Advances in Bioscience and Biotechnology
Vol.5 No.1(2014), Article ID:42364,8 pages DOI:10.4236/abb.2014.51011
Cell-type specific and non-redundant anti-proliferative effects of shRNA-mediated Galpha12- and Galpha13 knockdown in lung cancer cell lines
1Rudolf-Boehm-Institute for Pharmacology and Toxicology, University of Leipzig, Leipzig, Germany
2Biochemical Pharmacological Institute/bpc, Philipps-University Marburg, Marburg, Germany
3Present Address: Targos Molecular Pathology GmbH, Kassel, Germany
4Present Address: Institute for Immunology, Philipps-University Marburg, Marburg, Germany
5Walther Straub Institute of Pharmacology and Toxicology, University of Munich (LMU), Munich, Germany
6Comprehensive Pneumology Center Munich (CPC-M), German Center for Lung Research, Munich, Germany
Email: *achim.aigner@medizin.uni-leipzig.de
Copyright © 2014 Thomas R. H. Büch et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Thomas R. H. Büch et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 13 November 2013; revised 24 December 2013; accepted 13 January 2014
KEYWORDS
Galpha12; Galpha13; G12/13; Lung Cancer; SCLC; NSCLC; Gene Knockdown
ABSTRACT
In small cell lung cancer cells, various autocrine stimuli lead to the parallel activation of Gq/11 and G12/13 proteins. The contribution of the Gq/11-PLC-β cascade to the mitogenic effects in SCLC cells is well established, but the relevance of G12/13 signaling is less explored. While in prostate and breast cancer, G12/13 activation has been shown previously to promote invasiveness without being involved in cellular proliferation, previous data from our group indicate antiproliferative effects of G12/13 knockdown in small cell lung cancer (SCLC) cells. To further investigate the role of G12/13-dependent signaling in lung tumor cells, we employed shRNA-mediated targeting of Gα12, Gα13, or both, in SCLC and NSCLC cell lines. Lentiviral expression of shRNAs resulted in specific Gα12 and Gα13 knockdown. Of note, upon single knockdown of one family member, no counter-upregulation of the other one was observed. Interestingly, inhibition of proliferation was cell line dependent. In cell lines where knock-down led to antiproliferation, single knockdown of either Gα12 or Gα13 was sufficient to impair proliferation and double knockdown of Gα12 and Gα13 tended not to further increase anti-proliferative effects. Likewise, when single knockdown was insufficient for an inhibition of proliferation, no effects were observed in double knockdowns. Taken together, these findings indicate that both Gα12 and Gα13 affect cellular proliferation individually and interference with one family member is sufficient for anti-tumor effects.
1. INTRODUCTION
Lung cancer represents the leading cause of cancer-related death in the industrialized countries. Cancers of the lung are subdivided into different histological subtypes, e.g., adenocarcinoma, squamous cell carcinoma, large cell carcinoma and small cell carcinoma (SCLC). The former three subtypes are often subsumed as non-small cell carcinoma (NSCLC). Lung cancer of the SCLC type shows a very aggressive behavior with early manifestation of metastases. Thus, local therapeutic options like surgery are usually inapplicable in this tumor entity at the time of diagnosis. While current chemotherapy regimens in SCLC tumors clearly show clinical benefit, the overall survival after 5 years is still only 7% - 12% [1]. In NSCLC, although the potential to develop metastases is generally somewhat lower as compared to SCLC, dissemination is frequent at the time of diagnosis, necessitating systemic chemotherapy. This clearly emphasizes the need for novel treatment strategies based on the detailed understanding of molecular events underlying SCLC and NSCLC tumorigenesis and tumor progression.
In both SCLC and NSCLC cancer cells, growth and survival is critically regulated by a number of G proteindependent pathways. In SCLC cells, these signaling pathways are constitutively activated by the secretion of diverse autocrine stimuli, e.g., bradykinin, galanin, bombesin, or acetyl choline. The auto-stimulatory ligands activate G protein-coupled receptors (GPCR), which signal via the parallel activation of Gq/11 and G12/13 proteins. Activation of Gq/11-dependent pathways has been shown to lead to the stimulation of the Ras-ERK1/2 cascade [2,3]. In contrast, the distal effector molecules of G12/13 proteins in SCLC cells are less characterized. However, an important role of G12/13 proteins in this tumor entity has been suggested by the fact that shRNA-mediated downregulation of Gα12 or Gα13 proteins in the SCLC cell lines H69 and H209 led to a marked growth inhibition [4].
In this study, we selectively disrupted G12 and G13 function by knockdown of Gα12 or Gα13, respectively, in order to decipher their individual relevance in different lung cancer cell lines. RNAi-mediated specific gene targeting of the α-subunits of G12 or G13 (Gα12 or Gα13), alone or in combination, was achieved by infection of the SCLC cell lines H510, H82 and the NSCLC cell lines A549 and H441 with shRNA constructs using a lentiviral system. While single targeting revealed, cell-specifically, a profound anti-proliferative effect in most cell lines in response to Gα12 or Gα13 downregulation, double targeting of both Gα12 and Gα13 did not yield any additive effects. Thus, we show that both Gα12 and Gα13 play a critical role in SCLC and NSCLC proliferation in vitro, and that selectively interfering with Gα12 and Gα13 signaling, e.g. through RNAi-mediated therapeutic blockage of both G proteins, may provide a promising new avenue in lung cancer treatment.
2. MATERIALS AND METHODS
2.1. Viral shRNA Construct, Cell Lines, and Stable Transfection
Specific shRNA expression plasmids were generated as described previously [4], Briefly, chemically synthesized (MWG Biotech) oligonucleotides designed according to [5,6] were cloned into the pSUPER vector using the BglII and HindIII restriction sites, and correct insertion was controlled by sequencing. Using ClaI and BamHI, the H1-cassettes of the pSUPER vectors were then cloned into the lentivirus vector pLVTHM. For recombinant virus generation, 20 μg/10 cm dish of the cis-vectors were co-transfected into HEK cells together with 15 μg psPAX2 (“trans plasmid”) and with 6 μg pMD2G (VSV-G) using the calcium phosphate precipitation method. 6 - 8 h after transfection, the medium was changed, and 2 d later the supernatant was collected and sterile-filtered. Viruses were purified and concentrated using sucrose-sedimentation (20% sucrose in TBS, 26,000 rpm, 2 h), and, upon resuspension in complete RPMI medium, the viral titer was determined by serial dilution in HEK cells.
Lung cancer cell lines H82, H510, A549, and H441 were obtained from the American type culture collection (ATCC) and cultivated under standard conditions (37˚C, 5% CO2) in RPMI medium (PAA, Cölbe, Germany) supplemented with 10% fetal calf serum (FCS). H82, H510, A549, and H441 cells were transduced with 20 TU (transducing units) of recombinant virus, and after 48 h selected for stable integration by supplementing the medium with G418.
