Journal of Environmental Protection
Vol.4 No.8(2013), Article ID:35474,15 pages DOI:10.4236/jep.2013.48097
High Contribution of Sea Salt Aerosols on Atmospheric Particles Measured at an Urban Tropical Location in Reunion Island
![]()
1Observatoire Réunionais de l’Air (ORA), Réunion Island, France; 2LE2P, University of Réunion Island, Réunion Island, France; 3Institut National de l’Environnement Industriel et des Risques (INERIS), Verneuil en Halatte, France.
Email: *chatrapatty.bhugwant@atmo-reunion.net
Copyright © 2013 Chatrapatty Bhugwant et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received December 26th, 2012; revised January 30th, 2013; accepted February 9th, 2013
Keywords: PM10; nitrogen dioxide; anthropogenic; sea salt aerosols; chemical characterization; regulatory values; human health; lung cancer; urban; tropical
ABSTRACT
PM10 was monitored during 2008-2011 period at LUT and BON, two urban tropical stations close to each other at Saint-Pierre city, in Reunion Island (south-western Indian Ocean). During the study period, notable PM10 concentrations are observed at BON close to the coasts. At LUT, a more inland site, the daily PM10 concentration range between 13 and 70 µg/m3. Importantly, the limit value for the protection of the human health is systematically exceeded at BON while it is never exceeded at LUT. Also, the quality objective (QO: 30 µg/m3, on annual average) and the limit value for the human health protection (LV: 40 µg/m3, on annual average) are exceeded at BON each year during 2008-2011, while at LUT no regulatory values are exceeded. Nitrogen dioxide, an atmospheric tracer of anthropogenic activities was also monitored at LUT and BON. The mean diurnal NO2 variation is of the same level and order of magnitude and exhibits a similar pattern at both stations suggesting that distinct sources influence the PM10 at LUT and at BON. Chemical analysis was performed on daily filters sampled in September-November 2011 at the two stations to determine the sea salt contribution on PM10 across Saint-Pierre city. It showed that the sea salt contribution to the PM10 is 55% at BON in September 2011. The sea salt particles are therefore the main cause for the exceedances of the regulatory values of PM10 recorded at BON. The results importantly suggest that the notable PM10 concentrations measured at this urban marine site might have some but minor impact on human health.
1. Introduction
The study of particulate matter is of increasing interest to the scientific community (epidemiologists, researchers…) and to decision makers, in particular to determine the impact of this pollutant on human health [1-3]. It is thus important to conduct the monitoring of this pollutant especially in densely inhabited regions, in order to take adequate measures (e.g. prevent the surrounding population, in particular sensible persons such as children and old ones) if regulatory values are exceeded and also to study its long-term trend and effects. In this sense, since a few decades, decision makers have established regulatory values for the PM10 concentration levels, following the severity of the impact of this pollutant on human health and on the environment [4-6]. European directives, have set air quality standards (limit values, quality objectives) to be achieved for more than a dozen pollutants, among which the PM10 [7].
The European regulations for PM10 (e.g. Decree of 15th February 2002) is based on the following:
• Quality Objective (QO): 30 µg/m3 on annual average;
• Limit values (LV): 90.4% of the daily average must be below 50 µg/m3 (i.e., 35 days of exceedances allowed per calendar year) ;
• Limit values (LV): 40 µg/m3 on annual average.
Importantly, air quality organizations which provide air quality index daily to the population are confronted with the contribution of natural aerosols on particles measurements, especially in marine urban regions, which may in part hamper the air quality index.
Anthropogenic activities such as traffic circulation emit important amounts of particles and gases [8]. Several epidemiological studies indicate that lung cancer and other health effects are possibly associated with atmospheric particles [2,9-14]. In this sense, in June 2012, the International Agency for Research on Cancer (IARC) classified diesel engine exhaust as carcinogenic to humans (Group 1), based on sufficient evidence that this exposure is associated with an increased risk for lung cancer [3].
Natural sources are also a major cause for atmospheric particles [15-18]. In particular, marine aerosols contribute significantly to the global aerosols load and consequently influence the Earth’s radiative budget [19]. Some studies suggest that on coastal regions, sea salt aerosols may notably contribute to the annual mean particulate mass such as PM10 [20-22]. They emit a large amount of halogens, which are highly reactive and thus have a notable impact on the chemistry of hydrocarbons and/or ozone in the atmosphere [23]. Furthermore, sea salt plays an important role in a number of physical and chemical atmospheric processes [24,25]. For example, the reaction of sea salt particles with nitric acid forms sodium nitrate, which may be integrated in the particulate phase of the nitrogen budget [26]. The halogens released by the reaction of acidic gases with sea salt contribute, via heterogeneous reactions, to ozone destruction [27,28]. Sea salt may also indirectly impact vulnerable ecosystems via acid deposition [29,30]. Besides, sea salt aerosols contribute to corrosion of materials (e.g. metals) in coastal regions which cost billions for their rehabilitation [31]. Sea salt aerosols also contribute to cloud condensation nuclei (CCN), especially in marine regions [32,33]. Consequently, sea salt aerosols are essential components of atmospheric constituents at local, regional and global scales [34,35].
A number of studies have been dedicated to the modeling of atmospheric particles in general and sea salt particles in particular [36-38]. Many studies of marine aerosols and their role in the climate system were performed on the global scale [34,39-42]. For air quality assessment on local-to-regional scales, it is important to determine the gradients in sea salt levels and a number of regional models include a large description of sea salt [43-46]. The validation of these models is notably concerned by the limited number of available measurements. However, up to now, few atmospheric measurements have been undertaken with links to mixed sources (e.g. urban and marine aerosols) in particular the southern hemisphere [47-49].
The objective of this study is to quantify the sea salt contribution on the PM10 particles measured at two marine urban locations close to each other. Continuous PM10 measurements and chemical analysis of particles sampled daily on filters to determine the major ions (specific tracers of sea salt aerosols) was undertaken, to establish the distribution of sea salt aerosols on PM10 measured across Saint-Pierre city. These data were analyzed in conjunction with meteorological parameters in order to confirm the air masses origin. Finally, an assessment of the sea salt contribution based on the retrieval of the sea salt contribution on the PM10 measurements is proposed for the recalculation of the air quality index more realistic of anthropogenic activities.
2. Experimental Procedures—Measurement Locations
Réunion Island is a French department located in the South-Western Indian Ocean (21 S; 55 E). This tiny (area: ~2500 km2) and mountainous island (highest point: ~3075 m asl (above sea level)) holds two agglomerations with ~100,000 inhabitants: Saint-Denis, located to the North and Saint-Pierre located to the South of it (see Figure 1). Figure 1(a) is a zoom on Saint-Pierre city illustrating the geographical context of the measurement locations, in particular with respect to the coasts.
Saint-Pierre city is exempt of light/heavy industries susceptible emitting any atmospheric pollution and a significant proportion (~49%) of private cars as well as quite all trucks and buses operate with diesel at Reunion Island [50].
Figure 1(b) shows the configuration of Bons Enfants (BON hereafter) station, highlighting in particular:
• Its position, located about 200 m from the south coast of Reunion Island;
• Its proximity (about 1.5 km) with another urban station Luther King (LUT hereafter), the latter not showing any limit value of PM10 exceeded since 2008 (see Section 4.1.4 here after);
• The two measurement stations are located in the same urban environment, with light traffic circulation and at more than 0.5 to 1 km away from heavy highways. Hence, they are deemed representative of the urban background pollution level linked to anthropogenic activities (mainly traffic circulation);
• The presence of shallow water between the coast and the coral reefs which contributes to an efficient daily sea spray via wind flows and a potential sea salt source over this area. This is not the case for LUT which is downstream the coast too but where there is deep sea water with quite no coral reef and thus much lower sea spray emissions than at BON.
