Journal of Environmental Protection
Vol. 3 No. 7 (2012) , Article ID: 21135 , 16 pages DOI:10.4236/jep.2012.37076
Development of Immunochemical Methods for Purification and Detection of the Steroid Drug Medroxyprogesterone Acetate
![]()
1Department of Entomology, Institute of Plant Protection, the Volcani Center, Bet Dagan, Israel; 2Analyst Research Laboratories, Rehovot, Israel.
Email: vinnie2@agri.gov.il
Received March 12th, 2012; revised April 17th, 2012; accepted May 19th, 2012
Keywords: Medroxyprogesterone acetate; ELISA; Immunoaffinity chromatography; Sol-gel; Pharmaceutical residues; Residue monitoring
ABSTRACT
An immunochemical sol-gel-based immunoaffinity purification (IAP) method for purification and detection of the progestin drug medroxyprogesterone acetate (MPA) was developed. A polyclonal antibody (Ab) for MPA was generated, and two competitive (indirect and direct) sensitive enzyme-linked immunosorbent assays (ELISAs) for its detection were developed and implemented to determine the recovery and efficiency of the sol-gel based IAP method. The detection limits of the assays were 1.4 ± 0.2 ng∙mL−1 (n = 4) and 4.0 ± 0.4 ng∙mL−1 (n = 25) for the indirect and direct ELISAs, respectively. The Abs did not exhibit cross-reactivity with any other progestin or steroid hormone, with the exception of megestrol acetate, with which the Ab exhibited 76% cross-reactivity. The sol-gel IAP method successfully eliminated serum interference to a degree that enabled ELISA analysis of spiked serum samples. This method was also found fully compatible with subsequent chemical analytical methods, such as liquid chromatography followed by mass spectrometry (LC-MS/MS). The approaches developed in this study form a basis for analysis of MPA in biological samples and may be further used to study population exposure to MPA and to monitor MPA contamination in water samples.
1. Introduction
Studies in the past decade have shown that exposure to environmental chemicals influences human developmental and reproductive endpoints (for review see [1,2] and references therein). Hitherto, most studies on adverse health effects of environmental contaminants have focused mainly on agricultural pesticides, heavy metals, and toxic industrial waste [3,4]. One large class of chemicals that has received little attention for many years comprises residues of pharmaceutical products (PPs) that are used in human and veterinary medicine in quantities comparable to those of agrochemicals.
In recent years the presence of PP residues in food and the environment, and inadvertent exposure of populations to those substances via contaminated food have raised public and scientific awareness to the problem. This, together with the extensive food-safety legislation that limits drug residue levels in food, stimulated studies that focus on occurrence of PPs in food and the environment, and on possible correlations between food and environmental contamination and their adverse health effects. These studies revealed that numerous PPs and their metabolites do, indeed, contaminate aquatic environments (for review see [1,5-10] and references therein). However, currently there is very little information on the environmental fate of these substances, and our understanding of the possible transmission of PPs into the food chain and of potential human exposure is also very limited. Even less is known regarding whether and how such contaminants, once ingested, affect human health.
In order to evaluate the extent of the problem it is necessary to carry out large-scale monitoring programs that will enable determination of small quantities of PP residues in environmental, food, and biological samples. In the past few years we have conducted, within an EUFP-6 project on Food and Fecundity (F & F), a detailed review of pharmaceuticals that have the potential to affect human fecundity by exposure via the human food chain. Pharmaceuticals were reviewed, especially with regard to mechanisms of action, production and usevolumes, persistence in the environment, and severity of identified adverse health effects in humans. In light of an extensive literature survey, eight potentially endocrinedisrupting PPs were selected (for review see [8]); they comprised: four steroid hormones, i.e., levonorgestrel (LNG), ethinylestradiol (EE2), nortestosterone (NT), and medroxyprogesterone acetate (MPA, Figure 1(a)), which are the main components of contraceptive drugs and are also used as anabolic steroids; a representative nonsteroid anti-inflammatory drug (NSAID)—indomethacin (IMT); a representative selective serotonin re-uptake inhibitor (SSRI)—fluoxetine (FLX); the antibiotic, trimethoprim (TMP); and the beta-blocker atenolol (ATL). All of these compounds exhibit high stability in the environment, are used in large amounts and, most importantly, have been reported to affect fecundity [8].
In previous studies we developed enzyme-linked immunosorbent assays (ELISA) and immuno-affinity purification methods for the steroid hormone, LNG [11,12] and the NSAID, IMT [13]. In the present study we focused on the progestin drug, MPA. So far, there is very limited information available about the occurrence, fate and bioaccumulation of MPA in the environment or in the food chain, but its massive current use, and its high environmental stability and consequently long environmental half-life raise the possibility that it potentially could pose a high risk to human health.
MPA is a synthetic hormone and is the main ingredient of a long-acting hormonal contraceptive used by millions of women in over 90 countries worldwide since 1967 [14]. The primary contraceptive action mechanism of MPA is inhibiting secretion of gonadotropins—follicular steroid hormone, (FSH) and luteinizing hormone releasing hormone (LHRH)—thereby blocking follicular development and ovulation, and reducing ovarian production of estradiol [15]. MPA is also effective in treatment of endometriosis [16], and is used in veterinary medicine as an anabolic steroid.
Similarly to many other drugs, MPA elicits side effects: several studies have found it associated with a variable increase in insulin levels, particularly in diabetic or obese women; it reduces high-density lipoproteins and changes the elasticity of the arterial endothelium; and, through its hypoestrogenic affect it causes imbalance between bone resorption and formation which results in bone-mineral density decline (for review see [17]). A recent study demonstrated that use of MPA results in impairment of endometrial capillary integrity, because of its apoptotic effect on endometrial endothelial cells [18]; MPA also has been known for many years as an inducer of mammary cancer (for review see [19]). The adverse health effects of MPA, the obvious effect it has on women’s fertility, and the prohibitions that have been issued by the European Commission, China, and other countries on its use as an anabolic steroid in veterinary medicine, all indicate the need to maintain accurate identification and monitoring of small amounts of MPA in environmental, water, and food samples, in order to prevent exposure of “non-target” populations to the drug.
Currently, the most common methods for determination of MPA are high-performance liquid chromatography (HPLC) or gas chromatography (GC) (which requires analyte derivatisation) followed by tandem mass spectrometry (MS/MS) for example see ([20-22] and references therein). These methods were used to monitor MPA in wastewater and surface water, as well as in muscle tissue and kidney fat (which represent to a good extent presence of the synthetic hormone in food of animal origin).