2.2. RNA Preparation and Quantitative RT-PCR
Total RNA from tumor cells was isolated using the Tri reagent (PEQLAB, Erlangen, Germany) according to the manufacturer’s protocol. Reverse transcription was performed using the RevertAid H Minus First Strand cDNA Synthesis Kit from Fermentas (St. Leon-Rot, Germany) as follows: 2 μg total RNA and 1 μl random hexamer primer (0.2 μg/μl) were diluted in DEPC-treated water ad 11 μl, incubated at 70˚C for 5 min and chilled on ice prior to adding 4 μl 5X reaction buffer, 0.5 μl RNAse inhibitor (20 u/μl), 2 μl 10 mM dNTP mix and 1.5 μl DEPC-treated water. After incubation at 25˚C for 5 min, 1 μl Reverse Transcriptase (200 u/μl) was added, and the mixture was incubated for 10 min under the same conditions and for 60 min at 42˚C, prior to stopping the reaction by heating at 70˚C for 10 min and chilling on ice. Quantitative PCR was performed in a Light Cycler from Roche (Penzberg, Germany) using the Absolute QPCR SYBRGREEN Capillary Mix (Abgene) according to the manufacturer’s protocol with 4 μl cDNA (diluted 1:100), 1 μl primers (5 μM each) and 5 μl SYBR Green master mix. A pre-incubation for 15 min at 95˚C was followed by 55 amplification cycles: 10 sec at 95˚C, 10 sec at 55˚C and 10 sec at 72˚C. The melting curve for PCR product analysis was determined by rapid cooling down from 95˚C to 65˚C, and incubation at 65˚C for 15 sec prior to heating to 95˚C. To normalize for equal mRNA/ cDNA amounts, PCR reactions with Gα12- or Gα13-specific and with actin-specific primer sets were always run in parallel for each sample, and Gα12/Gα13 levels were determined by the ΔΔCt method with β-actin as reference housekeeping gene.
2.3. Proliferation Assays
Cell lines were plated into 96 well plates at 2000 - 4000 cells/well and 5 wells/time point and cultivated in RPMI/ 10% FCS in a humidified incubator under standard conditions. At the time points indicated, the number of viable cells was determined using a colorimetric assay. Briefly, the medium was aspirated and 50 µl of a 1:10 dilution of Cell proliferation Reagent WST-1 (Roche Molecular Biochemicals, Mannheim, Germany) in medium was added to the cells prior to incubation for 1 h at 37˚C. The absorbance at 450 nm was measured using a Dynex MRX microplate reader (Pegasus Scientific Inc., Rockville, MD).
3. RESULTS
3.1. Stable Expression of shRNAs Leads to Specific Knockdown of Gα12 or Gα13
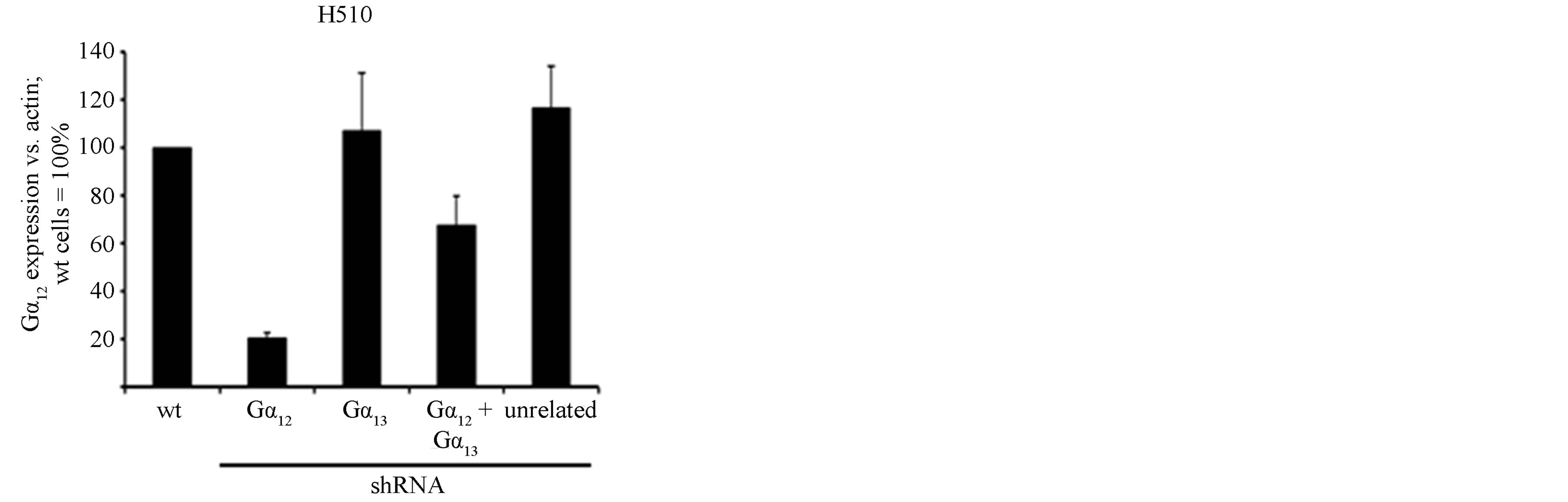
Using a lentiviral expression system, the expression of shRNAs directed against Gα12 or Gα13 in H510 SCLC cells led to a stable 70% (Gα13) to 80% (Gα12) downregulation of the respective target mRNA as compared to negative control-transfected (“unrelated”) H510 cells (Figures 1(a) and (b)). In this cell line, the double transfection with simultaneous expression of both Gα12- and Gα13-specific shRNAs resulted in a lower downregulation efficiency of Gα12 mRNA as compared to cells expressing only Gα12-specific shRNA (Figure 1(a)). Importantly, in single-transfected cells expressing either Gα12 or Gα13-directed shRNAs, no counter upregulation of the other G protein was discernible (Figures 1(a) and (b)). This finding suggests that an upregulation of Gα12 cannot compensate for the knockdown of Gα13 and vice versa, thus indicating a non-redundancy in the function of both proteins. This was further explored in proliferation assays.
3.2. Anti-Proliferative Effects Differ between Gα12 or Gα13 Knockdown and Are Cell Line Specific
Regarding the effects of Gα12- and Gα13 knockdown on proliferation in H510 cells, a ~50% inhibitory effect was found after single transfection with Gα13 shRNA. The double transfection with Gα12 and Gα13 shRNAs yielded similar rates of growth inhibition (Figure 1(c)). The most pronounced effect, however, was observed upon Gα12 single knockdown with a >90% reduction in tumor cell proliferation, suggesting a more dominant role of Gα12 in this cell model. The difference between Gα12 single vs. Gα12 + Gα13 double knockdown also indicates that the degree of Gα12 knockdown is critical, since the double infection had yielded higher residual Gα12 levels as compared to the single knockdown.