3. Experimental Set-Ups and Measurements
The PM10 and NO2 (nitrogen dioxide) concentrations
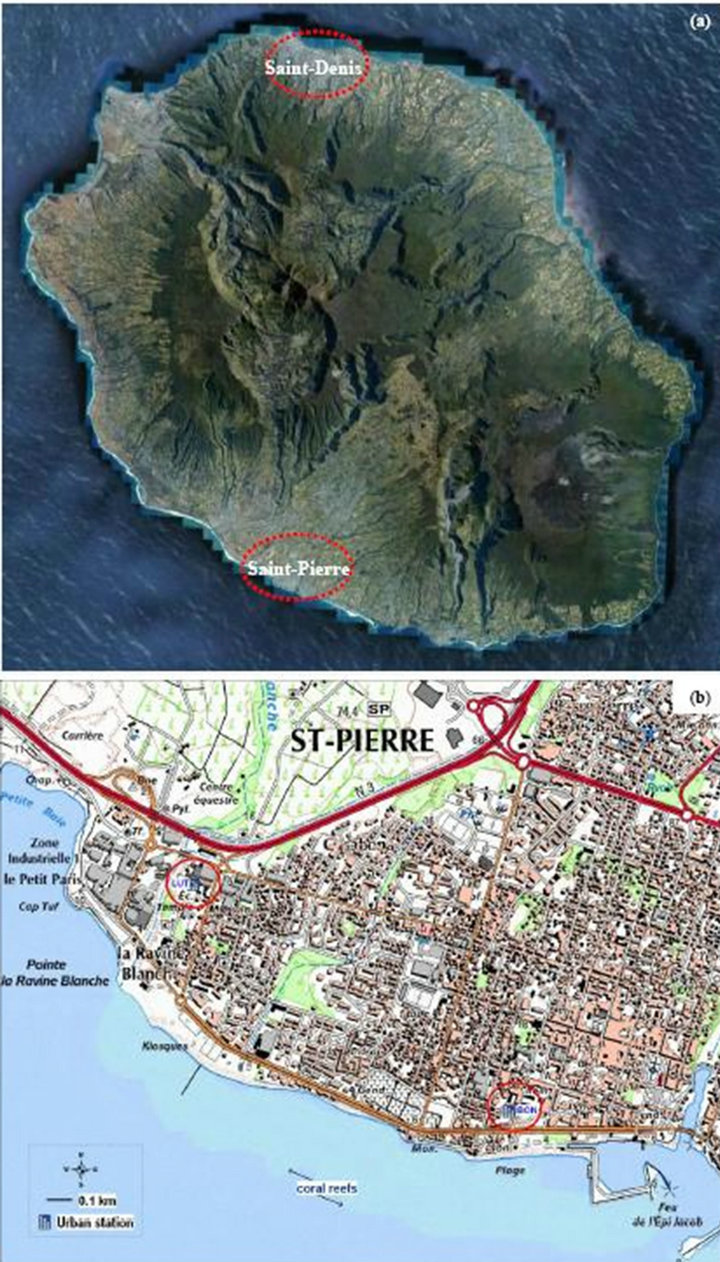
Figure 1. Map of Réunion Island. (a) (Source: Google Earth, 2011) and a zoom on Saint-Pierre agglomeration; (b) With location of Bons Enfants (BON) and Luther King (LUT) stations (Source: ©IGN-®authorization N˚ 9876).
have been recorded continuously at LUT and BON since August 2007.
The PM10 measurements are subsequently compared with meteorological data. In September 2011, atmospheric samples were also collected on filter on a daily basis for subsequent laboratory analysis in order to characterize the chemical composition of the PM10 particles.
3.1. Atmospheric Sampling Strategy
Figure 2 illustrates BON (upper left) and LUT (upper right) stations implemented at Saint-Pierre since mid- 2007. It may be seen that these two stations have the same characteristics and a similar environment.
3.1.1. Particles (PM10) Continuous Measurement
The PM10 concentration has been measured at LUT and BON using Tapered Element Oscillating Microbalances (TEOM, 1400ab models, commercialized by Thermo Environmental Instruments) equipped with Sample Equi-

Figure 2. Photography of the BON (upper left) and LUT (upper right) measurement stations.
libration and Filter Dynamic Measurements Systems (8500c models). The latter two devices (respectively referred to as SES and FDMS) allow limiting and taking into account Semi-Volatile Matter (SVM) losses inside the instrument. Indeed, the SES allows the reduction of heating at the inlet from 50 to 30˚C, and maintaining sample RH (relative humidity) below 25%, by means of a Nafion dryer. On the other hand, volatilization of SVM at 30˚C is resolved by the use of the FDMS system, which provides a full determination of volatile mass through a self-referencing gas conditioning scheme. PM measurements obtained by a TEOM equipped with both SES and FDMS systems have shown to compare very well with other real-time automatic analyzers accounting SVM [51-53]. The resulting measurement device is referred to as TEOM-FDMS hereafter. It allows continuous monitoring of the PM10 concentrations at a 15-mins time-base at the two Saint Pierre measurement locations.
3.1.2. NO2 Continuous Measurement
In order to determine the origin and main causes of the PM10 concentration variability recorded at LUT and BON, nitrogen dioxide (NO2) concentration, an atmospheric tracer of anthropogenic origin (mainly traffic circulation, in absence of any other source such as heavy industries and/or volcanic eruption) [49], was also monitored in parallel with the PM10 measurements by a AC31M analyzer commercialized by Environnement SA.
The principle of the NOx measurement, by the chemiluminescence’s technique is described in Bhugwant and Hoareau (2003) [54].
3.1.3. Determination of Aerosols Composition from Filter Samples
PM10 samples were collected daily on filters at LUT and BON in parallel with continuous atmospheric (PM10 and NO2) measurements during experiments carried out over a half-year, from July 2011 to December 2011.
The PM10 sampling was performed continuously and simultaneously at both LUT and BON sites by ORA using Partisol samplers type 2025 provided by the LCSQA (Laboratoire Central de Surveillance de la qualité de l’Air). The sampling was performed for two weeks sequences interspersed with a day off.
The filters used are of Tissuquartz Qat-Up 2500 type (diameter: 47 mm) commercialized by Pall, chosen for their low sodium and other mineral species levels (except silica). Filters were initially pre-fired at 500˚C during 2 hours to minimize filter blanks. Sampling was performed at a constant air-flow rate of 16 l/hr, and for a duration of 24-hour per filter. They were then stored at a temperature below 20˚C, including during transport (in cooler).
Chemical analyzes of selected filters sampled at LUT and BON were performed by LCSQA/INERIS. The analyses were performed in accordance with the CEN/TR 16269 and 16243 technical reports for the quantification of anions/cations (Cl−, NO3−, 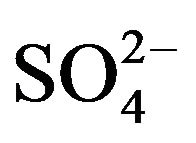 , Na+,
, Na+, 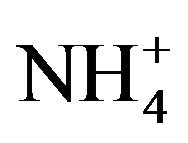 , K+, Mg2+ and Ca2+) respectively using ion chromatography, and carbon fractions (elemental carbon: EC and organic carbon: OC) using a thermal-optical protocol [55] implemented on a Sunset Lab. Instrument [56].
, K+, Mg2+ and Ca2+) respectively using ion chromatography, and carbon fractions (elemental carbon: EC and organic carbon: OC) using a thermal-optical protocol [55] implemented on a Sunset Lab. Instrument [56].
3.2. Complementary Data
Meteorological Data
The local meteorological parameters (winds speed and direction, etc.) are measured from the instruments onboard a tower at 10 m above the ground level in meteorological stations located close to LUT and BON locations. These instruments are part of the French Meteorological Service [57]. The meteorological data was collected from January 2008 to December 2011 at Pierrefonds (PIE), a station located in the vicinity of Saint Pierre city. This meteorological station is close to LUT and BON (distance: ~3.4 and 4.9 km respectively, and located to the north-west from them) and representative of the meteorological conditions prevailing over Saint-Pierre.
The meteorological data measured continuously with a 1/2-hour time-base were subsequently brought to a 1- hour basis in order to be compared with the atmospheric (PM10 and NO2) data.
4. Results and Discussion
The analysis of the atmospheric and meteorological datasets is first focused on their mean diurnal variation from one year to another, in order to study their trend and levels and also to look for possible links between them. The datasets are subsequently analyzed on a daily basis, in order to look closely at their variability and also to compare with the PM10 regulatory values. The daily PM10 variability at LUT and BON are then compared with the sea salt (cations and anions, in particular Na+ and Cl−) concentrations analyzed on the daily filter samples.
4.1. Diurnal Variation during the Study Period
4.1.1. Mean Diurnal PM10 Concentration Variation
The mean diurnal variation of PM10 concentration has been calculated at LUT and BON from 2008 to 2011, period during which data are available for a whole year (see Figure 3).
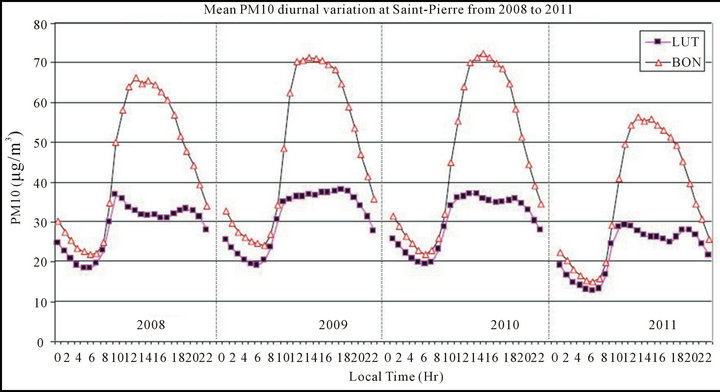
Figure 3. Mean diurnal PM10 concentration calculated annually at LUT and BON stations from 2008 to 2011.
The PM10 diurnal variation exhibits a particular pattern at BON and at LUT, with maximum levels at daytime (during significant anthropogenic activities, mainly traffic circulation) and minimum levels at night-time (absence of anthropogenic activities). However, no peak, corresponding to the morning and evening traffic circulation peaks is noticed except for LUT in 2008, 2010 and 2011, where such a tendency is slightly noticed, suggesting some influence of traffic circulation on the PM10 concentration at this site. Some studies show that PM10 measurements in urban locations mainly influenced by anthropogenic activities (in absence of industrial and natural/marine sources) exhibit a good correlation with traffic density [47,58]. Importantly, the PM10 levels are systematically and notably higher at BON than at LUT, especially during daytime (e.g. at 2:00 p.m., mean PM10 at BON: ~65 µg/m3 and at LUT: ~35 µg/m3).
