. doi:10.1093/annonc/mdm492
- N. Personeni, S. Fieuws, H. Piessevaux, et al., “Clinical Usefulness of EGFR Gene Copy Number as a Predictive Marker in Colorectal Cancer Patients Treated with cetuximab: A Fluorescent in Situ Hybridization Study,” Clinical Cancer Research, Vol. 14, 2008, pp. 5869-5876. doi:10.1158/1078-0432.CCR-08-0449
- M. Scartozzi, I. Bearzi, A. Mandolesi, et al., “Epidermal Growth Factor Receptor (EGFR) Gene Copy Number (GCN) Correlates with Clinical Activity of IrinotecanCetuximab in K-RAS Wild-Type Colorectal Cancer: A Fluorescence in Situ (FISH) and Chromogenic in Situ Hybridization (CISH) Analysis,” BMC Cancer, Vol. 9, 2009, p. 303. doi:10.1186/1471-2407-9-303
- M. Moroni, A. Sartore-Bianchi, S. Veronese and S. Siena, “EGFR FISH in Colorectal Cancer: What Is the Current Reality?” Lancet Oncology, Vol. 9, No. 5, 2008, pp. 402- 403. doi:10.1016/S1470-2045(08)70109-8
- P. Laurent-Puig, A. Cayre, G. Manceau, et al., “Analysis of PTEN, BRAF, and EGFR Status in Determining Benefit from Cetuximab Therapy in Wild-Type KRAS Metastatic Colon Cancer,” Journal of Clinical Oncology, Vol. 27, No. 35, 2009, pp. 5924-5930. doi:10.1200/JCO.2008.21.6796
- A. Algars, M. Lintunen, O. Carpén, R. Ristamäki and J. Sundström, “EGFR Gene Copy Number Assessment from Areas with Highest EGFR Expression Predicts Response to anti-EGFR Therapy in Colorectal Cancer,” British Journal of Cancer, Vol. 105, No. 2, 2011, pp. 255-262. doi:10.1038/bjc.2011.223
- L. J. Layfield, P. S. Bernard and N. S. Goldstein, “Color Multiplex Polymerase Chain Reaction for Quantitative Analysis of Epidermal Growth Factor Receptor Genes in Colorectal Adenocarcinoma,” Journal of Surgical Oncology, Vol. 83, No. 4, 2003, pp. 227-231. doi:10.1002/jso.10272
- G. Milano, M. C. Etienne-Grimaldi, L. Dahan, et al., “Epidermal Growth Factor Receptor (EGFR) Status and K-Ras Mutations in Colorectal Cancer,” Annals of Oncology, Vol. 19, No. 12, 2008, pp. 2033-2038. doi:10.1093/annonc/mdn416
- R. L. Rego, N. R. Foster, T. C. Smyrk, M. Le, et al., “Prognostic Effect of Activated EGFR Expression in Human Colon Carcinomas: Comparison with EGFR Status,” British Journal of Cancer, Vol. 102, 2010, pp. 165-172. doi:10.1038/sj.bjc.6605473
- L. B. Saltz, N. J. Meropol, P. J. Loehrer Sr., et al., “Phase II Trial of Cetuximab in Patients with Refractory Colorectal Cancer That Expresses the Epidermal Growth Factor Receptor,” Journal of Clinical Oncology, Vol. 22, No. 4, 2004, pp. 1201-1208. doi:10.1200/JCO.2004.10.182
- D. Cunningham, Y. Humblet, S. Siena, et al., “Cetuximab Monotherapy and Cetuximab Plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer,” New England Journal of Medicine, Vol. 351, 2004, pp. 337-345. doi:10.1056/NEJMoa033025
- F. Cappuzzo, M. Varella-Garcia, G. Finocchiaro, et al., “Primary Resistance to Cetuximab Therapy in EGFR FISH-Positive Colorectal Cancer Patients,” British Journal of Cancer, Vol. 99, 2008, pp. 83-89. doi:10.1038/sj.bjc.6604439
- K. Y. Chung, J. Shia, N. E. Kemeny, et al., “Cetuximab Shows Activity in Colorectal Cancer Patients with Tumors That Do Not Express the Epidermal Growth Factor Receptor by Immunohistochemistry,” Journal of Clinical Oncology, Vol. 23, No. 9, 2005, pp. 1803-1810. doi:10.1200/JCO.2005.08.037
- T. D. Barber, B. Vogelstein, K. W. Kinzler and V. E. Velculescu, “Somatic Mutations of EGFR in Colorectal Cancers and Glioblastomas,” New England Journal of Medicine, Vol. 351, 2004, p. 2883. doi:10.1056/NEJM200412303512724
- M. Moroni, S. Veronese, S. Benvenuti, G. Marrapese, et al., “Gene Copy Number for Epidermal Growth Factor Receptor (EGFR) and Clinical Response to anti EGFR Treatment in Colorectal Cancer: A Cohort Study,” Lancet Oncology, Vol. 6, 2005, pp. 279-286. doi:10.1016/S1470-2045(05)70102-9
- F. K. Doger, I. Meteoglu, P. Tuncyurek, et al., “Does the EGFR and VEGF Expression Predict the Prognosis in Colon Cancer?” European Surgical Research, Vol. 38, 2006, pp. 540-544. doi:10.1159/000096774
- D. Patel, X. Guo, S. Ng, M. Melchior, et al., “IgG Isotype, Glycosylation, and EGFR Expression Determine the Induction of Antibody-Dependent Cellular Cytotoxicity in Vitro by Cetuximab,” Human Antibodies, Vol. 19, 2010, pp. 89-99.