To further evaluate the role of Gα12 or Gα13 signalling in SCLC cells, we tested another SCLC cell line. H82 belongs to the variant type of SCLC cells as opposed to H510 cells which represent a classic type SCLC cell
 (a)
(a) (b)
(b)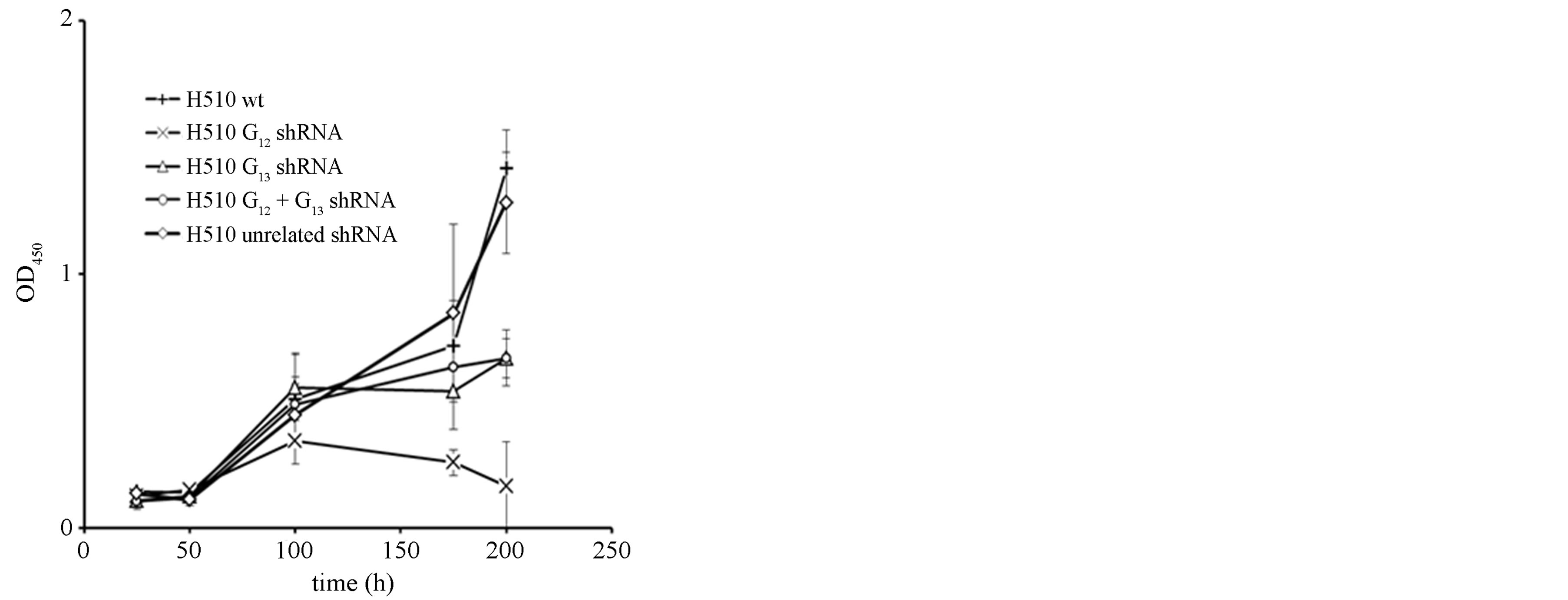 (c)
(c)
Figure 1. mRNA expression levels of Gα12 (a) and Gα13 (b) in relation to β-actin, as determined by quantitative RTPCR in the parental SCLC cell line H510 (wt, set to 100%), in H510 cells stably expressing shRNA directed against Gα12 or Gα13, respectively, in H510 cells expressing both Gα12 and Gα13 specific shRNA or, as a control, expressing unrelated shRNA. (c) Cell viability as determined by WST-1 assay in parental H510 cells (wt) or in H510 cells expressing unrelated shRNA or shRNA directed against Gα12 or Gα13.
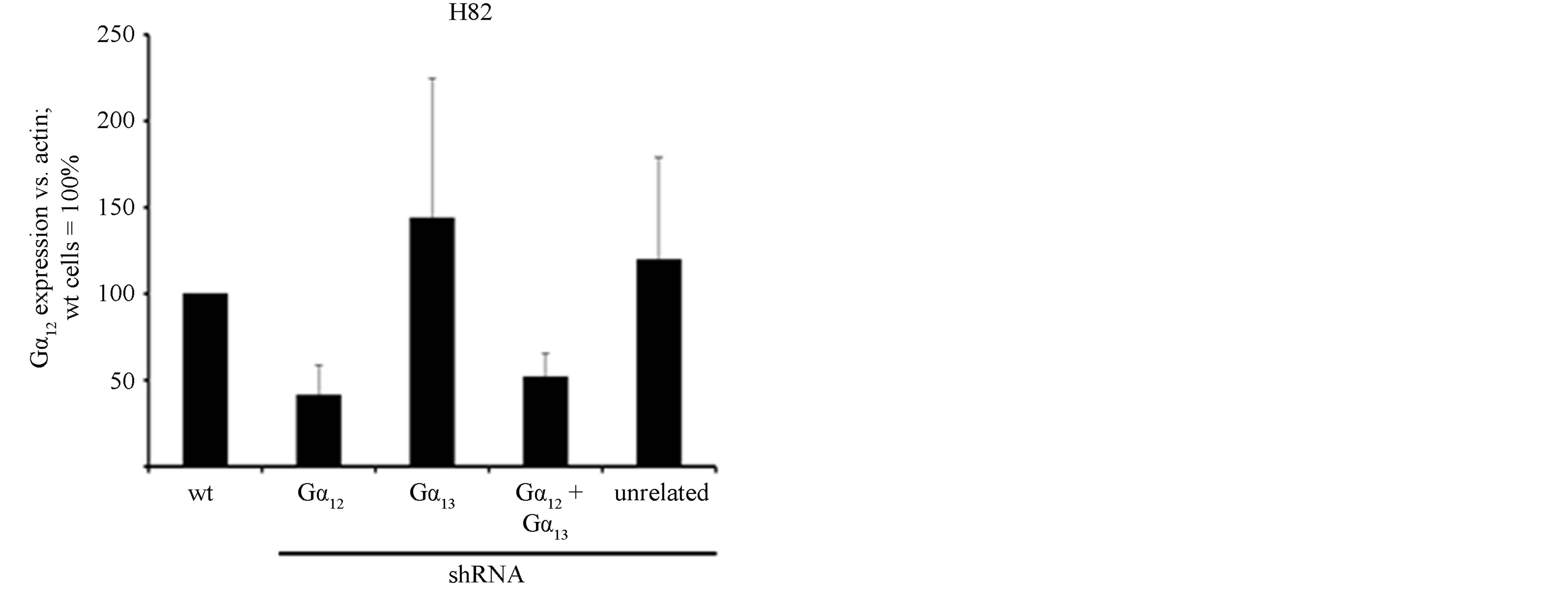
line. As in H510 cells, the expression of Gα12 or Gα13- specific shRNAs led to a specific at least 60% downregulation of the target G protein without affecting the non-targeted G protein (Figures 2(a) and (b)). Again, no compensatory (up-) regulation of the non-targeted G protein was observed, thus confirming the previous finding in H510 cells.
However, in contrast to H510 cells and despite the efficient knockdown, the anti-proliferative effect after shRNA expression was generally lower and showed a different pattern regarding the contributions of the individual G proteins. In fact, the strongest anti-proliferative effects occurred after Gα13 inhibition, either in the single or the double knockdown setting, whereas Gα12 inhibition (which had the strongest impact in H510 cells; see above) did not induce growth inhibition when compared to cells expressing unrelated shRNA (Figure 2(c)). Thus, the effects of interference with Gα12 or Gα13 appear to be cell-type specific.