Moreover, the mean diurnal evolution exhibits a notable variation from one year to another. Indeed, higher PM10 levels are observed in 2009 and 2010 (mean: 48 and 47 µg/m3 at BON and 31 and 30 µg/m3 at LUT respectively) while moderate levels are noticed in 2008 (mean: 44 µg/m3 at BON and 28 µg/m3 at LUT respectively) and low levels are measured in 2011 (mean: 36 µg/m3 at BON and 21 µg/m3 at LUT respectively). Nev ertheless, as the two locations are representative of the urban pollution over Saint Pierre city and close to each other, they are expected to record the same levels, tends and order of magnitude of PM10 concentrations, which is not the case. This variability may in part be due to the influence of meteorological conditions and other particles sources. Importantly, there is not any industry emitting atmospheric pollutions at Saint Pierre, especially in the vicinity of LUT and BON and both stations are quite equally distant from light/heavy highways (see Figure 1(b)).
4.1.2. Mean Diurnal NO2 Concentration Variation
It is essential to look for the influence of the anthropogenic activities (i.e., mostly traffic circulation) at SaintPierre city in order to establish links with the PM10 diurnal variability. In this sense, the mean diurnal variation of NO2 concentration measured at LUT and BON has been calculated each year from 2008 to 2011 (see Figure 4).
Firstly, the diurnal NO2 concentration variation exhibits the same level and trend at the two sites. It exhibits systematically a bimodal pattern, with a maximum peak at 7:00 a.m. and a secondary peak at 8:00 p.m. at both LUT and BON, attributable to morning and evening traffic peaks [48,58,59]. The hourly NO2 concentration is of the same order of magnitude at both sites but is in general slightly higher at BON than at LUT suggesting that the former site is a little more influenced by atmospheric pollutants induced by the surrounding traffic circulation. It may also be noticed that the mean diurnal evolution exhibits an annual variation, with however a difference with the PM10 diurnal profile (see Figure 3).
As observed for the PM10, high NO2 levels are also observed for 2010 (mean: 17 µg/m3 at BON and 14 µg/m3 at LUT respectively) while moderate levels are noticed for 2008, 2009 and 2011 (mean: 13, 14 and 13 µg/m3 at BON and 13, 11 and 12 µg/m3 at LUT respectively). The difference on the PM10 variability observed at the two sites is not noticeable on the NO2 concentration variation. The results imply that the anthropogenic activities contribution on the PM10 is quite similar and of the same order at both LUT and BON. Therefore, the potential factors which might explain the differences on the PM10 observed at LUT and BON, mainly during daytime, might be the conjugate effect of other (natural sources such as marine aerosols) contribution and dynamical processes. Indeed, as described earlier, BON is closer to the sea side (~200 m away from it) than LUT (~500 m away from it). Moreover, coral reefs are located directly downwind to the south of BON while they are quite far from LUT (see Figure 1(b)).
Due to the geographic configuration of the island (important relief) and the existence of coral reefs especially downwind BON station, the sea waves break constantly over the coral reefs and continuously generate marine aerosols via sea spray. This process might explain the higher PM10 levels systematically observed at BON than at LUT, especially during daytime when sea breezes prevail.
4.1.3. Mean Diurnal Winds (Direction and Speed) Variation
We tried to gain more information about the origin of air samples attaining Saint-Pierre agglomeration during the study period. We therefore analyzed meteorological data collected at PIE meteorological station. However, due to technical problems during the study period (from 2008 to 2011), only meteorological data measured in 2008 and 2009 are available. The mean winds (direction and speed) diurnal variation were calculated from winds data measured at PIE in 2008 and 2009 (see Figure 5).
The analysis being focused on the diurnal variation for each year, the change from one year to another on the diurnal pattern may only be on the winds amplitude of variation. The two years data available provide sufficient information about the daily variation and may be extrapolated to the 2010-2011 period. The mean winds (direction and speed) diurnal variation shows similar trends, patterns and levels in 2008 and 2009. High wind speeds (~5.7 m/s) are noticed at daytime (9:00 a.m. to
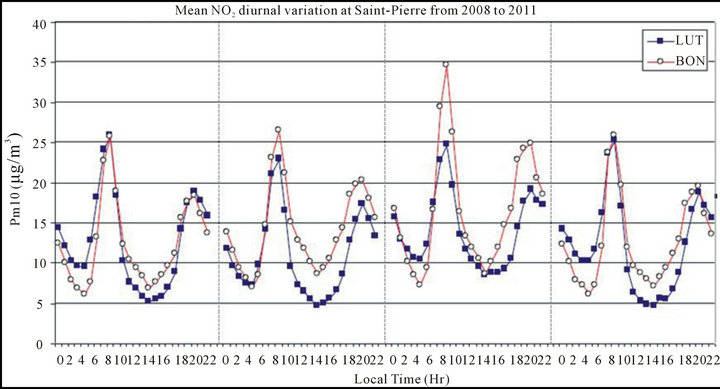
Figure 4. Mean diurnal NO2 concentration calculated annually at LUT and BON stations from 2008 to 2011.
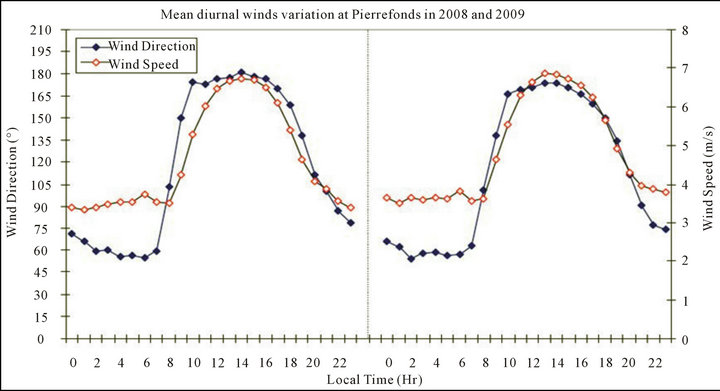
Figure 5. Annual winds diurnal variation calculated at Pierrefonds (PIE) meteorological station in 2008 and 2009.
8:00 p.m.) while at night-time (from 9:00 p.m. to 8:00 a.m.) they are low (~3.6 m/s). This difference is mainly due to the conjugate effect of prevailing easterlies and the land/sea breeze processes.
During night-time, Saint-Pierre city is under the inland airflow (~70˚), with a north-east origin of air masses. During daytime however, it is mainly of marine origin and thus under sea-spray flow, as the wind is in a south-east direction (~150˚). In addition to the large scale trade winds prevailing over the region, the daily abrupt change in the morning and afternoon airflow is typical of a land/sea-sea/land breezes process and is experienced by day-to-day variation at Saint-Pierre throughout the year [57]. Thus, a conjunction effect of winds (breezes and easterlies) and human activities occurs simultaneously at Saint-Pierre at daytime. The analysis points out that at night-time when the anthropogenic activities are quite negligible and the air masses prevailing over Saint-Pierre originate on average from inland (north-east), the PM10 and NO2 concentration levels are lowest and correspond to the background atmospheric pollution level over Saint-Pierre city. Conversely, at daytime when the anthropogenic activities are notable and the air masses prevailing over Saint-Pierre originate from south-east to south (mainly marine), the PM10 and NO2 concentration levels are highest and variable. The results suggest that at daytime, while the NO2 concentration variation is mainly due to local anthropogenic activities, the PM10 concentration is influenced by local anthropogenic activities and sea spray flow and thus marine sources as well, mostly at BON, as the latter site is closest to the coast than LUT.
4.1.4. Mean Monthly and Daily PM10 Concentration Variation
The monthly mean PM10 concentration calculated for 2008-2011 (not shown) indicates that during austral winter (August to October), the PM10 concentration is highest, with a mean value of 30 µg/m3 and 47 µg/m3 at LUT and BON respectively. Then, during austral summer (January to March), moderate PM10 concentration levels are observed (monthly mean: 28 µg/m3 and 43 µg/m3 at LUT and BON respectively). However, from April to July then from November to December, minimum PM10 values are measured at both sites (monthly mean: 27 and 41 µg/m3 at LUT and BON respectively). This PM10 seasonal variability may in part be explained by whitecaps and bursts, injecting seawater drops into the atmosphere, in particular when strong easterlies prevail in this region [42]. Also, the monthly PM10 evolution is corroborated with the swell seasonal variation, which exhibits high wave levels, in general in February and in October over the South-western Indian Ocean region [49,60]. Besides, during austral summer, the trade winds are stronger and thus the sea spray production due to wind shear on the sea surface is higher [57].
The mean daily PM10 concentration calculated at LUT and BON from 2008 to 2011 was also analyzed (not shown).We notice that the daily PM10 concentration exhibits distinct trend and level at both sites, from one year to another.
Indeed, during 2008, highest daily PM10 concentrations are observed from January to March then from September to November, while they are lowest from May to August at both sites. In 2009 however, while the daily PM10 concentration is high at BON from January to March, it exhibits moderate levels at LUT. Higher daily PM10 levels are observed both at BON and at LUT from April to September. Then, From October to December, lowest daily PM10 levels are observed at LUT while they remain high at BON. In 2010, the daily PM10 concentration is highest at BON from January to May while it is at moderate levels at LUT during this period. From June to October, the daily PM10 concentration is at moderate levels at BON while it is highest at LUT. Then, from November to December the daily PM10 levels are low both at BON and at LUT. In 2011, from January to March the daily PM10 concentration is highest at LUT while it exhibits moderate levels at BON. Then, from May to December, no clear trend is observed at both sites, with systematically higher PM10 levels at BON than at LUT.
The differences in the daily PM10 concentration seasonal and annual variation at BON and LUT may be explained by the conjugate effect of changes in meteorological conditions (winds, insolation, rainfall etc.), in source effects (anthropogenic activities and marine sources) and also in regional events such as swell and sea waves activities.