- J. García-Foncillas and E. Díaz-Rubio, “Progress in Metastatic Colorectal Cancer: Growing Role of Cetuximab to Optimize Clinical Outcome,” Clinical and Translational Oncology, Vol. 12, 2010, pp. 533-542. doi:10.1007/s12094-010-0551-3
- C. S. Karapetis, S. Khambata-Ford, D. J. Jonker, et al., “K-Ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer,” New England Journal of Medicine, Vol. 359, No. 8, 2008, pp. 1757-1765. doi:10.1056/NEJMoa0804385
- A. Lièvre, J. B. Bachet, V. Boige, et al., “KRAS Mutations as an Independent Prognostic Factor in Patients with Advanced Colorectal Cancer Treated with Cetuximab,” Journal of Clinical Oncology, Vol. 26, No. 15, 2008, pp. 374-379. doi:10.1200/JCO.2007.12.5906
- A. F. Sobrero, J. Maurel, L. Fehrenbacher, et al., “EPIC: Phase III Trial of Cetuximab Plus Irinotecan after Fluoropyrimidine and Oxaliplatin Failure in Patients with Metastatic Colorectal Cancer,” Journal of Clinical Oncology, Vol. 26, No. 14, 2008, pp. 2311-2319. doi:10.1200/JCO.2007.13.1193
- F. Molinari, L. Felicioni, M. Buscarino, S. De Dosso, et al., “Increased Detection Sensitivity for KRAS Mutations Enhances the Prediction of Anti-EGFR Monoclonal Antibody Resistance in Metastatic Colorectal Cancer,” Clinical Cancer Research, Vol. 17, No. 14, 2011, pp. 4901- 4914. doi:10.1158/1078-0432.CCR-10-3137
- H. J. Andreyev, A. R. Norman, D. Cunningham, et al., “Kirsten Ras Mutations in Patients with Colorectal Cancer: The ‘RASCAL II’ Study,” British Journal of Cancer, Vol. 85, 2001, pp. 692-696. doi:10.1054/bjoc.2001.1964
- W. De Roock, B. Claes, D. Bernasconi, et al., “Effects of KRAS, BRAF, NRAS, and PIK3CA Mutations on the Efficacy of Cetuximab plus Chemotherapy in Chemotherapy-Refractory Metastatic Colorectal Cancer: A Retrospective Consortium Analysis,” Lancet Oncology, Vol. 11, No. 8, 2010, pp. 753-762. doi:10.1016/S1470-2045(10)70130-3
- S. D. Richman, M. T. Seymour, P. Chambers, et al., “KRAS and BRAF Mutations in Advanced Colorectal Cancer Are Associated with Poor Prognosis but Do Not Preclude Benefit from Oxaliplatin or Irinotecan: Results from the MRC FOCUS Trial,” Journal of Clinical Oncology, Vol. 27, No. 35, 2009, pp. 5931-5937. doi:10.1200/JCO.2009.22.4295
- F. Di Nicolantonio, M. Martini, F. Molinari, et al., “WildType BRAF Is Required for Response to Panitumumab or Cetuximab in Metastatic Colorectal Cancer,” Journal of Clinical Oncology, Vol. 26, No. 35, 2008, pp. 5705-5712. doi:10.1200/JCO.2008.18.0786
- J. Tol, M. Koopman, A. Cats, et al., “Chemotherapy, Bevacizumab, and Cetuximab in Metastatic Colorectal Cancer,” New England Journal of Medicine, Vol. 360, 2009, pp. 563-572. doi:10.1056/NEJMoa0808268
- J. Tol, J. R. Dijkstra, M. Klomp, et al., “Markers for EGFR Pathway Activation as Predictor of Outcome in Metastatic Colorectal Cancer Patients Treated with or without Cetuximab,” European Journal of Cancer, Vol. 46, No. 11, 2010, pp. 1997-2009. doi:10.1016/j.ejca.2010.03.036
- L. Simi, N. Pratesi, M. Vignoli, R. Sestini, F. Cianchi, et al., “High-Resolution Melting Analysis for Rapid Detection of KRAS, BRAF, and PIK3CA Gene Mutations in Colorectal Cancer,” American Journal of Clinical Pathology, Vol. 130, 2008, pp. 247-253. doi:10.1309/LWDY1AXHXUULNVHQ
- A. Ličar, P. Cerkovnik and S. Novaković, “Distribution of Some Activating KRAS and BRAF Mutations in Slovene Patients with Colorectal Cancer,” Medical Oncology, 2010, Ahead of Print.