3.3. Inhibition of Gα12 or Gα13 Expression in the NSCLC Cell Lines
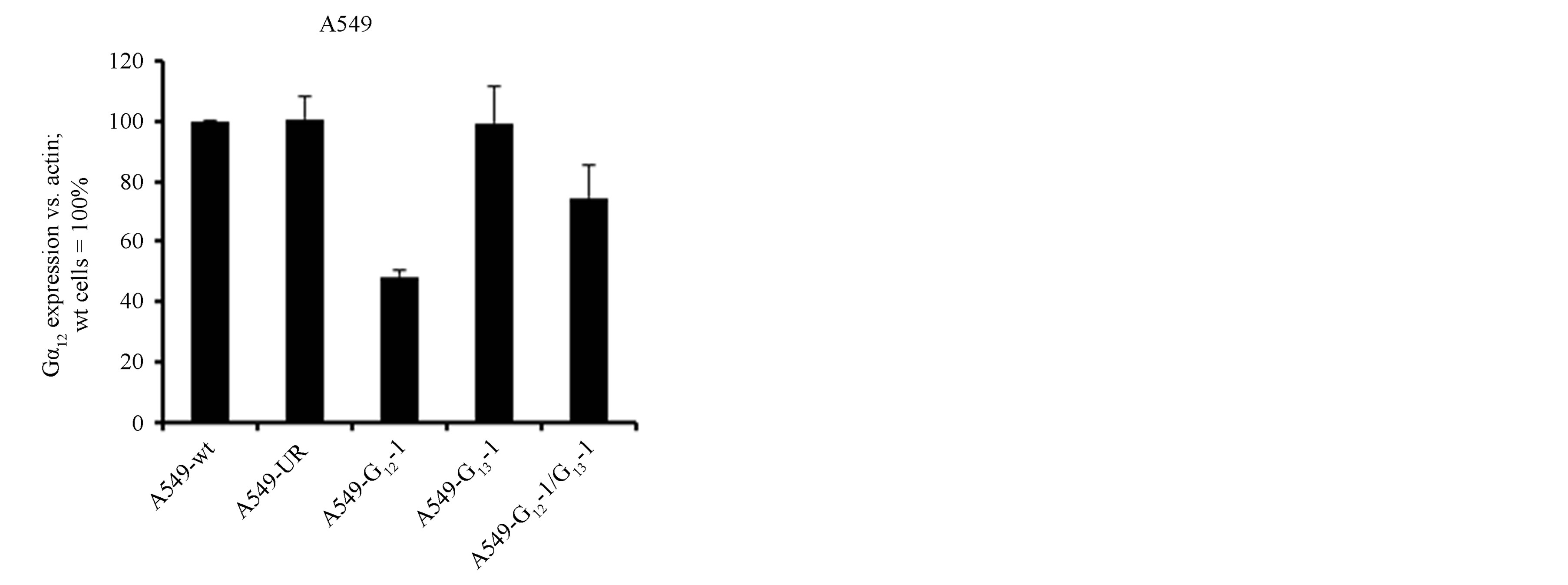
Extending our studies towards NSCLC, we tested the role of Gα12 and Gα13 in A549 cells. This cell line shows characteristics of adenocarcinoma cells and resembles type II alveolar cells. In these cells, the efficiency of shRNA-mediated knockdown of the Gα subunits was overall lower (albeit still significant) as compared to H510 or H82 cells, with a ~50% (Gα12) to ~60% (Gα13) residual levels (Figures 3(a) and (b)). Again, the double knockdown yielded somewhat less Gα12 knockdown efficacy as compared to the single targeting of Gα12. In this cell line, despite profound overall proliferation, no antiproliferative effect of the shRNAs was discernible (Figure 3(c)). Even the parallel Gα12 and Gα13 double knockdown did not show a trend towards growth inhibition. One possibility is that cell-type specific differences were responsible for the missing shRNA effects. Alternatively, this lack of anti-proliferative effects could be potentially attributable to the lower knockdown efficiency in these cells as compared to the SCLC cell lines. To further test these assumptions and to further analyse if Gα12 or cells Gα13 knockdown can lead to antiproliferation in NSCLC cells, another NSCLC cell line with a different biological background, H441, was employed.