Importantly, the quality objective (30 µg/m3, on annual average) as well as the limit value for the protection of the human health (Centile 90.4 = 50 µg/m3, on daily average not to be exceeded more than 35 days/year and the limit value (LV = 40 µg/m3, on annual average) are exceeded each year at BON, when the daily PM10 datasets are analyzed on a civil year.
The number of days exceeding the limit value and the annual average for the PM10 concentration at BON and LUT from 2008 to 2011 were calculated (see Table 1). It may be seen that the quality objective (QO = 30 µg/m3, on annual average) as well as the limit value for the protection of the human health (LV = 40 µg/m3, on annual average) are systematically exceeded every year at BON (except for the VL which is not exceeded in 2011), while at LUT only the QO is exceeded in 2010.
Also, the centile 90,4 (i.e., the number of days exceeding the limit value of 50 µg/m3, not to be exceeded more than 35 days/year) is systematically exceeded at BON each year, while at LUT the number of days exceeding the limit value of are quite lower than 35 days.
The results point out that the origin and main causes of the PM10 concentration at BON is quite different than at LUT, although both urban sites are close to each other.
4.2. Determination of the Chemical Composition of the PM10
The knowledge of the particles composition measured
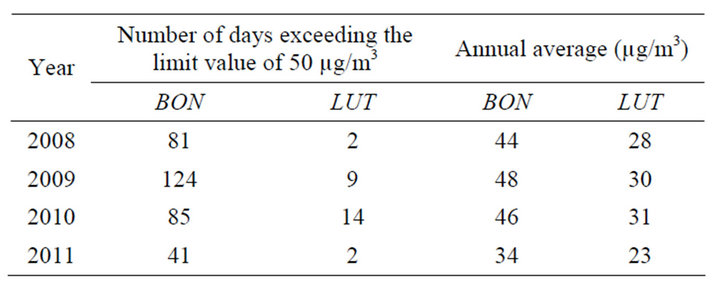
Table 1. Number of days exceeding the limit value and the annual average calculated for the PM10 concentration measured at BON and LUT from 2008 to 2011.
in Saint-Pierre is a key to confirm and quantify the contribution of sea salt in the PM10 concentration exceedances. In a preliminary study undertaken in late 2008, it has been suggested that one of the main causes of the PM10 concentration variability at BON may be an additional contribution of sea salt aerosols [60]. Based on in-situ observations, such as large deposits of sea salt crystals inside the samples head (see Figure 6), piecemeal analysis of the chemical composition of filters collected on the automatic PM10 (TEOM-FDMS) analyzers, and analysis of the waves and swell data with the PM10 concentrations in late 2008, the probable role played by the sea salts in the occurrence of exceedances of limit values on PM was highlighted [60].
In this sense, since July 2011, particles sampled daily on filters in parallel with continuous PM10 concentration measurements at BON and LUT were analyzed in laboratory by INERIS. The analyses were performed for 39 days of filter samples during which the daily PM10 concentration mostly exceeded the limit value of 50 µg/m3 between September 2011 and November 2011.
The aim of this chemical characterization was to determine the fraction of daily sea salt particles measured in the PM10 measurements at LUT and BON stations.
The sea salt concentrations are determined following the recommendations of the European guide for estimating the contributions from natural sources, as follows:
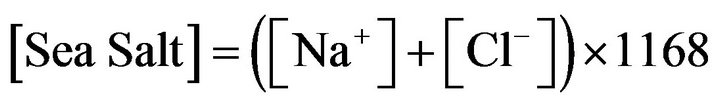 (1)
(1)
and after verification of the correct correlation and compliance reports characteristics of sea droplets between these two species and between Na+ and Mg2+, magnesium being also a good tracer of sea salt.
The sea salt sulfate (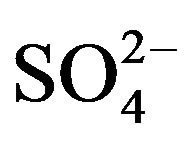 ) concentrations contribution is determined using the
) concentrations contribution is determined using the 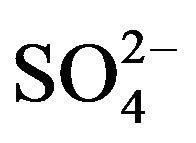 /Na+ ratio of seawater (~0.25), allowing then to determine the concentra-
/Na+ ratio of seawater (~0.25), allowing then to determine the concentra-

Figure 6. Photography showing the deposition of sea salt crystals inside the PM10 sampling head at BON.
tions of non sea salt sulfate (nss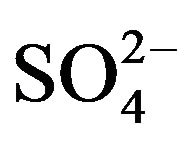 ). Finally, particulate organic matter (OM) concentrations are obtained using a conversion factor of 1.6 * OC à OM, [61,62].
). Finally, particulate organic matter (OM) concentrations are obtained using a conversion factor of 1.6 * OC à OM, [61,62].
The daily PM10 concentrations recorded at BON and LUT stations have been calculated from August 2011 to mid-December 2011, period during which the particles sampled on filters were available for chemical analysis (see Figure 7).
We have focused more specifically the chemical analysis on the September 2011 results, during which there is a marked episode of daily exceeded PM10 values (between 05/09/2011 and 10/09/2011). During this month, there are 12 exceedances of the daily limit value threshold on the PM10 at BON. It may be noted that during this period, LUT has not exceeded any daily threshold value.
4.2.1. Sea Salt Contribution on the PM10 Concentrations in September 2011
The average chemical composition was performed for September 2011 (from 1st to 13th then from 15th to 27/09) from daily filters sampled at LUT and BON stations (see Figure 8). It may be noted that the sea salt appears clearly as the main fraction of PM10 sampled on each site (~50% at LUT and ~55% at BON).
We also note the large proportion of chemical species other than the cat-ions/anions and carbonaceous species, which can not be explained only by other components such as heavy metals (not shown), the latter being a minor/negligible contribution. This part is reasonably, primarily, attributable to the presence of liquid water on marine aerosols. Indeed, sea salts are known to be highly hydrophilic and are in fact present in the atmosphere as saline suspension, in particular over coastal regions [16]. The validity of this assumption is confirmed by the quite good correlation established between the sea salt concentration and the difference between PM10 and the sum of the other chemical species {EC + OM + 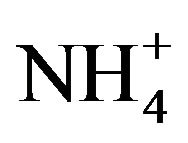 +
+ 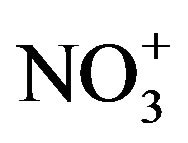 + nss
+ nss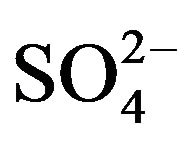 } (see Figure 9).
} (see Figure 9).
The value of the slope obtained for the linear regression suggests that the sea salt contribution to the PM10 concentrations recorded at LUT and BON stations is about 1.34 times larger than the suggested estimate of the sea salt concentrations in the non-hydrated solid form.
This hypothesis implies that a mass of water vapor (denoted ssH2O hereafter) corresponding to 1/3 of the mass of dry sea salt is also measured by the TEOMFDMS.
It is in good agreement with the modeling results of hygroscopic properties of NaCl crystals placed at a relative humidity of 10% corresponding to the relative humidity in the dryer outlet of the FDMS modules [63]. It may also be noted that the intercept (~3.7 µg/m3), obtained for the correlation presented in Figure 9 might be
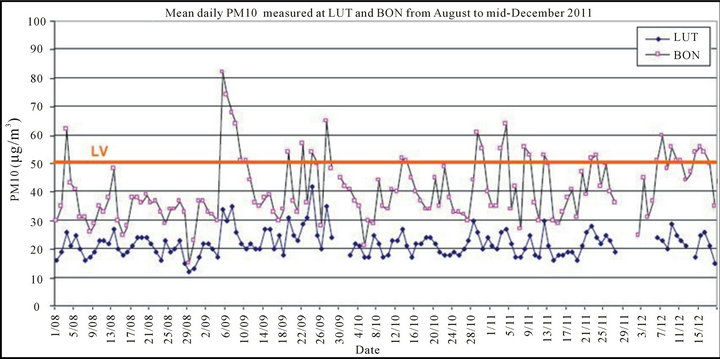
Figure 7. Mean daily PM10 concentrations measured at BON and LUT stations from August 2011 to mid-December 2011.
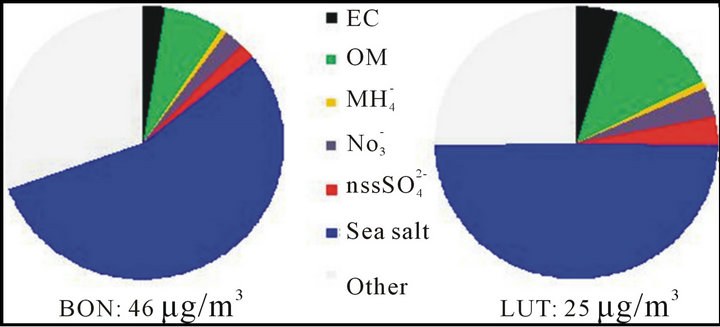
Figure 8. Average chemical composition of PM10 sampled daily on filters at LUT and BON in September 2011.