Sample-preparation methods for the above analyses involve combinations of multistep analytical procedures that include hydrolysis, extensive solid/liquid extraction (SLE) and/or liquid/liquid extraction (LLE), followed by defatting of the resulting extracts, and final cleanup by various solid-phase extraction (SPE) procedures. Additional reported procedures include accelerated solvent extraction (ASE) and supercritical fluid extraction (SFE) [20-22] and references therein. Application of the above procedures results, in many cases, in very low recovery of the analyte.
In order to be able to perform large-scale monitoring and to be able to detect small amounts of analytes it is necessary to develop simple, fast and cost-effective diagnostic methods, and also sample-preparation protocols that provide high recovery. This is especially true for environmental and food samples, which contain, in most cases, components that might interfere with the analysis —either chemical or immunochemical. Indeed, a wide variety of immunochemical methods have been developed and used to monitor MPA in human and animal serum, animal muscles, kidney and fat tissue. They include: radio-immunoassays (RIAs, [23,24]); and homologous and heterologous ELISAs that use polyclonal or monoclonal antibodies (Abs), and a variety of reporting methods (colorimetric, luminescent, gold nanoparticles [25-28]); time-resolved fluoroimmunoassays (TRFIA, [29]); capillary electrophoresis immunoassays [30]; and quantum dot fluoroimmunoassays [31]. However, the problem of sample preparation has not been resolved. Although immunochemical methods are more tolerant to matrix interference than instrumental chemical methods, it is still necessary to purify the analyte from the samples before it can be tested by ELISA, and in all of the above studies multistep extraction and sample cleanup were employed. Thus, it is urgently necessary to focus on simple, fast, and cost-effective sample preparation methods that will be applicable to both chemical instrumental (LC-MS) and immunochemical analysis (e.g., ELISA).
For many years immunoaffinity purification (IAP) has been regarded as one of the leading technologies for purification, concentration and isolation of chemical and biological compounds. In the past two decades our laboratory has developed an IAP method that uses Abs entrapped in a SiO2 ceramic matrix. This method has been successfully applied by many laboratories, including ours, for purification of many compounds in the analysis of a variety of environmental, food, and biological samples. For review see [11-13,32-44]) and references therein.
In the present study we developed a sol-gel-based IAP for MPA. A polyclonal Ab was generated, and two sensitive and highly specific ELISAs were developed. The antiserum was used to develop a sol-gel-based IAP method for purification and concentration of MPA, and several sol-gel formats, containing various amounts of antiserum, were examined under diverse experimental conditions. The sol-gel-IAP method was tested for its capability to eliminate serum interference with ELISA, and its compatibility with chemical analytical procedures was tested by means of LC-MS/MS. The approaches developed in this study form a basis for determination of MPA in biological samples in order to monitor their pharmacokinetic properties, and these approaches could be further used to study population exposure to MPA and also to monitor the occurrence of MPA contamination in food, soil, and other environmental samples.
2. Materials and Methods
2.1. Immunochemical Methods
2.1.1. Antiserum
Polyclonal anti-MPA antiserum was generated in rabbits by using MPA conjugated to bovine serum albumin (BSA) (Sigma, St. Louis, MI) as an immunogen, as described in section 2.1.2., below (Figure 1(c)). Each injection was carried out with 0.93 mg/0.5 mL of the MPA-BSA conjugate.
2.1.2. Preparation of MPA-BSA Conjugate for Immunization
The 4-pregnen-16α-methyl-17-ol-3,20-dione acetate-3-carboxymethyloxime (MPA-3-CMO; Figure 1(b)) (Steraloids, Newport, RI, USA) was first converted to its active ester form as follows: 30 mg (60 µmol) of MPACMO were mixed with 34 mg (300 µmol) of N-hydroxysuccinimide (Sigma) and 124 mg (600 µmol) of N’,N’- dicyclohexylcarbodiimide (DCC) (Sigma), and dissolved in 2600 μL of dimethylformamide (DMF) (Labscan, Dublin, Ireland). The reaction was allowed to proceed at room temperature for 4 h and then was further incubated at 4˚C for 12 h. The mixture was centrifuged at 4000 × g
 (a)
(a)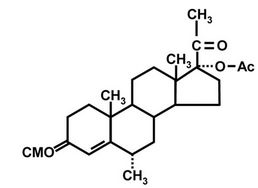 (b)
(b)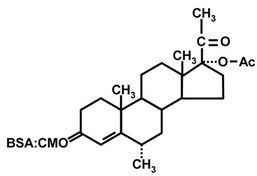 (c)
(c)
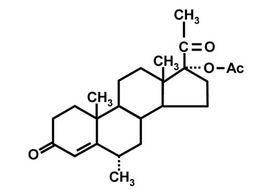
Figure 1. Structures of MPA (a); MPA-3-CMO (b); and MPA-BSA conjugate used for immunization (c). OVA and HRP were coupled to MPA in the same position as BSA.
at room temperature (25˚C) for 15 min, and 2460 μL of supernatant, containing 30 mg of activated MPA, i.e., 12.2 mg∙mL−1, were recovered. Ten milligrams (820 µL) of the active ester were added, drop-wise, to 10 mg of BSA dissolved in 4.5 mL of 0.13 M NaHCO3 at pH 8.5. The reaction was allowed to proceed for 1 h at room temperature and the solution was then dialyzed against 4 L of 0.13 M NaHCO3 at pH 8.5 for 3 days at room temperature. The solution was changed three times daily. The hapten MPA-BSA conjugate was stored as aliquots at –20˚C pending injection into rabbits.
Prior to immunization, 0.5 mL of the conjugate was mixed with Complete Freund’s Adjuvant (1st injection) or with incomplete Adjuvant (2nd to 4th injections). Two rabbits were injected at each time point. Bleeds were collected after each boost and were tested for activity with checker-board experiments. The 3rd and 4th bleeds were almost equally active towards the antigen, but only the 3rd bleed was used for ELISA and sol-gel IAP experiments.
2.1.3. Preparation of MPA-OVA Coating Antigen
The method was similar to that described above for preparation of the MPA-BSA conjugate, except that 4.16 µL of the supernatant, containing 50 µg (0.11 µmol) of activated MPA, was added, drop-wise, to 5 mg (0.11 µmol) of ovalbumin from egg white (OVA) (Sigma), dissolved in 750 µL of 0.13 M NaHCO3 at pH 8.5 (i.e., hapten:carrier-protein molar ratio of 1:1). The reaction was allowed to proceed at 4˚C for 1 h, and the unbound hapten and other small-molecular-weight components were separated from the protein/hapten conjugate by size exclusion with a Centricon 30 (Amicon, Millipore, Billerica, MA). The reaction mixture was spun at 4000 × g at room temperature for 20 min, and washed twice with 0.75 mL of 0.13 M NaHCO3 at pH 8.5. The final volume was adjusted to 750 µL by adding 0.13 M NaHCO3 at pH 8.5, and the mixture was then kept at –20˚C pending use. Protein content of the MPA-OVA conjugate was determined by means of the Bradford reaction and was found to be 3 mg∙mL−1.