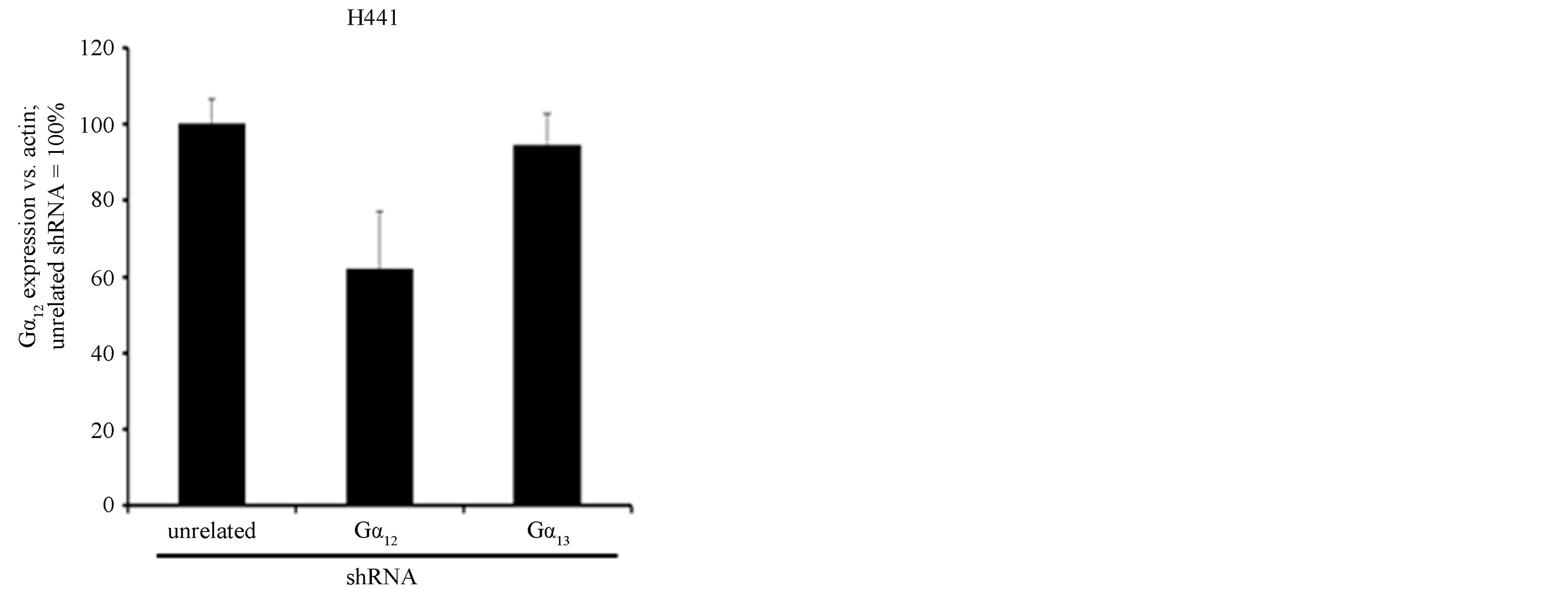
This cell line has features of adenocarcinoma cells but, in contrast to A549 cells, does not resemble alveolar type II cells while rather showing differentiation markers of club cells. Comparable to A549 cells, the efficiency of shRNA-mediated downregulation of Gα12 and Gα13 in H441 was generally lower when compared to the SCLC cell lines (Figures 4(a) and (b)). Despite the only 30% - 35% knockdown of Gα12 and Gα13, however, marked ~50% anti-proliferative effects were observed (Figure 4(c)). Thus, the relatively lower efficiency of shRNAs against Gα12 and Gα13 does not account (alone) for the lack of effects on proliferation in A549 cells. Rather, cell inhibi-
 (a)
(a)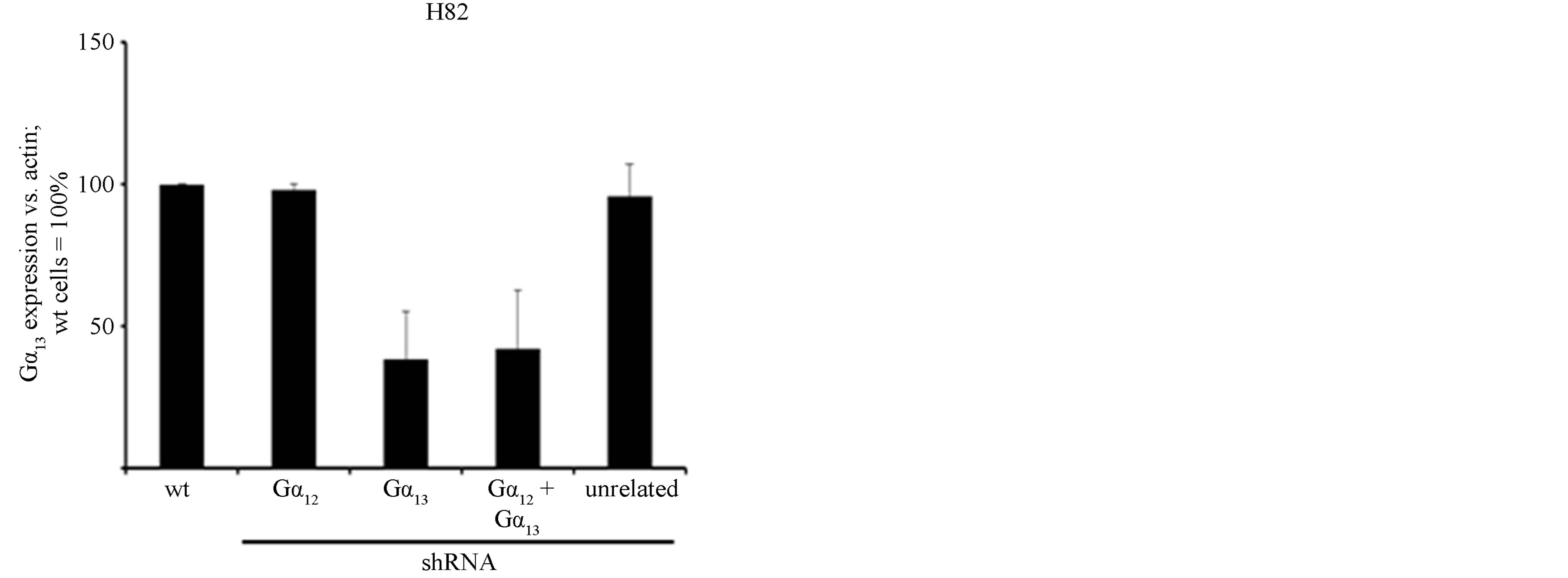 (b)
(b)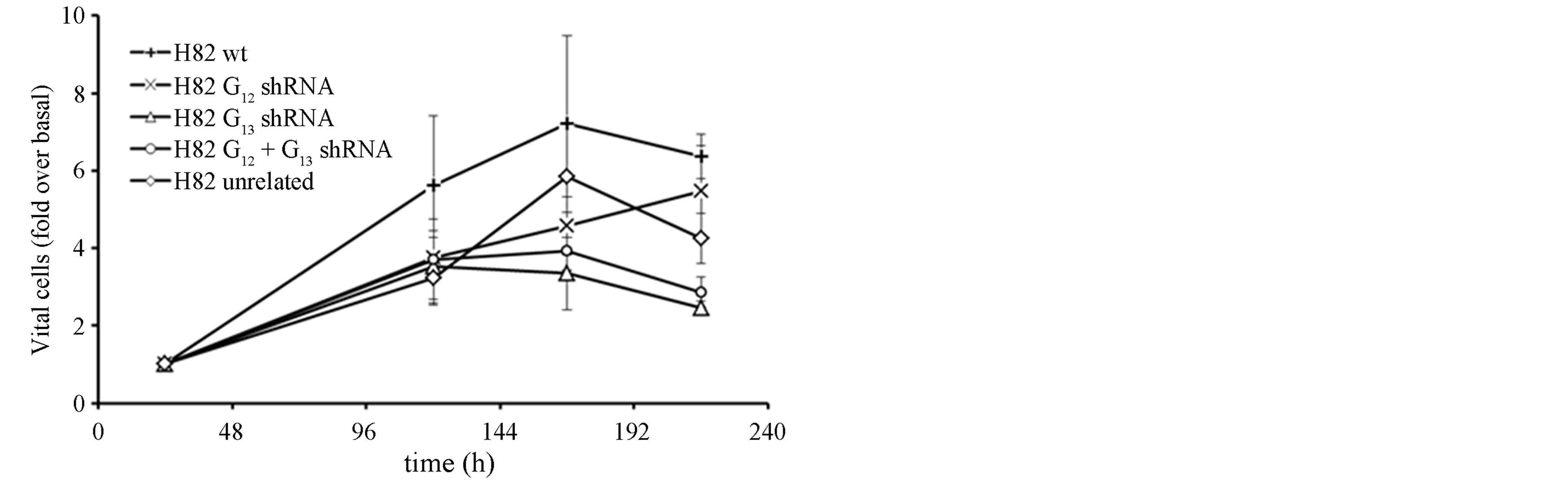 (c)
(c)
Figure 2. mRNA expression levels of Gα12 (a) and Gα13 (b) in relation to β-actin, as determined by quantitative RTPCR in the parental SCLC cell line H82 (wt, set to 100%), in H82 cells stably expressing shRNA directed against Gα12 or Gα13, respectively, in H82 cells expressing both Gα12 and Gα13 specific shRNA or, as a control, expressing unrelated shRNA. (c) Cell viability as determined by WST-1 assay in parental H82 cells (wt) or in H82 cells expressing unrelated shRNA or shRNA directed against Gα12 or Gα13; the start value (24 h after seeding) was set = 1.