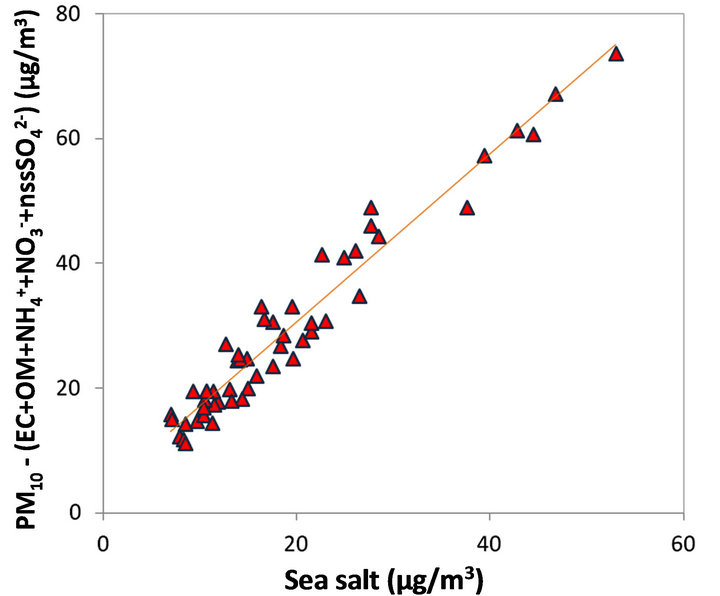
Figure 9. A proportional relationship obtained between the sea salt (non-hydrated) concentrations and an additional amount (other than EC, OM, 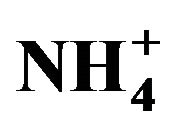 ,
, 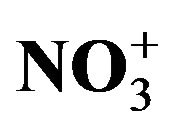 and nss
and nss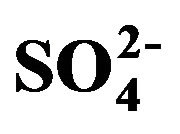 ).
).
primarily attributable to the presence of terrigenous particles in the PM10 as well as the instrumental TEOMFDMS blank.
As shown in Figure 9, the results allow a quite exhaustive “daily chemical closure” of PM10 concentrations measured on the two study sites during September 2011. It therefore appears that the sea salt contribution is maximal on days corresponding to exceedances of the 50 μg/m3 daily threshold at BON, while this contribution is quite less at LUT, and therefore the notable differences observed between both sites can mainly be attributed to sea salt aerosols.
The daily concentrations of major chemical species which constitute the PM10 fraction at LUT and BON stations during September 2011 were calculated (see Figure 10).
The results presented in Figure 10 suggest not only that the daily threshold exceedances recorded at BON station are mainly due to sea salt aerosols but also that almost all other chemical species studied present relatively homogeneous concentrations at both LUT and BON sites.
This result of this analysis (see Figure 11(a)) indicating a very good correlation between the sum of {EC + OM + 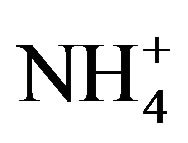 +
+ 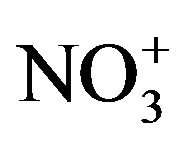 + nss
+ nss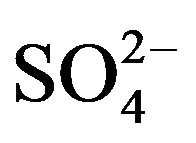 } on the two stations for September 2011. It is also noteworthy that the difference of PM10 concentrations between these two stations can largely be attributed to the difference in concentrations of sea salt hydrates attaining each site (see Figure 11(b)).
} on the two stations for September 2011. It is also noteworthy that the difference of PM10 concentrations between these two stations can largely be attributed to the difference in concentrations of sea salt hydrates attaining each site (see Figure 11(b)).
We can therefore reasonably consider that there is an almost linear relationship between the difference of
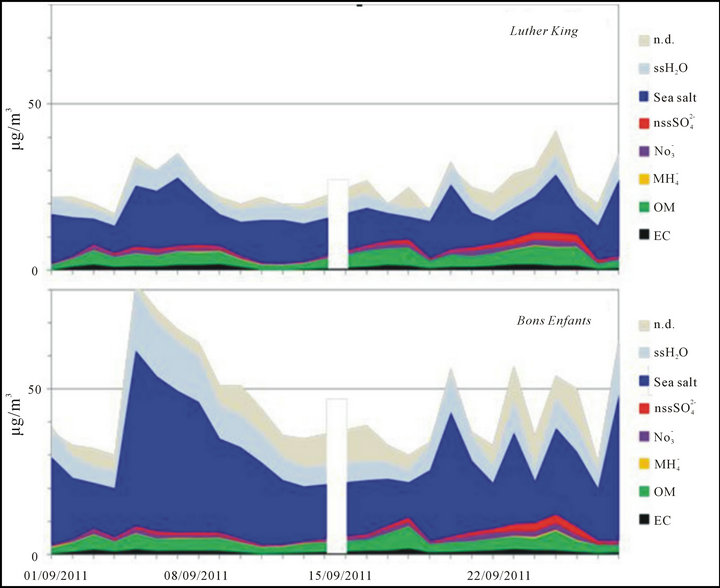
Figure 10. Daily concentration of major chemical species which form the PM10 fraction at LUT and BON stations during September 2011.
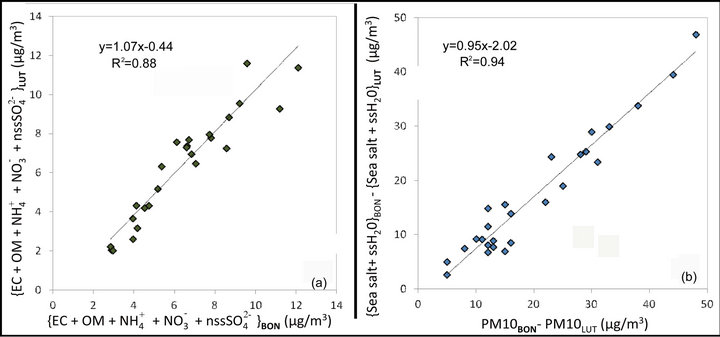
Figure 11. Homogeneity of particle concentrations mainly of anthropogenic origin observed at BON and LUT (a) and the major role played by sea salts on the difference in PM10 concentrations measured at the two stations (b).
PM10 concentration measured at the two stations and the sea salt contribution at BON station. This relationship is illustrated by Figure 12, suggesting the following equation:
 …(2)
…(2)
As mentioned earlier, the intercept of this equation is partially related to the presence of terrigenous particles and values of the instrumental PM10 analyzers blank used.
However, its temporal variability and the uncertainty estimation it induces (possible risk of overestimation) can be considered as an offset, in the case of daily exceedances related to sea salt, by not taking into account the liquid water (ssH2O) proposed in equation (2).
4.2.2. Differences between PM10 Concentrations Measured at Both Sites
Due to technical problems (related to the malfunction of the samplers as a result of high ambient relative humidity) and analytical costs, it was not possible to analyze in such detail all daily PM10 exceedances recorded in late 2011. Nevertheless, based on the results described in Section 4.2.1, it is possible to specify that all the exceedances observed in October and November 2011, can be mainly attributed to sea salt contribution (see Table 2).
The equation (1) proposed previously was applied to each daily threshold exceedances that could be investigated during late 2011. This application was performed in order to “re-calculate” the sea salt contribution at BON station from differences of PM10 concentrations between BON and LUT during days on which the daily exceedances were measured at BON. The data obtained were then compared to the sea salt concentrations esti-
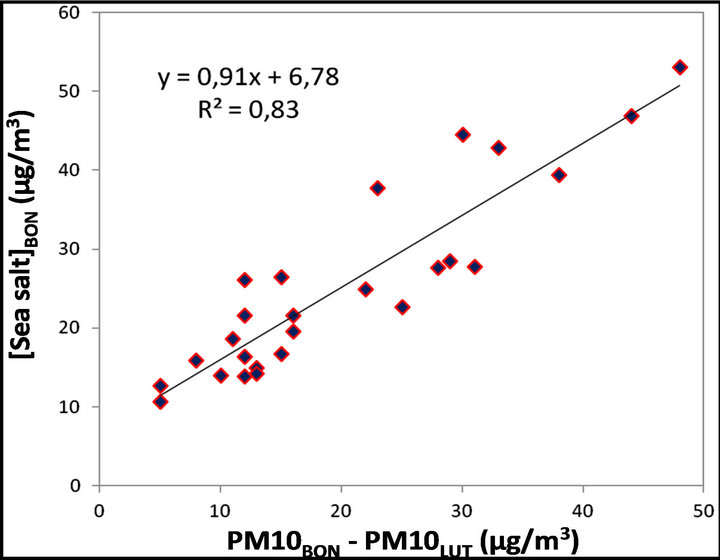
Figure 12. Correlation between the sea salt concentrations measured at BON and difference in PM10 concentrations recorded between LUT and BON stations.