- E. Van Cutsem, I. Lang, G. Folprecht, et al., “Cetuximab Plus FOLFIRI: Final Data from the CRYSTAL Study on the Association of KRAS and BRAF Biomarker Status with Treatment Outcome,” Journal of Clinical Oncology, Vol. 28, 2010.
- M. Frattini, P. Saletti, E. Romagnani, V. Martin, et al., “PTEN Loss of Expression Predicts Cetuximab Efficacy in Metastatic Colorectal Cancer Patients,” British Journal of Cancer, Vol. 97, 2007, pp. 1139-1145. doi:10.1038/sj.bjc.6604009
- M. Jhawer, S. Goel, A. J. Wilson, et al., “PIK3CA Mutation/PTEN Expression Status Predicts Response of Colon Cancer Cells to the Epidermal Growth Factor Receptor Inhibitor Cetuximab,” Cancer Research, Vol. 68, 2008, pp. 1953-1961. doi:10.1158/0008-5472.CAN-07-5659
- F. Perrone, A. Lampis, M. Orsenigo, et al., “PI3KCA/ PTEN Deregulation Contributes to Impaired Responses to Cetuximab in Metastatic Colorectal Cancer Patients,” Annals of Oncology, Vol. 20, No. 1, 2009, pp. 84-90. doi:10.1093/annonc/mdn541
- A. Sartore-Bianchi, M. Martini, F. Molinari, et al., “PIK3CA Mutations in Colorectal Cancer Are Associated with Clinical Resistance to EGFR-Targeted Monoclonal Antibodies,” Cancer Research, Vol. 69, 2009, pp. 1851- 1857. doi:10.1158/0008-5472.CAN-08-2466
- H. Prenen, J. De Schutter, B. Jacobs, W. De Roock, et al., “PIK3CA Mutations Are Not a Major Determinant of Resistance to the Epidermal Growth Factor Receptor Inhibitor Cetuximab in Metastatic Colorectal Cancer,” Clinical Cancer Research, Vol. 15, 2009, pp. 3184-3188. doi:10.1158/1078-0432.CCR-08-2961
- S. Ogino, K. Nosho, G. J. Kirkner, et al., “PIK3CA Mutation Is Associated with Poor Prognosis among Patients with Curatively Resected Colon Cancer,” Journal of Clinical Oncology, Vol. 27, No. 9, 2009, pp. 1477-1484. doi:10.1200/JCO.2008.18.6544
- N. Itoh, S. Semba, M. Ito, et al., “Phosphorylation of Akt/PKB Is Required for Suppression of Cancer Cell Apoptosis and Tumor Progression in Human Colorectal Carcinoma,” Cancer, Vol. 94, No. 12, 2002, pp. 3127- 3134. doi:10.1002/cncr.10591
- Y. Baba, K. Nosho, K. Shima, et al., “Phosphorylated AKT Expression Is Associated with PIK3CA Mutation, Low Stage, and Favorable Outcome in 717 Colorectal Cancers,” Cancer, Vol. 117, No. 7, 2011, pp. 1399-1408. doi:10.1002/cncr.25630
- F. Loupakis, L. Pollina, I. Stasi, et al., “PTEN Expression and KRAS Mutations on Primary Tumors and Metastases in the Prediction of Benefit from Cetuximab Plus Irinotecan for Patients with Metastatic Colorectal Cancer,” Journal of Clinical Oncology, Vol. 27, No. 16, 2009, pp. 2622-2629. doi:10.1200/JCO.2008.20.2796
- J. A. McCubrey, L. S. Steelman, S. L. Abrams, et al., “Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT Pathways in Malignant Transformation and Drug Resistance,” Advances in Enzyme Regulation, Vol. 46, No. 1, 2006, pp. 249-279. doi:10.1016/j.advenzreg.2006.01.004
- A. Sartore-Bianchi, F. Di Nicolantonio, M. Nichelatti, et al., “Multi-Determinants Analysis of Molecular Alterations for Predicting Clinical Benefit to EGFR Targeted Monoclonal Antibodies in Colorectal Cancer,” PLoS One, Vol. 4, 2009, p. e7287. doi:10.1371/journal.pone.0007287