tory effects upon Gα12 or Gα13 gene targeting appear to be cell line specific, with at least in some cases rather
 (a)
(a)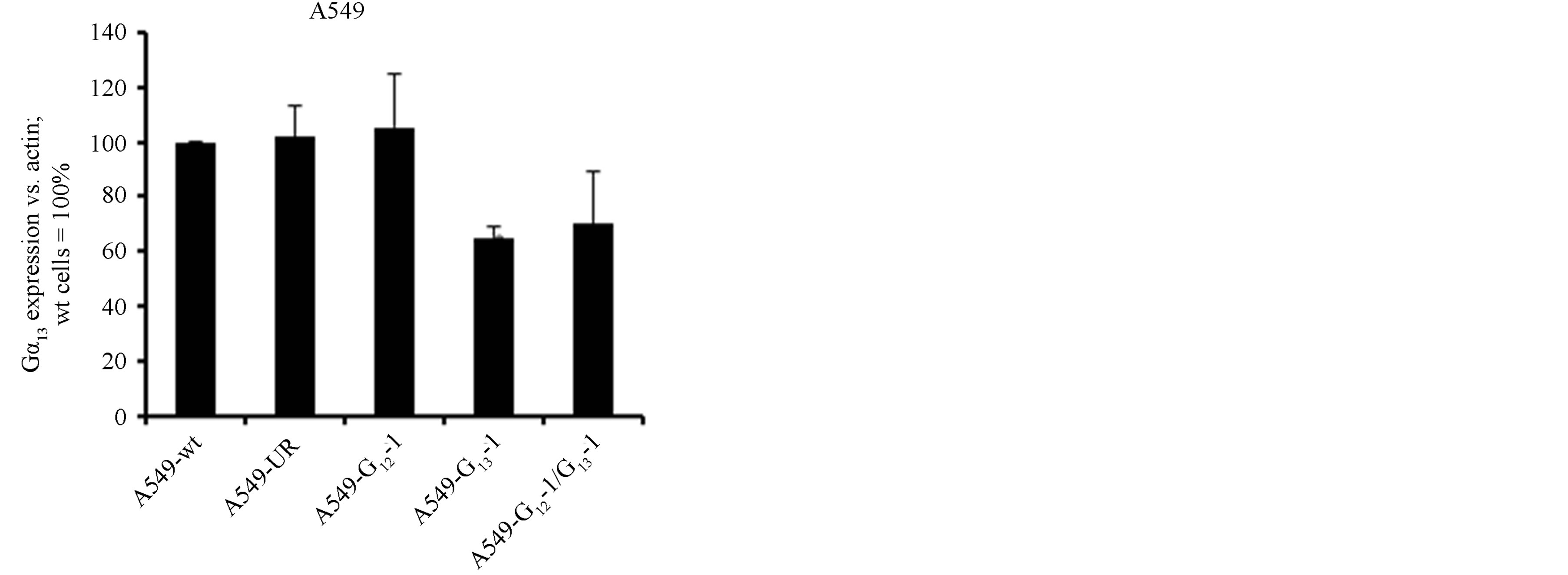 (b)
(b)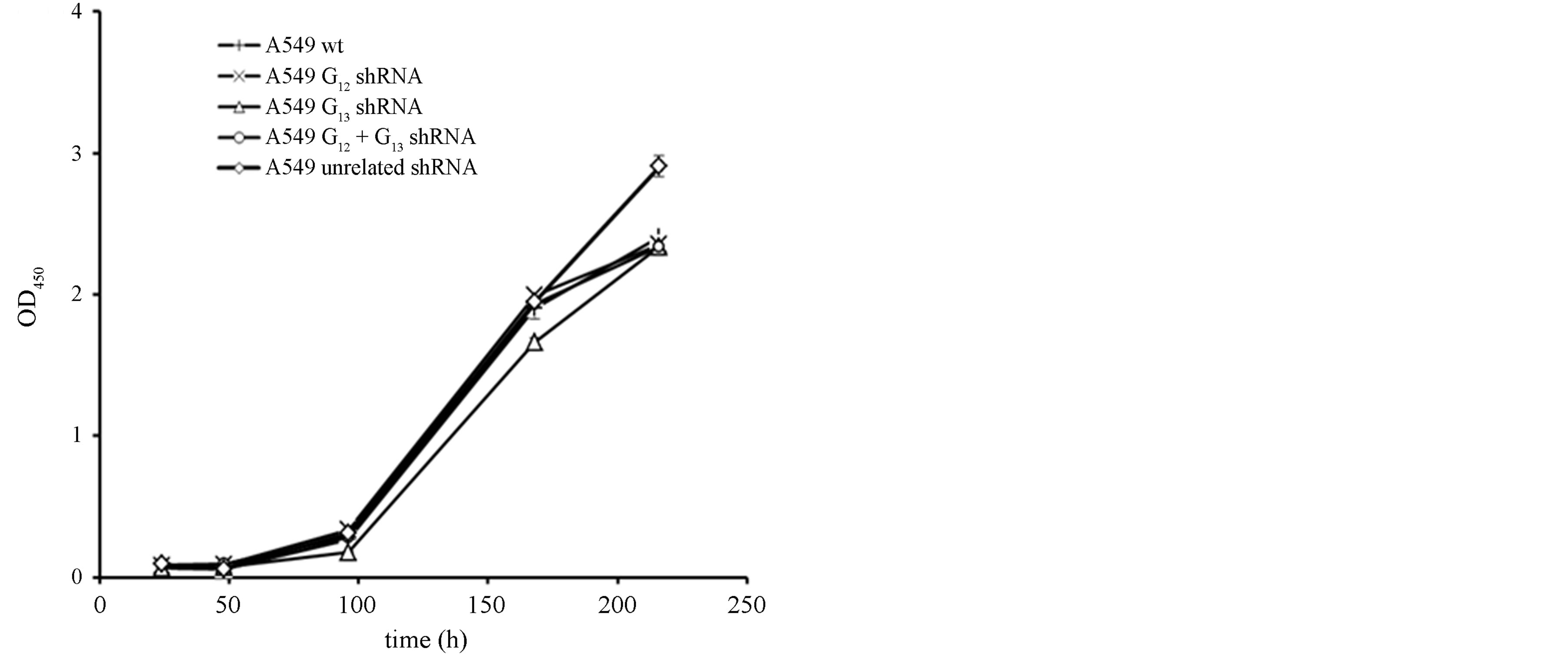 (c)
(c)
Figure 3. mRNA expression levels of Gα12 (a) and Gα13 (b) in relation to β-actin, as determined by quantitative RTPCR in the parental NSCLC cell line A549 (wt, set to 100%), in A549 cells stably expressing shRNA directed against Gα12 or Gα13, respectively, in A549 cells expressing both Gα12 and Gα13 specific shRNA or, as a control, expressing unrelated shRNA. (c) Cell viability as determined by WST-1 assay in parental A549 cells (wt) or in A549 cells expressing unrelated shRNA or shRNA directed against Gα12 or Gα13.
 (a)
(a) (b)
(b)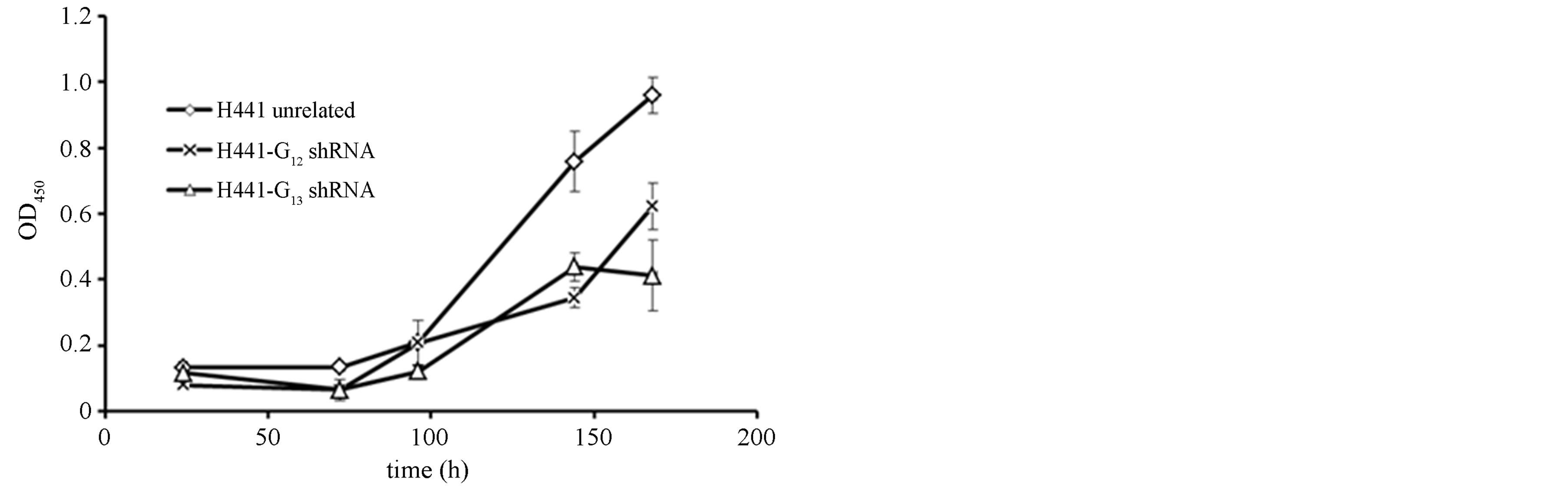 (c)
(c)
Figure 4. mRNA expression levels of Gα12 (a) and Gα13 (b) in relation to β-actin, as determined by quantitative RTPCR in H441 NSCLC cells stably expressing shRNA directed against Gα12 or Gα13 or, as a control, in H441 cells expressing unrelated shRNA. (c) Cell viability as determined by WST-1 assay in H441 cells expressing unrelated shRNA orshRNA directed against Gα12 or Gα13.