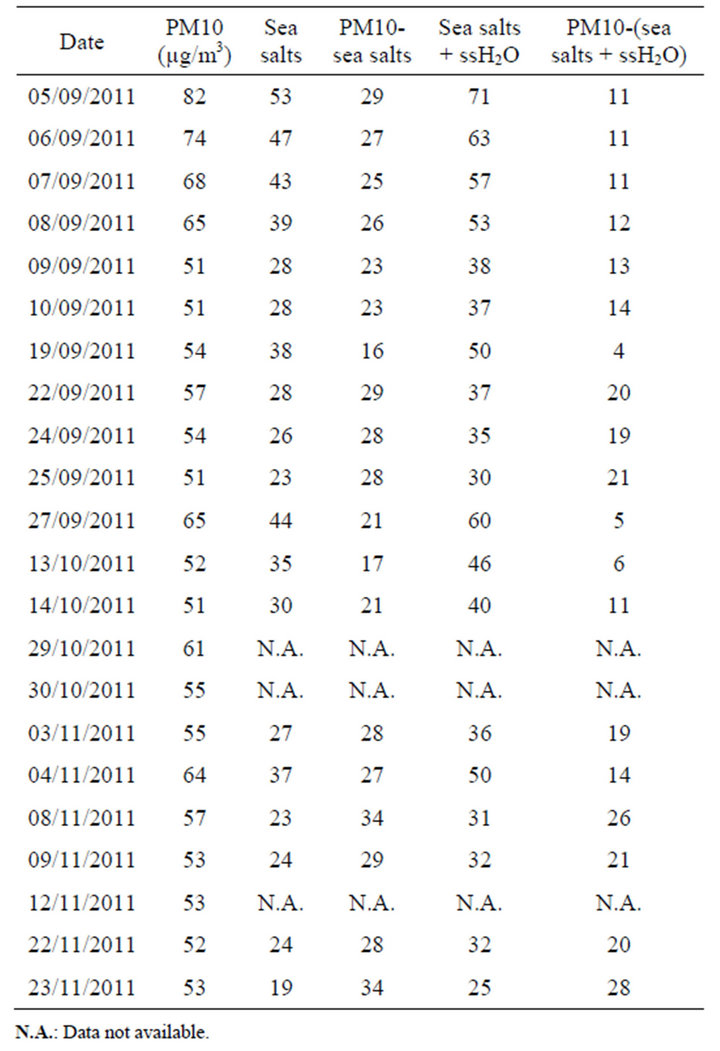
Table 2. Subtraction of daily sea salt contribution to PM10 concentrations >50 µg/m3 at BON in September, October and November 2011.
mated from chemical analyses during this period.
The results of this analysis (see Figure 13) show that there is good agreement between sea salt concentrations measured at BON and those estimated from differences in PM10 concentrations at the two stations. These results suggest an overall uncertainty <50% for the empirical methodology. It may be emphasized that the latter estimate does not account for the influence of the amount of water associated with the presence of sea salts on the total PM10 concentrations. Thus, a possible overestimation due to the application of this methodology is partially offset by the non-inclusion of water concentrations.
Finally, as a test, this methodology was applied to all close to each other. The mean diurnal winds data measured at Pierrefonds a close meteorological station showed that at daytime the air samples with high wind speeds are mainly of south-eastern to southern (mainly marine) origin. At night-time, the air samples with moderate wind speeds are mainly of north-eastern to eastern (urban/terrigenous) origin. The proximity of coral reefs with
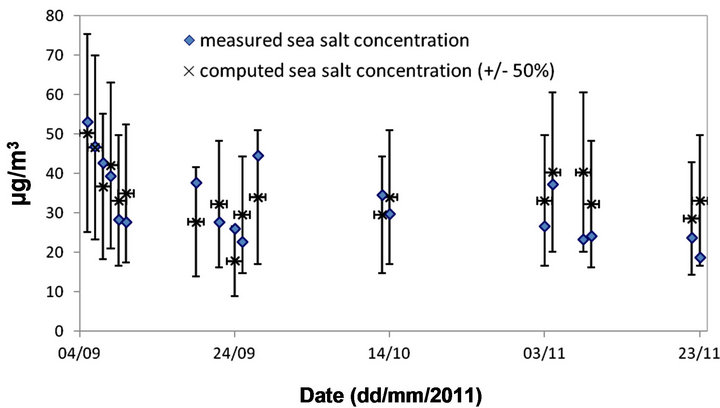
Figure 13. Comparison between sea salt concentrations measured and “re-calculated” at BON for which the daily PM10 >50 μg/m3 during September-November 2011 period.
the BON station and the conjugate effect of winds points out the notable influence of marine aerosols on the PM10 concentration variability observed at BON than at LUT.
5. Conclusions
The daily PM10 concentration profiles calculated at both sites also show that their trend and pattern are different. The PM10 concentration recorded at LUT and BON was compared with the regulatory values (daily and annual average). Importantly, the number of days exceeding the limit value for the protection of the human health (centile 90.4, i.e., the mean daily value of 50 µg/m3 not to be exceeded more than 35 days/year) is systematically exceeded at BON while at LUT it is never exceeded. Also, the quality objective (QO: 30 µg/m3 on annual average), the limit value for the human health protection (LV: 40 µg/m3 on annual average) and the centile 90,4 are exceeded at BON each year during 2008-2011 period, while at LUT no other regulatory values are exceeded except for the QO value which is exceeded in 2010.
The main source of the PM10 was determined by the analysis of the composition of the sampled air. In September 2011, particles sampled daily on filters in parallel with continuous PM10 concentration measurements at BON and LUT were analyzed in laboratory in order to determine the presence of major ions (tracers of marine origin) and carbonaceous (elemental and organic) species (tracers of anthropogenic origin).
The analyses were performed for 39 days of filter samples during which the daily PM10 concentration mostly exceeded the limit value of 50 µg/m3, between September and November 2011. The analysis results showed that the sea salt appears clearly as the main fraction of PM10 sampled on each site (~50% at LUT and ~55% at BON).
The results suggest not only that the daily threshold exceedances recorded on the BON station are mainly due to sea salts but also that all other chemical species studied present relatively homogeneous concentrations at both LUT and BON sites.
A calculation based on the sampled filter analysis results was applied to the entire PM10 data set. This methodology was applied to all exceedances of 50μg/m3 recorded at BON station and compared with the PM10 recorded at LUT station over the 2008-2011 period. The results showed that over this period, the daily PM10 concentration measured at BON station is quite below the limit value of 35 days/year for the threshold of 50μg/m3, after subtraction of the sea salt contribution. It may therefore be assessed that the notable PM10 levels recorded at BON have minor impact on human health at this urban marine site.
Finally, this preliminary study points out the necessity of in-situ measurements to assess the origin and main causes of PM10 concentration variability at two close urban measurement stations, in response with local-toregional scale processes (e.g. sea salt emissions and anthropogenic activities), under ‘particular’ geographical (notable relief) and meteorological conditions (land/sea breezes, recirculation, etc.). It constitutes an adequate device for decision makers to take necessary steps, especially to inform the surrounding population, in order to give an appropriate air quality index to the population. It may also avoid giving misleading information pertaining to the potential health and environmental impacts of PM10, when QO and LV for human health are exceeded. This work can be further extended by conducting epidemiological studies to assess the health impact associated with atmospheric particles in urban tropical regions. However, it requires detailed investigation of other species (thorough chemical analysis of filter samples) in parallel with continuous monitoring of other atmospheric pollutants (ex. heavy metals and SO2) for at least a civil year, especially “during” and “off” significant particular local/regional (e.g. swell) events. Also, this experiment must be undertaken at other more inhabited marine locations in order to confirm these results.
6. Acknowledgements
We acknowledge the French “Ministry of Environment” (MEDDE), ADEME, CINOR, TCO and CIVIS, for their financial support to the ORA air quality network at Réunion Island.
We gratefully acknowledge the ORA staff for technical support and for the atmospheric data sampling.
INERIS/LCSQA is acknowledged for the analysis of the particles sampled on filters in the frame of the CARA program.
We also gratefully acknowledge all the technical teams who have contributed to the sampling, the filter management and transportation of equipments and the implementation and validation of chemical analyses, especially C. Guadagno and G. Peris from ORA, and S. Verlhac, R. Aujay, V. Minguet, Y. Chatellier and N. A. Papin from INERIS/LCSQA.
Thanks also to Météo-France for providing the meteorological data.
REFERENCES
- M. S. O’Neill, D. Loomis, V. M. Torres-Meza, A. Retama and D. Gold, “Estimating Particle Exposure in the Mexico City Metropolitan Area,” Journal of Exposure Analysis and Environmental Epidemiology, Vol. 12, No. 2, 2002, pp. 145-156.
- M. D. Attfield, P. L. Schleiff, J. H. Lubin, A. Blair, P. A. Stewart, R. Vermeulen, J. B. Coble and D. T. Silverman, “The Diesel Exhaust in Miners Study: A Cohort Mortality Study with Emphasis on Lung Cancer,” Journal of the National Cancer Institute, Vol. 104, No. 11, 2011, pp. 869-883. doi:10.1093/jnci/djs035
- IARC (International Agency for Research on Cancer), “Diesel Engine Exhaust Carcinogenic,” Press Release No 213, 2012.
- C. Dir, “Council Directive 1999/30/EC of 22nd April 1999 Relating to Limit Values for Sulphur Dioxide, Nitrogen Dioxide and Oxides of Nitrogen, Particulate Matter and Lead in Ambient Air,” Official Journal of the European Union, L163/41, 1999.
- WHO, “Health Aspects of Air Pollution with Particulate Matter, Ozone and Nitrogen Dioxide,” Report on a WHO Working Group, 2003.
- WHO, “Air Quality Guidelines for Particulate Matter, Ozone, Nitrogen Dioxide and Sulphur Dioxide, Global Update 2005,” Summary of risk Assessment, Geneva, 2006.
- E. Dir, “European Directive 2008/50/EC of the European Parliament and the Council of 21st May 2008 on Ambient Air Quality and Cleaner Air for Europe,” Official Journal of the European Union, L152/1, 2008.
- IPCC (Intergovernmental Panel on Climate Change), S. Solomon, D. Qin, M. Manning, Z. Chen, M. Marquis, K. B. Averyt, M. Tignor and H. L. Miller, “Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change,” Cambridge University Press, Cambridge, 2007.