2.1.4. Preparation of MPA-HRP Conjugate
The method was similar to those described in the previous two subsections, except that 4.16 µL of the supernatant, containing 50 µg (0.11 µmol) of activated MPA, were added, drop-wise, to 0.5 mg (0.011 µmol) of horseradish peroxidase (HRP) dissolved in 750 µL of 0.13 M NaHCO3 at pH 8.5 (i.e., carrier protein:hapten molar ratio of 1:10). The reaction was allowed to proceed at room temperature for 1 h, and the unbound hapten and other small-molecular-weight components were separated from the protein/hapten conjugate by size exclusion as described in section 2.1.3., above. The final volume was adjusted to 750 µL by adding 0.13 M NaHCO3 at pH 8.5, and 750 µL of ethylene glycol (Sigma) were added to the conjugate solution, which was then kept at –20˚C pending use.
2.1.5. MPA-Competitive ELISA
Two competitive ELISAs were developed (indirect and direct formats). All of the experiments in the present study used a standard MPA compound prepared from stock solution dissolved in ethanol at 1 mg∙mL−1.
For the indirect ELISA, transparent F96 Maxisorp microtiter plate wells (Nunc, Roskilde, Denmark) were coated with 100 μL of MPA-OVA conjugate, diluted 1:6000 (i.e., containing 0.5 μg∙mL−1) in 0.5 M carbonate buffer, pH 9.6. Six wells (designated as background— Bg-wells) were coated with an equivalent amount of OVA, and served as controls to determine nonspecific binding. After incubation overnight (ON) at 4˚C, the wells were washed three times with 0.05 M phosphate buffer containing 0.15 M NaCl and 0.1% Tween-20 (PBST). Then 50 μL of each of 12 serial dilutions of MPA (in PBS + 20% ethanol—PBS-E) ranging from 0.049 to 100 ng per 50 μL were added to the wells, together with 50 μL of anti-MPA antiserum diluted 1:2500 (final dilution of 1:5000) in PBS-2 × T (i.e., PBS containing 0.2% Tween 20, pH 7.2). All the additions were made in duplicate. The Bg wells and another six wells received only PBS-E without MPA, and served to determine nonspecific binding, or maximal binding (designated as 100%) in the absence of competing analyte. The plates were incubated overnight at 4˚C and washed as above with PBST, and 100 μL of secondary Ab conjugated to HRP (goat antirabbit HRP conjugated; Sigma), diluted 1:30,000 in PBST were added to the plates. The plates were incubated for 2 h at room temperature, rinsed with PBST, and tested for HRP activity by addition of 100 μL of colorimetric 3,3’,5,5’-tetramethyl benzidine (TMB) substrate (Chromogen, Dako North America Inc. Carpenteria, CA, USA). The color reaction was stopped after 10 min by addition of 50 μL of 4 M H2SO4, and the absorbance was measured with an ELISA reader at 450 nm.
The tolerance of the assay to various concentrations of organic solvents was tested similarly, except that the MPA standard was made up in PBS or (PBS + 20% methanol), and the Bg and maximal-binding wells were tested in the presence of each of these solvents instead of PBS-E.
For the competitive direct ELISA, transparent F96 Maxisorp microtiter plate wells (Nunc, Roskilde, Denmark) were coated with 100 μL of protein A (Sigma) at 1 µg per 100 µL, made up in 0.5 M carbonate buffer, pH 9.6, and were incubated overnight at 4˚C. The plates were washed three times with PBST, and 100 µL of anti-MPA antiserum, diluted 1:8000 or 1:16,000 in 0.5 M carbonate buffer, pH 9.6, were added to the plates, which were then incubated overnight at 4˚C. The plates were washed three times with PBST, and 12 serial dilutions of MPA standard diluted in PBS-E at concentrations ranging from 0.0049 to 10 ng/50 µL or any other tested compound or sample (i.e., tap water, normal human serum, NHS or low fat milk diluted in PBS-E) were added to the plates, together with 50 μL of MPA-HRP conjugate diluted 1:1000 (i.e., a final dilution of 1:2000) in PBS-2 × T. The whole procedure was performed in duplicate. Six wells which were not coated with the primary Ab, and received just PBS-E, served as a reaction Bg control. Other six wells, that were coated with the primary Ab with no competing MPA (to which 50 μL of PBS-E were added), served to determine maximal binding (designated as 100%). The reaction was incubated for 2 h at room temperature, the plates were washed three times with PBST, and 100 µL of a colorimetric TMB substrate was added. The color reaction was stopped after 10 min by addition of 50 μL of 4 M H2SO4, and the absorbance at 450 nm was measured with an ELISA reader.
The tolerance of the assay to various concentrations of organic solvents was tested similarly, except that the MPA standard was made up in either PBS or (PBS + 50% ethanol), and the Bg and maximal-binding wells were tested in the presence of each of these solvents instead of PBS-E.
Cross reactivity (CR) of the Abs with a variety of steroid hormones was determined by adding the MPA or the tested compounds at 12 serial dilutions in PBS-E, all ranging from 0.0049 to 10 ng per 50 µL, and testing their ability to compete with the MPA-HRP conjugate for binding to a limited amount of the MPA antiserum adsorbed to the microplate. All these additions and tests were performed in duplicate.
2.1.6. Sample Preparation and Analysis
Samples comprising 1 mL of tap water, low-fat (3%) milk, or NHS were spiked at 50 ng∙mL−1 with MPA made up in PBS-E. The MPA content in the samples was tested with the competitive direct ELISA as described above, using five successive twofold dilutions, ranging from 1:4 to 1:64, made up in each respective matrix (diluted 1:1.3 in PBS-E). The Bg and maximal-binding wells received 50 µL of the un-spiked matrix (tap water, milk, or NHS diluted 1:1.3, in PBS-E) instead of PBS-E. The MPA content in tested samples was determined by reference to an MPA calibration curve, after linearization of the data by transformation to a logit-log plot by means of the Origin software, Version 6.0 (Microcal Software, Northampton, MA, USA). Slopes of the curves obtained for all the samples were tested for parallelism with the standard curve by testing for homogeneity of regression slopes, according to Sokal and Rohlf [45].