mild knockdowns being sufficient. In line with this, the individual contributions of Gα12 and Gα13 also seem to depend on the cell line, since in H441 cells a largely identical growth inhibition upon Gα12 or Gα13 single knockdown was observed, unlike in H510 (Gα12 effects more profound) or H82 cells (Gα13 knockdown more important).
4. DISCUSSION
Heterotrimeric G proteins are critically involved in the regulation of virtually every physiologic and pathophysiologic process. Since G protein-dependent signaling plays a pivotal role in the regulation of cellular growth, survival, motility, and differentiation, it does not come as a surprise that these proteins are important signaling molecules in cancer formation and progression and thus represent promising drug targets [7-10]. According to the different α subunits, these proteins are subdivided into the Gs, Gi/o, Gq/11, and G12/13 subfamily [11].
Of note, one of the first identified biological functions of the G12/13 subfamily was its propensity for neoplastic transformation, as shown after overexpression of wildtype Gα12 in mouse fibroblasts [12]. Moreover, in a number of studies a marked oncogenic potential of the Gα12 and Gα13 subunits has been described upon overexpression of GTPase deficient (and therefore constitutively active) variants [13,14]. Additionally, it has been shown that the stimulation of G12/13-coupled receptors can also induce neoplastic transformation [15,16]. Thus, even in the presence of endogenous expression levels of wildtype G12/13 proteins, the functional overactivation of these signaling molecules can contribute to a malignant phenotype. However, the tumorigenic effects of G12/13-dependent signaling may well be dependent on the tumor entity and cell-type [17]. Thus, the role of the individual protein (Gα12 or Gα13) has to be elucidated separately, i.e. apart from the other in every tumor entity, and many studies so far have relied on the somewhat non-natural ectopic overexpression of Gα12 or Gα13. The latter requirements make the RNAi-mediated knockdown a particularly well-suited approach for more detailed functional analyses.
One problem in the understanding of tumor signaling via G12/13 proteins is that a multitude of downstream effector molecules exist (see [18,19] for review). For this reason, the mechanistic basis of oncogenic signaling of G12/13 proteins is still widely unsettled. Besides a classic pathway that is regulated by G12/13-dependent cascades and affects the actin cytoskeleton via stimulation of Rho family proteins [20], thus leading to alterations in cell motility and consequently in their metastatic and invasive potential [21], a role of G12/13 proteins in proliferation or cellular survival has been emphasized by a number of studies [4,22]. Indeed, with regard to lung cancer cells, we demonstrated previously that the knockdown of Gα12 or Gα13 severely impairs the proliferation in the SCLC cell lines H69 and H209 [4]. This is in line with reports on the relevance of G12/13-coupled neuropeptide receptors [23] and on the constitutive secretion of autostimulatory agonists for G12/13-coupled receptors (for review: [2]).
Given the aforementioned cell-type specific roles of G12/13 signaling, however, it was critical to determine whether these previous findings were also attributable to other SCLC cell lines and could even be extended towards NSCLC. Indeed, the results presented herein suggest that an important role of G12/13 proteins in the regulation of proliferation can be also found in NSCLC cells like H441. However, the findings in the NSCLC cell line A549, where inhibition of G12/13 did not produce a growth inhibiting effect, underpin the importance of the cellular context for G12/13-dependent mitogenic effects.
Since G12 and G13 signaling affects overlapping target molecules, potentially redundant functions of these two G proteins are an important issue. Besides parallel, overlapping activities, this also includes the question if an enhancement in Gα12 signaling could compensate for the inhibition of Gα13 in tumor cells and vice versa. If so, the therapeutic efficacy of any intervention targeting only one of the two G proteins would be greatly diminished. The highly specific, RNAi-based knockdown in stable cell lines provides an excellent model to test the presence or absence of counter-regulatory effects. Importantly, the shRNA-mediated downregulation of Gα12 or Gα13 mRNA did not produce an upregulation of the non-targeted G protein. Moreover, despite the in different degrees of functional relevance of both G proteins in different cancer cells as outlined above, this was true in any of the investigated cell lines. It should also be noted that, unlike seen in other oncogene families (see e.g. [24] for HER receptor), the double targeting of both Gα12 and Gα13 did not lead to synergistic anti-proliferative effects. In tumor cells insensitive to a single knockdown of Gα12 or Gα13 (see A549 experiments), the double targeting of both G proteins had no anti-proliferative effect either. This demonstrates that the absence of a cellular dependence on Gα12 or Gα13 cannot simply be explained by the compensatory signaling through the other, and implies that other cellular or molecular features determine the cellular dependence on Gα12 and Gα13. More importantly from a possible therapeutic viewpoint, in cells sensitive to inhibition of G12/13 signaling like H510, H82, and H441, the targeting of one of the two G proteins is sufficient to impair proliferation.
Finally, one has to bear in mind that many G proteincoupled receptors constitutively activated in tumor cells propagate signaling by activating G12/13 and Gq/11 proteins in parallel [25,26]. Therefore, the downregulation of Gα12 or Gα13 may lead to a relative overactivation of Gq/11-dependent pathways. Thus, dysbalanced G protein signaling rather than the parallel activation of G12/13 and Gq/11 proteins may lead to an inhibition of proliferation, and may thus contribute to the antitumor effects and their cell line-specificity observed here upon downregulation of Gα12 or Gα13.
DISCLOSURE STATEMENT
The authors declare no competing financial interests.
ACKNOWLEDGEMENTS
We acknowledge the skillful technical assistance of Bärbel Obst. The project was funded by a grant from the Deutsche Forschungsgemeinschaft (DFG; Gu265/6-1, Ai 24/10-1) and by the German Center for Lung Research and the BMBF (German Ministry of Education and Research).