- R. Bhatia, P. Lopipero and A. H. Smith, “Diesel Exhaust Exposure and Lung Cancer,” Epidemiology, Vol. 9, No. 1, 1998, pp. 84-91. doi:10.1097/00001648-199801000-00017
- EPA, “Fourth External Review Draft of Air Quality Criteria for Particulate Matter,” 2003.
- U. Gehring, J. Heinrich, U. Krämer, V. Grote, M. Hochadel, D. Sugiri, M. Kraft, K. Rauchfuss, H. G. Eberwein and H.-E. Wichmann, “Long-Term Exposure to Ambient Air Pollution and Cardiopulmonary Mortality in Women,” Epidemiology, Vol. 17, No. 5, 2006, pp. 545- 551. doi:10.1097/01.ede.0000224541.38258.87
- L. Carbajal-Arroyo, A. Barraza-Villarreal, R. DurandPardo, H. Moreno-Macias, R. Espinoza-Lain, P. ChiarellaOrtigosa and I. Romieu, “Impact of Traffic Flow on the Asthma Prevalence among School Children in Lima, Peru,” Journal of Asthma, Vol. 44, No. 3, 2007, pp. 197- 202. doi:10.1080/02770900701209756
- C. A. Pope, R. T. Burnett, M. J. Thun, E. E. Calle, D. Krewski, K. Ito and G. D. Thurston, “Lung Cancer, Cardiopulmonary Mortality, and Long-Term Exposure to Fine Particulate Air Pollution,” Journal of the American Medical Association, Vol. 288, No. 9, 2002, pp. 1132- 1141.
- C. A. Pope, M. Ezzoti and D. W. Dockery, “Fine Particulate Air Pollution and Life Expectancy in the United States,” The New England Journal of Medicine, Vol. 360, 2009, pp. 376-386. doi:10.1056/NEJMsa0805646
- M. O. Andreae and P. J. Crutzen, “Atmospheric Aerosols: Biogeochemical Sources and Role in Atmospheric Chemistry,” Science, Vol. 276, No. 5315, 1997, pp. 1052- 1058. doi:10.1126/science.276.5315.1052
- C. D. O'Dowd, M. H. Smith, I. E. Consterdine and J. A. Lowe, “Marine aerosol, Sea-Salt, and the Marine Sulphur Cycle: A Short Review,” Atmospheric Environment, Vol. 31, No. 1, 1997, pp. 73-80. doi:10.1016/S1352-2310(96)00106-9
- B. J. Finlayson-Pitts, “The Tropospheric Chemistry of Sea-Salt: A Molecular-Level View of the Chemistry of NaCl and NaBr,” Chemical Reviews, Vol. 103, No. 12, 2003, pp. 4801-4822. doi:10.1021/cr020653t
- E. R. Lewis and S. E. Schwartz, “Sea Salt Aerosol Production: Mechanisms, Methods, Measurements, and Models—A Critical Review,” Geophysical Monograph Series, Vol. 152, American Geophysical Union, Washington DC, 2004. doi:10.1029/GM152
- C. D. O’Dowd and G. De Leeuw, “Marine Aerosol Production: A Review of the Current Knowledge,” Philosophical Transactions of the Royal Society A, Vol. 365, No. 1856, 2007, pp. 1753-1774. doi:10.1098/rsta.2007.2043
- N. W. Tindale and P. P. Pease, “Aerosols over the Arabian Sea: Atmospheric Transport Pathways and Concentrations of Dust and Sea Salt,” Deep-Sea Research Part II, Vol. 46, No. 8-9, 1999, pp. 1577-1595. doi:10.1016/S0967-0645(99)00036-3
- J. Putaud, F. Raes, R. VanDingenen, E. Bruggemann, M. Facchini, S. Decesari, S. Fuzzi, R. Gehrig, C. Hueglin, P. Laj, G. Lorbeer, W. Maenhaut, N. Mihalopoulos, K. Mueller, X. Querol, S. Rodriguez, J. Schneider, G. Spindler, H. Ten Brink, K. Torseth and A. Wiedensohler, “European Aerosol Phenomenology-2: Chemical Characteristics of Particulate Matter at Kerbside, Urban, Rural and Background Sites in Europe,” Atmospheric Environment, Vol. 38, No. 16, 2004, pp. 2579-2595. doi:10.1016/j.atmosenv.2004.01.041
- A. M. M. Manders, M. Schaap, X. Querol, M. F. M. A. Albert, J. Vercauteren, T. A. J. Kuhlbusch and R. Hoogerbrugge, “Sea Salt Concentrations across the European Continent,” Atmospheric Environment, Vol. 44, No. 20, 2010, pp. 2434-2442. doi:10.1016/j.atmosenv.2010.03.028
- M. Hess, U. K. Krieger, C. Marcolli, T. Huthwelker, M. Ammann, W. A. Lanford and Th. Peter, “Bromine Enrichment in the Near-Surface Region of Br-Doped NaCl Single Crystals Diagnosed by Rutherford Backscattering Spectrometry,” The Journal of Physical Chemistry A, Vol. 111, No. 20, 2007, pp. 4312-4321. doi:10.1021/jp0674120
- R. C. Easter, S. J. Ghan, Y. Zhang, R. D. Saylor, E. G. Chapman, N. S. Laulainen, H. Abdul-Razzak, H. R. Leung, X. D. Bian and R. A. Zaveri, “MIRAGE: Model Description and Evaluation of Aerosols and Trace Gases,” Journal of Geophysical Research, Vol. 109, No. D20, 2004, Article ID: D20210. doi:10.1029/2004JD004571
- N. Mahowald, J.-F. Lamarque, X. X. Tie and E. Wolff, “Sea Salt Aerosol Response to Climate Change: Last Glacial Maximum, Preindustrial, and Doubled Carbon Dioxide Climates,” Journal of Geophysical Research, Vol. 111, No. D5, 2006, Article ID: D05303. doi:10.1029/2005JD006459
- S. Tamm and M. Schulz, “Open-Ocean Aerosol Composition Obtained during 15 Months on a North Sea Ferry,” Atmospheric Environment, Vol. 37, Suppl. 1, 2003, pp. 133-143. doi:10.1016/S1352-2310(03)00241-3
- E. M. Knipping and D. Dabdub, “Impact of Chlorine Emissions from Sea-Salt Aerosol on Coastal Urban Ozone,” Environmental Science & Technology, Vol. 37, No. 2, 2003, pp. 275-284. doi:10.1021/es025793z
- A. Cohan, W. Chang, M. Carreras-Sospedra and D. Dabdub, “Influence of Sea-Salt Activated Chlorine and Surface-Mediated Renoxification on the Weekend Effect in the South Coast Air Basin of California,” Atmospheric Environment, Vol. 42, No. 13, 2008, pp. 3115-3129. doi:10.1016/j.atmosenv.2007.11.046
- S. C. Pryor and R. J. Barthelmie, “Particle Dry Deposition to Water Surfaces: Processes and Consequences,” Marine Pollution Bulletin, Vol. 41, No. 1-6, 2000, pp. 220-231.
- M. Van Loon, R. Vautard, M. Schaap, R. Bergström, B. Bessagnet, J. Brandt, P. Builtjes, J. H. Christensen, J. H. Cuvelier, A. Graf, J. E. Jonson, M. Krol, J. Langner, P. Roberts, L. Rouil, R. Stern, L. Tarrasón, P. Thunis, E. Vignati, L. White and P. Wind, “Evaluation of LongTerm Ozone Simulations from Seven Regional Air Quality Models and Their Ensemble Average,” Atmospheric Environment, Vol. 41, No. 10, 2007, pp. 2083-2097. doi:10.1016/j.atmosenv.2006.10.073
- T. H. Muster and I. S. Cole, “Attachment Efficiencies of Salt Aerosols onto Infrastructure and Implications for Atmospheric Corrosion,” Journal of the Electrochemical Society, Vol. 152, No. 3, 2005, pp. B125-B131. doi:10.1149/1.1859813
- Y. J. Yoon and P. Brimblecombe, “Modelling the Contribution of Sea Salt and Dimethyl Sulfide Derived Aerosol to Marine CCN,” Atmospheric Chemistry and Physics, Vol. 2, No. 1, 2002, pp. 17-30. doi:10.5194/acp-2-17-2002
- V. Vinoj and S. K. Satheesh, “Direct and Indirect Radiative Effects of Sea-Salt Aerosols over Arabian Sea,” Current Science (India), Vol. 86, No. 10, 2004, pp. 1381- 1390.
- S. L. Gong, L. A. Bartie, J.-P. Blanchet, “Modeling SeaSalt Aerosols in the Atmosphere—1. Model Development,” Journal of Geophysical Research, Vol. 102, No. D3, 1997, pp. 3805-3818.
- E. Athanasopoulou, M. Tombrou, S. N. Pandis and A. G. Russell, “The Role of Sea-Salt Emissions and Heterogeneous Chemistry in the Air Quality of Polluted Coastal Areas,” Atmospheric Chemistry and Physics, Vol. 8, No. 19, 2008, pp. 5755-5769. doi:10.5194/acp-8-5755-2008
- B. Finlayson-Pitts and J. N. Pitts Jr., “Chemistry of the Upper and Lower Atmosphere, Theory, Experiments and Applications,” Academic Press, Waltham, 1999.
- H. Hass, M. van Loon, C. Kessler, R. Stern, J. Matthijsen, F. Sauter, Z. Zlatev, J. Langner, V. Foltescu and M. Schaap, “Aerosol Modelling: Results and Intercomparison from European Regional-Scale Modelling Systems,” Special Rep. EUROTRAC-2 ISS, Munchen, 2003.