2.2. Sol-Gel IAP
2.2.1. Sol-Gel Entrapment of Anti-MPA Antiserum
Entrapment involved a two-step procedure in which hydrolysis was followed by polymerization of tetramethylsilane (TMOS; Aldrich) as described previously [46]. Briefly, an acidic silica sol-solution was obtained by mixing TMOS with 2.5 mM HCl in double-distilled water (DDW) at a molar ratio of 1:12 (unless otherwise indicated). The mixture was stirred for 1 min until a clear solution was obtained; it was then sonicated for 30 min in a Model T-460/H, 285 W, 2.75-L ultrasonicator bath (ELMA, Singen-Hohentwiel, Germany). The reaction was performed under a well-ventilated fume hood. AntiMPA antiserum (125 μL of 4 × concentrated antiserum— equivalent to 28 mg protein—unless otherwise indicated) was premixed with 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer (HEPES, 99.99%, Sigma) at pH 7.6, to a final volume of 0.5 mL, and added to 0.5 mL of the prehydrolyzed TMOS mixture. Other formats (1:8, 1:6, and 1:4) were made up similarly with different TMOS:HCl ratios. In cases where gels were prepared in the presence of polyethylene glycol (PEG) 10% of PEG- 400 (Merck, Darmstadt, Germany), with average molecular weight of 400 g∙mol−1, corresponding to approximately seven methylene units in the chain, was added to the TMOS:HCl mixture. Gels in which no antiserum was entrapped (termed “empty”) were prepared by mixing 0.5 mL of hydrolyzed TMOS with 0.5 mL of HEPES buffer, pH 7.6. The solution was mixed quickly for 5 s, and gelation occurred within 1 - 2 min. After 30 min, the gels, each of total volume of 1 mL, were washed with 2 mL of HEPES buffer at pH 7.6 and stored wet under 2 mL of HEPES at 4˚C, pending use. The gels exhibited high stability and could be used more than a month after preparation; however, in most cases they were used within 24 h.
2.2.2. Binding and Elution of MPA from Sol-Gel IAP Columns
Wet gels were thoroughly crushed with 2 mL of 50 mM PBS, pH 7.2 and packed into 20-mL (1.5 × 12) cm, Econo-Pac disposable chromatography columns (Bio Rad, Philadelphia, PA, USA). These sol-gel columns were washed with 50 mL of PBS, pH 7.2, prior to sample application. To ensure optimal binding, the columns were kept under buffer at all times during the experiment. Aliquots of 1000 ng of MPA standard were spiked into 1 mL of PBS, pH 7.2, or 10 mL of NHS (diluted 1:10 in PBS, pH 7.2) and kept for 1 h at room temperature. Samples were applied to “empty” sol-gel columns or to columns doped with 500 μL of anti-MPA antiserum (unless otherwise indicated). The eluate was collected and applied to the column once again to ensure better binding. Unbound MPA was removed by washing the columns with 20 mL of DDW. Elution was performed with 10 mL of PESTI-S absolute ethanol (Bio-Lab, Jerusalem, Israel). The eluted fraction was subjected to vacuum evaporation to remove the eluting solvent, and the eluate was reconstituted in 1000 μL of PBS-E for ELISA. Binding experiments were performed with pairs of sol-gel columns, comprising (A) an experimental column containing antiMPA antiserum (total binding), and (B) an empty control column without antiserum (nonspecific binding). Specific binding was defined as the difference between total and nonspecific binding. The MPA content in sol-gel IAP eluates was determined by means of the direct ELISA. All samples were tested in duplicate at five successive twofold dilutions in PBS-E that were within the range of the standard curve. The dilutions are listed in detail in the legend of each Figure or Table. Aliquots of 50 μL of the tested sample were added to the wells, together with 50 μL of MPA-HRP conjugate diluted 1:1000 (i.e., a final dilution of 1:2000) in PBS-2 × T, in duplicate, as described above for the direct ELISA (section 2.1.5). The MPA content in tested samples was determined by reference to an MPA calibration curve, after linearization of the data by transformation to a logit-log plot by means of the Origin software, Version 6.0 (Microcal Software, Northampton, MA, USA). Slopes of the curves obtained for all the samples were tested for parallelism with the standard curve as described above in section 2.1.6.
2.2.3. Solid-Phase Extraction (SPE): Sample Application and Elution
Solid-phase extraction was carried out when the sol-gel eluates were further subjected to LC-MS/MS analysis. Oasis SPE columns (Waters, Milford, MA, USA) were preconditioned by two consecutive washes with 10 mL of PESTI-S absolute ethanol (Bio-Lab, Jerusalem, Israel), followed by 10 mL of DDW. Sol-gel eluates (10 mL of absolute ethanol) were diluted 1:10 with DDW to bring the ethanol concentration in the sample to 10% before it was applied to the SPE columns. Sol-gel-eluted samples were loaded onto the columns, which were then washed with 10 mL of 10% ethanol. Elution was carried out with 1 mL of absolute ethanol, and the eluted fraction was subjected to vacuum evaporation to remove the eluting solvent. Samples were kept dry at −20˚C, pending use.
2.3. Chemical Analytical Methods
LC-MS/MS Analysis
An LC-MS/MS method was developed, to assess the compatibility of sol-gel IAP with chemical analyses. This was done by determining the precision with which MPA content in spiked sol-gel/SPE eluates could be determined. Empty sol-gel columns (1:6 format) and columns doped with 300 µL of anti-MPA antiserum were loaded with 1 mL of un-spiked DDW (instead of MPA) and the “samples” were eluted with 10 mL of ethanol and then diluted with DDW to 10% ethanol and passed through an SPE column as described in section 2.2.3. All samples were vacuum evaporated and were reconstituted, prior to LC-MS/MS analysis, with 150 µL of the diluent alone, which comprised 30% acetonitrile (ACN; J.T. Baker, Phillipsburg, N.J), made up in HPLC-grade DDW purified with the MiliQ system. Eluates from doped and empty columns were then spiked with MPA at 10 ng∙min−1 and subjected to LC-MS/MS analysis. Eluates from doped columns were also spiked with MPA standard made up from a stock solution of MPA in methanol at a series of concentrations ranging from 1 to 100 ng∙mL−1 made up from a stock solution of MPA in methanol at 100 µg∙mL−1 (J.T. Baker, Phillipsburg, N.J). These samples were used to generate a calibration curve.
Samples were analyzed by LC-MS/MS multiple reaction monitor (MRM) detection in the positive-ion mode, after separation on a reverse-phase C-18 column, attached to an Alliance model 2795 HT HPLC system (Waters, Milford, MA). The liquid chromatographic separation was carried out on a Gemini C-18 column (50 × 2.0) mm, 3 µm particle size, 110 Å pore size (Phenomenex, Torrance, CA), with an injected volume of 10 µL. Solvent A comprised 10% aqueous ACN containing 0.1% of ammonium hydroxide (J.T. Baker, Phillipsburg, NJ, USA); solvent B comprised 90% aqueous ACN containing 0.1% of ammonium hydroxide. For the analysis the solvent initially comprised 65% A and 35% B; after 0.5 min, the solvent was modified over 5 min, according to the Waters linear program, to 100% B; the flow rate was 0.3 mL∙min−1; the MPA retention time, tR was 4.1 min. Following the LC analysis, all samples were analyzed with a Micromass Quattro Pt triple-quadrupole mass spectrometer operating in the electrospray ionization mode. The data were processed with Masslynx v. 4.0 and Quantlynx v.4.0 software. The amount of MPA in the samples was determined by comparison with calibration curves based on the spiked samples, and constructed by plotting the concentrations in the spiked samples against the peak areas found in their chromatograms.