REFERENCES
- Govindan, R., Page, N., Morgensztern, D., Read, W., Tierney, R., Vlahiotis, A., Spitznagel, E.L. and Piccirillo, J. (2006) Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: Analysis of the surveillance, epidemiologic, and end results database. Journal of Clinical Oncology, 24, 4539-4544. http://dx.doi.org/10.1200/JCO.2005.04.4859
- Heasley, L.E. (2001) Autocrine and paracrine signaling through neuropeptide receptors in human cancer. Oncogene, 20, 1563-1569. http://dx.doi.org/10.1038/sj.onc.1204183
- Gudermann, T. and Roelle, S. (2006) Calcium-dependent growth regulation of small cell lung cancer cells by neuropeptides. Endocrine-Related Cancer, 13, 1069-1084. http://dx.doi.org/10.1677/erc.1.01302
- Grzelinski, M., Pinkenburg, O., Buch, T., Gold, M., Stohr, S., Kalwa, H., Gudermann, T. and Aigner, A. (2010) Critical role of G(alpha)12 and G(alpha)13 for human small cell lung cancer cell proliferation in vitro and tumor growth in vivo. Clinical Cancer Research, 16, 1402-1415. http://dx.doi.org/10.1158/1078-0432.CCR-09-1873
- Reynolds, A., Leake, D., Boese, Q., Scaringe, S., Marshall, W.S. and Khvorova, A. (2004) Rational siRNA design for RNA interference. Nature Biotechnology, 22, 326-330. http://dx.doi.org/10.1038/nbt936
- Elbashir, S.M., Harborth, J., Weber, K. and Tuschl, T. (2002) Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods, 26, 199- 213.
- Smrcka, A.V. (2013) Molecular targeting of Galpha and Gbetagamma subunits: A potential approach for cancer therapeutics. Trends in Pharmacological Sciences, 34, 290- 298. http://dx.doi.org/10.1016/j.tips.2013.02.006
- Wu, J., Xie, N., Zhao, X., Nice, E.C. and Huang, C. (2012) Dissection of aberrant GPCR signaling in tumorigenesis—A systems biology approach. Cancer Genomics & Proteomics, 9, 37-50.
- Entschladen, F., Zanker, K.S. and Powe, D.G. (2011) Heterotrimeric G protein signaling in cancer cells with regard to metastasis formation. Cell Cycle (Georgetown, Tex), 10, 1086-1091.
- Spiegelberg, B.D. and Hamm, H.E. (2007) Roles of Gprotein-coupled receptor signaling in cancer biology and gene transcription. Current Opinion in Genetics & Development, 17, 40-44. http://dx.doi.org/10.1016/j.gde.2006.12.002
- Hepler, J.R. and Gilman, A.G. (1992) G proteins. Trends in Biochemical Sciences, 17, 383-387. http://dx.doi.org/10.1016/0968-0004(92)90005-T
- Chan, A.M., Fleming, T.P., McGovern, E.S., Chedid, M., Miki, T. and Aaronson, S.A. (1993) Expression cDNA cloning of a transforming gene encoding the wild-type G alpha 12 gene product. Molecular and Cellular Biology, 13, 762-768.
- Xu, N., Bradley, L., Ambdukar, I. and Gutkind, J.S. (1993) A mutant alpha subunit of G12 potentiates the eicosanoid pathway and is highly oncogenic in NIH 3T3 cells. Proceedings of the National Academy of Sciences of the United States of America, 90, 6741-6745. http://dx.doi.org/10.1073/pnas.90.14.6741
- Xu, N., Voyno-Yasenetskaya, T. and Gutkind, J.S. (1994) Potent transforming activity of the G13 alpha subunit defines a novel family of oncogenes. Biochemical and Biophysical Research Communications, 201, 603-609. http://dx.doi.org/10.1006/bbrc.1994.1744
- Gardner, J.A., Ha, J.H., Jayaraman, M. and Dhanasekaran, D.N. (2013) The gep proto-oncogene Galpha13 mediates lysophosphatidic acid-mediated migration of pancreatic cancer cells. Pancreas, 42, 819-828. http://dx.doi.org/10.1097/MPA.0b013e318279c577
- Ha, J.H., Ward, J.D., Varadarajalu, L., Kim, S.G. and Dhanasekaran, D.N. (2014) The gep proto-oncogene Galpha12 mediates LPA-stimulated activation of CREB in ovarian cancer cells. Cellular Signalling, 26, 122-132. http://dx.doi.org/10.1016/j.cellsig.2013.08.012
- Offermanns, S. and Schultz, G. (1994) What are the functions of the pertussis toxin-insensitive G proteins G12, G13 and Gz? Molecular and Cellular Endocrinology, 100, 71-74. http://dx.doi.org/10.1016/0303-7207(94)90281-X
- Kelly, P., Casey, P.J. and Meigs, T.E. (2007) Biologic functions of the G12 subfamily of heterotrimeric g proteins: Growth, migration, and metastasis. Biochemistry, 46, 6677-6687. http://dx.doi.org/10.1021/bi700235f
- Dhanasekaran, N. and Dermott, J.M. (1996) Signaling by the G12 class of G proteins. Cellular signalling, 8, 235- 245. http://dx.doi.org/10.1016/0898-6568(96)00048-4
- Kozasa, T., Hajicek, N., Chow, C.R. and Suzuki, N. (2011) Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. The Journal of Biochemistry, 150, 357-369. http://dx.doi.org/10.1093/jb/mvr105
- Juneja, J., Cushman, I. and Casey, P.J. (2011) G12 signaling through c-Jun NH2-terminal kinase promotes breast cancer cell invasion. PloS one, 6, E26085. http://dx.doi.org/10.1371/journal.pone.0026085
- Radhakrishnan, R., Ha, J.H. and Dhanasekaran, D.N. (2010) Mitogenic signaling by the gep oncogene involves the upregulation of s-phase kinase-associated protein 2. Genes & cancer, 1, 1033-1043.
- Touge, H., Chikumi, H., Igishi, T., Kurai, J., Makino, H., Tamura, Y., Takata, M., Yoneda, K., Nakamoto, M., Suyama, H., Gutkind, J.S. and Shimizu, E. (2007) Diverse activation states of RhoA in human lung cancer cells: Contribution of G protein coupled receptors. Journal of Clinical Oncology, 30, 709-715.
- Rubinson, D.A., Hochster, H.S., Ryan, D.P., Wolpin, B.M., McCleary, N.J., Abrams, T.A., Chan, J.A., Iqbal, S., Lenz, H.J., Lim, D., Rose, J., Bekaii-Saab, T., Chen, H.X., Fuchs, C.S. and Ng, K. (2013) Multi-drug inhibition of the HER pathway in metastatic colorectal cancer: Results of a phase I study of pertuzumab plus cetuximab in cetuximab-refractory patients. Investigational New Drugs. http://dx.doi.org/10.1007/s10637-013-9956-5
- Beekman, A., Helfrich, B., Bunn, P.A., Jr. and Heasley, L.E. (1998) Expression of catalytically inactive phospholipase Cbeta disrupts phospholipase Cbeta and mitogenactivated protein kinase signaling and inhibits small cell lung cancer growth. Cancer Research, 58, 910-913.
- Wittau, N., Grosse, R., Kalkbrenner, F., Gohla, A., Schultz, G. and Gudermann, T. (2000) The galanin receptor type 2 initiates multiple signaling pathways in small cell lung cancer cells by coupling to G(q), G(i) and G(12) proteins. Oncogene, 19, 4199-4209. http://dx.doi.org/10.1038/sj.onc.1203777
NOTES
*Corresponding author.
#These authors contributed equally to the work.