- B. Bessagnet, A. Hodzic, H. Vautard, M. Beekmann, S. Cheinet, C. Honoré, C. Liousse and L. Rouil, “Aerosol Modeling with CHIMERE: Preliminary Evaluation at the Continental Scale,” Atmospheric Environment, Vol. 38, No. 18, 2004, pp. 2803-2817. doi:10.1016/j.atmosenv.2004.02.034
- S. L. Gong, L. A. Barrie, J.-P. Blanchet, K. von Salzen, U. Lohmann, G. Lesins, L. Spacek, L. M. Zhang, E. Girard, H. Lin, R. Leaitch, H. Leighton, P. Chylek and P. Huang, “Canadian Aerosol Module: A Size-Segregated Simulation of Atmospheric Aerosol Processes for Climate and Air Quality Models 1. Module Development,” Journal of Geophysical Research, Vol. 108, No. D1, 2003, pp. AAC 3-1-AAC 3-16. doi:10.1029/2001JD002002
- W. Guelle, M. Schulz, Y. Balkanski and F. Dentener, “Influence of the Source Formulation on Modeling the Atmospheric Global Distribution of Sea Salt Aerosol,” Journal of Geophysical Research, Vol. 106, No. D21, 2001, pp. 27509-27524. doi:10.1029/2001JD900249
- P. Stier, J. Feichter, S. Kinne, S. Kloster, E. Vignati, J. Wilson, L. Ganzeveld, I. Tegen, M. Werner, Y. Balkanski, M. Schulz, O. Boucher, A. Minikin and A. Petzold, “The Aerosol-Climate Model ECHAM5-HAM,” Atmospheric Chemistry and Physics, Vol. 5, No. 4, 2005, pp. 1125- 1156.
- P. Jiménez-Guerrero, O. Jorba, M. T. Pay, J. P. Montávez, S. Jerez, J. J. Gómez-Navarro and J. M. Baldasano, “Comparison of Two Different Sea-Salt Aerosol Schemes as Implemented in Air Quality Models Applied to the Mediterranean Basin,” Atmospheric Chemistry and Physics, Vol. 11, No. 10, 2011, pp. 4833-4850. doi:10.5194/acp-11-4833-2011
- V. L. Foltescu, S. C. Pryor and C. Bennet, “Sea Salt Generation, Dispersion and Removal on the Regional Scale,” Atmospheric Environment, Vol. 39, No. 11, 2005, pp. 2123-2133. doi:10.1016/j.atmosenv.2004.12.030
- B. Langmann, S. Varghese, E. Marmer, E. Vignati, J. Wilson, P. Stier and C. O’Dowd, “Aerosol Distribution over Europe: A Model Evaluation Study with Detailed Aerosol Microphysics,” Atmospheric Chemistry and Physics, Vol. 8, No. 6, 2008, pp. 1591-1607. doi:10.5194/acp-8-1591-2008
- M. Schaap, R. M. A. Timmermans, F. J. Sauter, M. Roemer, G. J. M. Velders, G. A. C. Boersen, J. P. Beck and P. J. H. Builtjes, “The LOTOS-EUROS Model: Description, Validation and Latest Developments,” International Journal of Environment and Pollution, Vol. 32, No. 2, 2008, pp. 270-290. doi:10.1504/IJEP.2008.017106
- A. S. Zakey, F. Giorgi and X. Bi, “Modeling of Sea Salt in a Regional Climate Model: Fluxes and Radiative Forcing,” Journal of Geophysical Research, Vol. 113, No. D14, 2008. doi:10.1029/2007JD009209
- C. Bhugwant, H. Cachier, M. Bessafi and J. Leveau, “Impact of Traffic on Black Carbon Aerosol Concentration at La Reunion Island (Southern Indian Ocean),” Atmospheric Environment, Vol. 34, No. 20, 2000, pp. 3464- 3473. doi:10.1016/S1352-2310(99)00405-7
- C. Bhugwant and P. Brémaud, “Simultaneous Measurements of Black Carbon, PM10, Ozone and NOx Variability at a Locally Polluted Island in the Southern Tropics,” Journal of Atmospheric Chemistry, Vol. 39, No. 3, 2001, pp. 261-280. doi:10.1023/A:1010692201459
- C. Bhugwant, M. Bessafi and B. Siéja, “The Potential Impact of Marine Aerosols via the Swell and the Oceanic Waves on the PM10 Concentration Measurements at Urban Marine Locations, Air Pollution Emissions,” Nova Publishers, New York, 2012.
- INSEE, “TER (Tableau Economique de La Réunion),” 2010, p. 3014. in French.
- W. E. Wilson, B. D. Grover, R. W. Long, N. L. Eatough and D. J. Eatough, “The Measurement of Fine-Particulate Semivolatile Material in Urban Aerosols,” Journal of the Air & Waste Management Association, Vol. 56, No. 4, 2006, pp. 384-397. doi:10.1080/10473289.2006.10464527
- B. D. Grover, N. L. Eatough, D. J. Eatough, J. C. Chow, J. G. Watson, J. L. Ambs, M. B. Meyer, P. K. Hopke, R. Al-Horr, D. W. Later and W. E. Wilson, “Measurement of Both Nonvolatile and Semi-Volatile Fractions of Fine Particulate Matter in Fresno, CA,” Aerosol Science and Technology, Vol. 40, No. 10, 2006, pp. 811-826. doi:10.1080/02786820600615071
- J. Wanjura, B. Shaw, C. Parnell, R. Lacey and S. Capareda, “Comparisons of Continuous Monitor (TEOM) and Gravimetric Sampler Particulate Matter Concentrations,” American Society of Agricultural and Biological Engineers, Vol. 51, No. 1, 2008, pp. 251-257.
- C. Bhugwant and J.-L. Hoareau, “Variability of NO2 in Different Environments at a Moderately Polluted Island over the Southwestern Indian Ocean,” Atmospheric Research, Vol. 66, No. 4, 2003, pp. 241-259. doi:10.1016/S0169-8095(03)00038-3
- F. Cavalli, M. Viana, K. E. Yttri, J. Genberg and J.-P. Putaud, “Toward a Standardised Thermal-Optical Protocol for Measuring Atmospheric Organic and Elemental Carbon: The EUSAAR Protocol,” Atmospheric Measurement Techniques, Vol. 3, No. 1, 2010, pp. 79-89. doi:10.5194/amt-3-79-2010
- M. E. Birch and R. A. Cary, “Elemental Carbon-Based Method for Monitoring Occupational Exposures to Particulate Diesel Exhaust,” Aerosol Science and Technology, Vol. 25, No. 3, 1996, pp. 221-241. doi:10.1080/02786829608965393
- O. S. Météo-France, “Atlas Climatique de La Réunion,” Bureau d’étude Climatologique, Direction Interrégionale de La Réunion, Annual Report No. 1657, 2000.
- N. Pérez, J. Pey, M. Cusack, C. Reche, X. Querol, A. Alastuey and M. Viana, “Variability of Particle Number, Black Carbon, and PM10, PM2.5, and PM1 Levels and Speciation: Influence of Road Traffic Emissions on Urban Air Quality,” Aerosol Science and Technology, Vol. 44, No. 7, 2010, pp. 487-499. doi:10.1080/02786821003758286
- M. T. Limon-Sanchez, P. Carbajal-Romero, L. Hernandez-Mena, H. Saldarriaga-Norena, A. Lopez-Lopez, R. Cosio-Ramirez, J. L. Arriaga-Colina and W. Smith, “Black Carbon in PM2.5, Data from Two Urban Sites in Guadalajara, Mexico during 2008,” Atmospheric Pollution Research, Vol. 2, No. 3, 2011, pp. 358-365. doi:10.5094/APR.2011.040
- V. Grondin and C. Bhugwant, “Internship Report, ORA, Etude de l’Impact des Embruns Marins Sur la Qualité de l’Air en Milieu Urbain à la Réunion,” 2008, in French.
- B. J. Turpin, P. Saxena and E. Andrews, “Measuring and Simulating Particle Organics in the Atmosphere: Problems and Prospects,” Atmospheric Environment, Vol. 34, No. 18, 2000, pp. 2983-3013. doi:10.1016/S1352-2310(99)00501-4
- J. C. Cabada, S. N. Pandis, R. Subramanian, A. L. Robinson, A. Polidori and B. Turpin, “Estimating the Secondary Organic Aerosol Contribution to PM2.5 Using the EC Tracer Method Special Issue of Aerosol Science and Technology on Findings from the Fine Particulate Matter Super Sites Program,” Aerosol Science and Technology, Vol. 38, Suppl. 1, 2004, pp. 140-155. doi:10.1080/02786820390229084
- E. Friese and A. Ebel, “Temperature Dependent Thermodynamic Model of the System H+-NH4+-Na+-
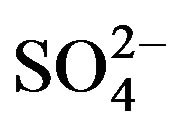 -
- 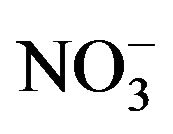 -Cl−-H2O,” Journal of Physical Chemistry A, Vol. 114, No. 43, 2010, pp. 11595-11631. doi:10.1021/jp101041j
-Cl−-H2O,” Journal of Physical Chemistry A, Vol. 114, No. 43, 2010, pp. 11595-11631. doi:10.1021/jp101041j
NOTES
*Corresponding author.