2.4. Statistics
Differences between the average values were subjected to Tukey-Kramer one-way ANOVA at p < 0.05 (95%).
3. Results and Discussion
3.1. Development of MPA ELISAs
3.1.1. Optimization and Sensitivity Determination
The present study focused on the development of a solgel-based IAP method for purification and concentration of the progestin drug, MPA. For this purpose a polyclonal Ab for MPA was generated, and immunochemical assays for evaluation of the efficiency of the sol-gel based IAP method were developed. The immunochemical detection assays were based on a microplate ELISA. Our previous experience in development of ELISAs revealed that different ELISA formats resulted in differing sensitivities and cross-reactivity patterns, and that different formats reacted differently to the presence of interfering factors in “real world” samples. Therefore, it is essential to develop more than one assay format and to choose the best one for each specific purpose. This is especially important in cases where biological, food, and environmental samples are analyzed, because such samples are not homogeneous; they may contain pigmentation and other factors that might interfere heavily and in differing ways with different assay formats. In light of the above considerations two immunochemical assays (competitive indirect and direct ELISAs) for monitoring the MPA were developed. In the competitive indirect ELISA microplates were coated with a hapten-carrier protein (OVA) conjugate and the analyte competed with the hapten conjugate adsorbed to the microplate for binding to a limited amount of Abs. In the competitive direct ELISA Abs were adsorbed onto the microplate via Protein A, and the analyte competed with a hapten-enzyme conjugate (HRP) for binding to a limited amount of the adsorbed Ab. Development of the MPA ELISA involved two sets of experiments: the first set was intended to determine the optimal dilutions of the coating conjugate MPA-OVA (indirect format), the MPA-HRP conjugate (direct format), the antiserum and the secondary Ab (checkerboard tests); the second set was intended to generate a standard curve, to determine the I50 value and the limit of detection (LOD, I20) of the assay, the tolerance of the Abs to organic solvents, and their cross-reactivity with other progestins and steroid hormones.
The first set of experiments revealed that for the competitive indirect ELISA dilutions of 1:6000 for the MPA-OVA conjugate and 1:5000 (final) of the anti-MPA antiserum resulted in high binding and a low background, i.e., nonspecific binding. In the direct ELISA dilutions of 1:8000 or 1:16,000 for the coating Ab and of 1:2000 (final) for the HRP-hapten conjugate resulted in a good signal-to-background ratio (data not shown). The second set of experiments determined the working ranges of the assay, i.e., 0.1 - 25 and 0.1 - 10 ng per 50 µL for the indirect and direct ELISAs, respectively (Figures 2 and 3), and the I50 and I20 values for both assay formats. Basically, there were no marked differences between the working ranges of the respective formats, and both exhibited high affinities and low detection limits, with I50 and I20 values of 34.0 ± 15.4 ng∙mL−1 and 1.4 ± 0.2 ng∙mL−1 (n = 4), respectively, in the indirect ELISA, and 20.2 ± 1.6 ng∙mL−1 and 4.0 ± 0.4 ng∙mL−1 (n = 25), respectively, for the direct ELISA (Table 1). The interand intra-assay accuracies were 93 and 87%, respectively. Because of the very low solubility of MPA in neutral aqueous buffers both assays were carried out in the presence of 10% ethanol. The presence of ACN or acetone (at 10%), or higher percentages (25%) of ethanol in the direct ELISA did not have a marked effect on either value. Methanol (at 10%) in the indirect ELISA slightly improved both values, and a similar trend was obtained in the absence of organic solvents (Table 1). Since the reproducibility of the direct ELISA in the presence of 10% ethanol in PBST was slightly better than that of the indirect ELISA, the direct format and this buffer were chosen for all subsequent experiments. The tolerance of
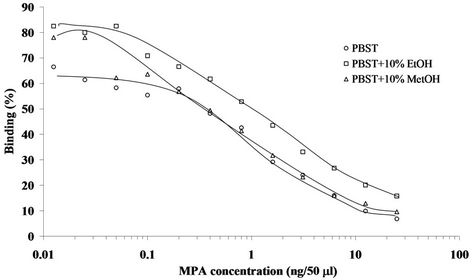
Figure 2. Representative standard curves of MPA in indirect competitive ELISA format. The assay was carried out in PBST, and PBST containing 10% ethanol (EtOH) or 10% methanol (MetOH). Assay conditions: 1:6000 dilution of the coating antigen and 1:5000 (final) dilution of the primary Ab.
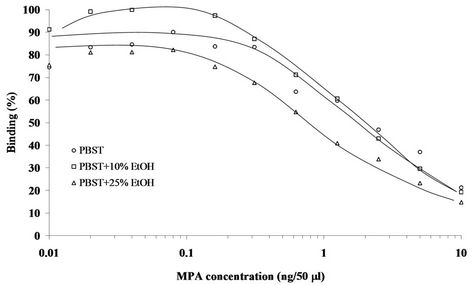
Figure 3. Representative standard curves of MPA in competitive direct ELISA format. The assay was carried out in PBST, or PBST containing 10% or 25% ethanol (EtOH). Assay conditions: 1:16,000 dilution of the coating primary Ab and 1:2000 (final) dilution of the MPA-HRP conjugate.
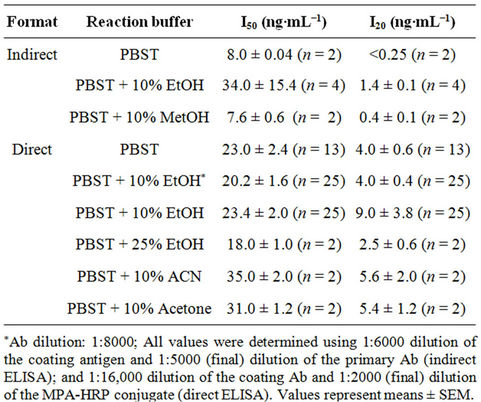
Table 1. I50 and detection limit (I20) values of indirect and direct ELISA formats.
the assay to both of these organic solvents was an important finding. The low solubility of MPA in aqueous buffers necessitates its extraction and analysis in the presence of organic solvents, therefore the immunochemical assays used to monitor it must tolerate the presence of such solvents.
3.1.2. Cross-Reactivity (CR)
Once the assay had been optimized and the sensitivity determined, the ELISA was used to characterize the antiserum for specificity and CR with other progestins and steroid hormones (Table 2). The CR with all of the listed compounds was determined by means of the direct ELISA format. As indicated in Table 2, under the tested conditions the MPA antiserum cross-reacted only with megestrol acetate, to an extent of 76%; no CR was detected with any of the other tested compounds. Thus, the results demonstrated high affinity towards MPA, with CR with only one other progesterone derivative.
Several immunoassays for monitoring MPA were reported previously [23-31], of which three were in an ELISA format [26-28]. All of these three assays were developed in the indirect competitive ELISA format (in which the coating antigen is adsorbed to the plate) and were carried out in PBS or PBST as the reaction buffer.

Table 2. Cross reactivity of the anti-MPA antiserum with various progestins and steroid hormones.
These studies exhibited I50 and LOD values in the ranges of 1.8 - 2.0 and 0.08 - 0.3 ng∙mL−1, respectively, similar to those obtained in our present study (i.e., 8.0 and <0.25 ng∙mL−1, respectively) in the indirect competitive ELISA format (when PBST was used as the reaction buffer, Table 1). Other immunoassays, based on capillary electrophoresis [30] or on the use of gold nanoparticles as a reporting system [25] exhibited similar detection limits (0.25 - 0.3 ng/ml). No other study used a competitive direct ELISA format, which offers the advantage of involving fewer steps in its performance, and no study demonstrated tolerance of the ELISA to organic solvents, which, as indicated above, offers an important advantage in monitoring MPA, which is only sparingly soluble in water. Only two studies [27,28] evaluated the CR, and in both cases the Abs (whether polyclonal or monoclonal) exhibited CR of 62% and 65% with megestrol acetate, similar to the value of 76% obtained in the present study (Table 2).
3.2. MPA Sol-Gel-Based IAP
The Abs that were developed in the present study were also used in development of a sol-gel-based IAP method for purification and concentration of MPA. For many years IAP has been regarded as one of the leading technologies for purification, concentration and isolation of chemical and biological compounds, and recently, IAP was revealed to be a very useful and reliable method for purification and concentration of agricultural and environmental residue samples [47-52]. The practical simplicity of the IAP method and its potential for reducing the levels of organic solvents that are often used to extract the tested analyte make IAP suitable for agricultural, environmental, and epidemiological applications.
Most studies of MPA monitoring in food or biological samples, e.g., animal tissue or human serum, found that such samples must undergo extensive multistep extraction and cleanup processes (e.g., liquid/liquid extraction, defatting, SPE) prior to analysis, to eliminate matrix interference, and this applies to both immunochemical and chemical instrumental analysis (for example see [20-22] and references therein). Employment of such protocols results in analyte loss, impairs accuracy, and limits the scope for employment of these methods in large-scale monitoring; these disadvantages highlight the general need to improve and simplify sample preparation methods and, simultaneously, to reduce their cost. Application of IAP to agricultural and environmental samples, e.g., food, soil extracts, effluent water, etc., and even to biological samples, e.g., human serum or breast milk, requires, among other things, the ability to protect the Abs against denaturing factors or organic solvents that are present in extracts of the tested samples and that might impair the activity of the Abs.
One of the main goals of the present study, therefore, was to develop a method that satisfies these requirements and is also highly effective in purification of the analyte obtained from biological, environmental and food samples, and to that end, we have focused on and developed an effective sol-gel-based IAP method for purification and concentration of MPA. The ability of the sol-gelentrapped Abs to bind the analyte was tested with several different sol-gel formats. The amounts of MPA recovered from the sol-gel columns were determined with the direct ELISA. The column binding capacity was examined with various amounts of entrapped Abs, ranging from 100 to 500 µL of antiserum (equivalent to 5.6 - 28 mg protein), in a 1:12 sol-gel format. Binding of MPA was dose dependent, gradually increasing to a maximum at an amount of 400 µL Abs (Figure 4).
The sol-gel composition and preparation conditions are known to greatly influence the structure of the polymer. Previous studies have shown that the properties of the biocomposite, i.e., a sol-gel in which a biomolecule is entrapped, can be drastically affected by changes in the TMOS:HCl ratio, and also by involvement of additives in the sol-gel process. Such additives include hydrophobic moieties; e.g., polyethylene glycol (PEG), glycerol, polyvinylimidazole, etc.; surfactants; liposomes; organic solvents, e.g., cyclohexane; polysaccharides, e.g., dextran, cellulose, or chitosan; cofactors, e.g., redox modifiers; or even biological or synthetic materials. These additives may alter the physical properties of the gel, e.g., its rigidity, mechanical stability, pore size, and optical or elec-
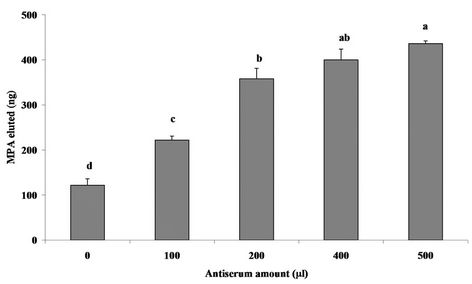
Figure 4. Dose response of MPA binding to various amounts of sol-gel-entrapped antiserum, ranging from 100 to 500 μL (equivalent to 5.6 - 28 mg protein). The sol-gel format used was 1:12. Binding in the absence of entrapped antiserum (0 μL, “empty” column) indicates nonspecific binding. Binding was tested with 1000 ng of MPA. Amounts of eluted MPA were determined with the direct ELISA format using five successive twofold dilutions, ranging from 1:2 to 1:32, in PBS-E. Each bar represents the mean ± SEM of four independent measurements. Means with the same letter do not differ significantly at p < 0.05.
trochemical clarity; they may affect the interactions of the gel with the entrapped biomolecules, and alter its overall activity and stability [53-55]. We therefore have examined the effects of several different sol-gel formats on the activity of the entrapped Abs. Four different TMOS:HCl ratios were used, in the presence and absence of PEG. Table 3 compares the performance of the various formats with regard to total binding, nonspecific binding (i.e., to columns that did not contain Abs, empty columns), and net binding. Examination of the nonspecific binding revealed relatively high values, some of which exceeded 20% of the applied analyte. Comparison of the levels of net binding in the presence and absence of PEG revealed significantly higher binding of MPA to the entrapped Abs in the absence of the porogen in the 1:4 and 1:6 formats. Similar results were obtained at 1:8 although the values did not differ significantly, because of high variability. At 1:12, binding in the presence and absence of PEG did not differ significantly. In almost all of our previous studies PEG significantly improved the binding capacity of a variety of analytes and reduced their nonspecific binding [56-58]. The exception was IMT, for which addition of PEG did not improve the binding capacity [13]. In general, all sol-gel formats were

Table 3. Effects of various sol-gel formats on the activity of entrapped anti-MPA antiserum.
highly effective in binding the analyte, although large amounts of antiserum (500 µL) were required for the 1:4 and 1:6 formats, compared with the 1:8 and 1:12 formats, in which, one-fifth of the amount of antiserum was entrapped (100 µL) and resulted in efficient binding of MPA which was only 1.2 to 1.7 or 1.6 to 2.1 lower than the amounts bound by the 1:4 and 1:6, respectively.
MPA binding was also tested with protein-A-purified IgG, which had been purified from the whole antiserum as described previously [56] and entrapped in a 1:8 sol-gel format. The results revealed dose-dependent net bindings of 45, 10, 169, 272, and 543 ng with 40, 80, 160, 300, and 400 µL of IgGs (equivalent to 80 - 800 µL of whole antiserum), respectively.
Interestingly, the binding of the purified IgGs was much lower than that of the whole antiserum, and 300 µL of the purified IgG bound less than 100 µL of the whole antiserum. Similar results were obtained previously, when IgG and whole antiserum of anti-di-nitrophenyl antiserum were used [56]. It is possible that the large amounts of nonspecific protein in the whole antiserum protected the entrapped IgGs from damage during gel formation.
The method was also optimized with respect to the eluting solvent. Four solvents were tested for their ability to dissociate MPA from the sol-gel IAP column: absolute ethanol, methanol, acetone, and ACN. The results clearly indicated that ethanol and methanol eluted MPA efficiently, and that the volume needed for full recovery was 5 mL. The other two solvents were ineffective as eluting solvents and resulted in a recovery of less than 1% of the applied analyte (data not shown).
3.3. Analysis of Spiked and Un-Spiked Samples
3.3.1. Evaluation of Unpurified Samples by ELISA
In order to test the ability of the ELISA to monitor MPA in “real world” samples, two sets of experiments were carried out: in the first, various percentages (0.5%, 2%, 4%, 8% and 16%) of un-spiked NHS and low fat milk were evaluated for their interference with the direct ELISA; in the second set, tap water, NHS and low-fat milk were spiked with MPA at 50 ng∙mL−1 and the ability of the immunoassay to determine the analyte content was monitored.
In the first experiment calibration curves were generated in the presence of NHS or low-fat milk at percentages of 0.5%, 2%, 4%, 8% and 16%, and the effects of the matrix on the reaction background (Bg, i.e., nonspecific binding of MPA-HRP to protein A adsorbed onto the plate), maximal binding, I50, and detection limit (I20) were determined by comparison with a calibration curve generated in PBST-E alone. The results (Table 4) revealed no interference of either matrix with the Bg of the ELISA, and almost no interference of milk content was observed with the maximal binding (i.e., binding of the MPA-
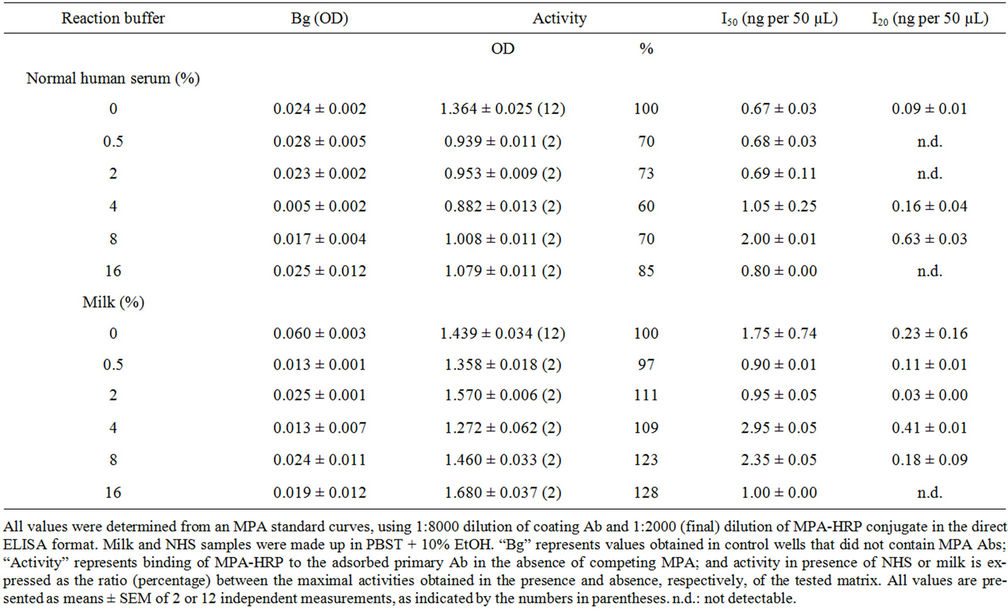
Table 4. Effect of normal human serum and low fat milk on MPA ELISA.
HRP conjugate to the Ab adsorbed to the microplate) up to 16%. The NHS interfered with the immunoassay, and the maximal binding obtained at the various percentages of serum dropped by 15% - 40% (Table 4). Evaluation of the I50 and I20 values indicated similar values in the presence and absence of the various percentages of milk and NHS, and, although the values were slightly higher at 8% serum, the trend was not consistent and the I50 value at 16% serum did not differ from that in the absence of the matrix. Attempts to monitor spiked MPA in NHS and milk samples did not succeed because of matrix interference, as indicated by inability to obtain a line parallel to the calibration curve. Tap water also interfered with the assay to a certain extent, and only 46% of the spiked amount was recovered (Table 5). It is important to note that occurrence of little or no interference of a tested matrix with the ELISA, as in the above cases of milk and serum, does not indicate the ability to evaluate the amount of spiked MPA in these matrices as the analyte could adhere to the serum proteins, or micelles or fatty components of the milk in a manner that does not enable their accurate determination.
3.3.2. Sol-Gel IAP of MPA from Spiked Human Serum Samples
The inability to determine MPA in spiked serum and milk samples indicated the need to purify samples prior to their immunochemical analysis. Although sample dilution is always a possible means of minimizing matrix interference, it might impair the detection of low levels of MPA residues in “real world” samples. Therefore, unspiked human serum samples and samples spiked with 1000 ng of MPA were applied on “empty” sol-gel columns and on columns that had been doped with 500 μL of Abs, and the MPA content of each eluate was moni-
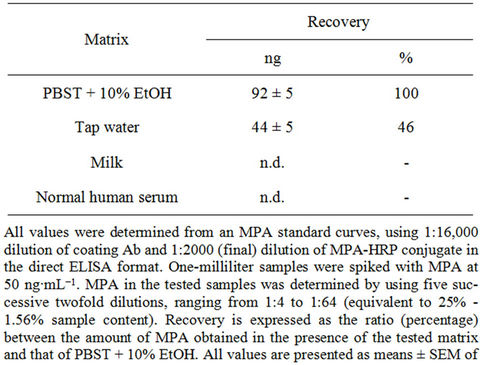
Table 5. Determination of spiked MPA in various matrices.
tored by competitive direct ELISA. Spiked serum samples that had not undergone sol-gel IAP served as controls to monitor the efficiency of the IAP process. The results presented in Table 6 reveal the high recovery rate (110%) of MPA from spiked serum samples, and clearly demonstrate the high efficiency of the sol-gel IAP method for removing interfering components from undiluted human serum samples, in a manner that enabled quantitative determination of analyte. Negligible amounts of MPA were detected in unspiked serum samples that had undergone IAP, and in spiked and unspiked samples that had passed through “empty” sol-gel columns. Untreated, i.e., “before IAP”, samples interfered with the assay, and the yields that were obtained from those spiked serum samples were above 3000 ng, which clearly indicated a false positive result. These findings reveal once again the high efficiency of the sol-gel method for analyte purification in single-step high recovery.
A wide variety of Abs—monoclonal, polyclonal, and purified IgGs—have been entrapped in sol-gel polymers, and their applications in IAP of samples of serum and other substances have been reported by many laborato-
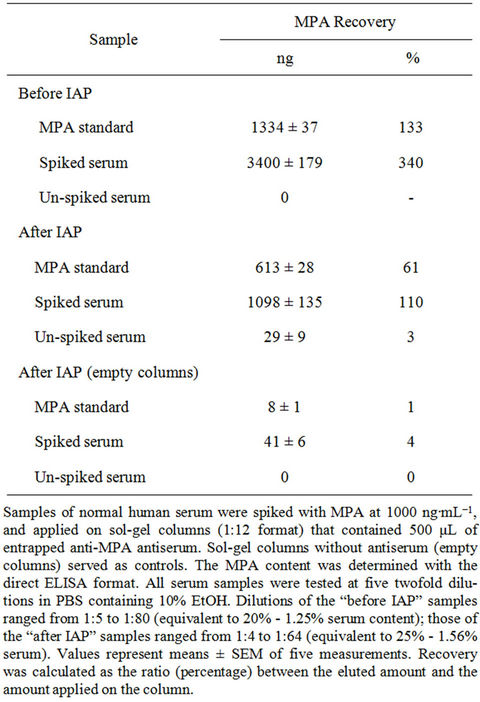
Table 6. Recovery of MPA from spiked and un-spiked samples of normal human serum, before and after sol-gel IAP.
ries, including ours [32,33,35,37]. The overall advantages of the sol-gel technique, and the successful entrapment of a wide variety of Abs, which enables development of efficient IAP protocols, indicate the generic nature of the method and the practicability of implementing it for purification of a wide range of analytes.
Although ELISA is known to tolerate matrix interference, even tap-water samples interfered with the immunoassay (Table 5), and human serum exhibits a much higher potential to interfere with the assay by affecting analyte-Ab interactions and thereby causing false-negative or false-positive results.
Indeed, the present findings indicate that maximal binding was affected by a serum content as low as 4% (Table 4), and MPA could not be detected in samples with serum contents of 25% or 20% (Tables 5 and 6, respectively). Pretreatment of the samples by IAP significantly reduced matrix interference, eliminated the need to adjust pH values to ensure compatibility with the assay, and provided concentrated, ready-to-use samples that enabled accurate monitoring of MPA (where the serum content was equivalent to 25%, Table 6). To the best of our knowledge, there is only one previous report on employment of ELISA for monitoring MPA in plasma; however, in that study the sample underwent extraction with organic solvents prior to analysis [26].
3.4. Compatibility of Sol-Gel IAP with Chemical Analysis
Sol-gel eluates were also tested for their compatibility with LC-MS/MS analysis, by examining the extent to which sol-gel eluates (that had been further concentrated by SPE) interfered with the analysis. This was done by spiking the eluates with MPA prior to the LC-MS/MS analysis and monitoring the accuracy by which the compound can be determined in the tested sample. For that purpose 1-mL of DDW was applied on empty and doped sol-gel columns; the eluates were subjected to SPE concentration, then spiked with MPA at 10 ng∙mL−1, and analyzed by LC-MS/MS. The results revealed high accuracy in determination of the MPA content in the spiked eluates from both the doped and empty sol-gel columns (12.09 and 13.60 ng∙mL−1, respectively), and, as indicated in Figure 5, showed that the MPA in the spiked eluate of the doped sol-gel column was identical with an MPA standard, and generated only a single peak with no impurities. The results indicate that the eluates did not interfere with the LC-MS/MS analysis, and also that sol-gel IAP/SPE samples could be analyzed directly by LC-MS/MS according to the above protocol, without any further treatment. This finding highlights a major advantage of the sol-gel IAP method over commonly used extraction and purification methods that involve tedious,
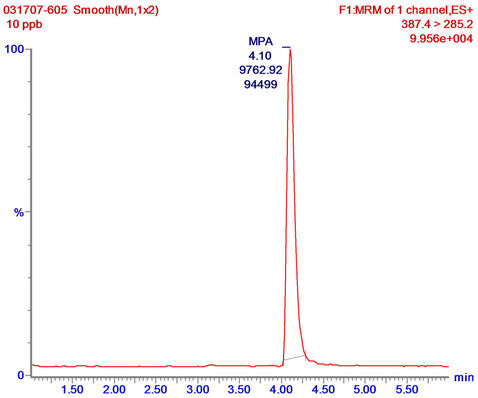 (a)
(a)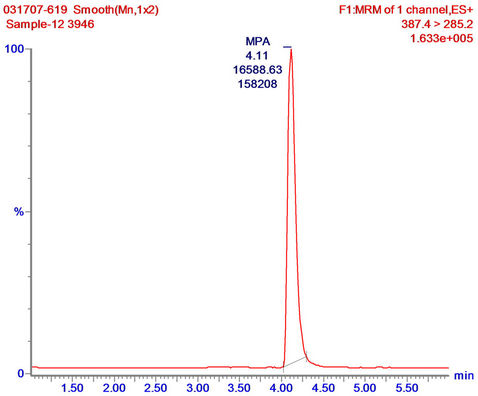 (b)
(b)
Figure 5. LC-MS/MS analysis of MPA standard in LC running buffer (a); and sol-gel/SPE eluates (concentrated from 1 mL of tap water) spiked with MPA at 10 ng∙mL−1 (b). Sample reconstitution and analysis were carried out as described in “Materials and methods”.
long, and expensive multistep processes that may result in low yields of the tested analyte and production of large volumes of toxic waste. The sol-gel IAP method described and discussed above eliminates the need for all of these steps, and enables rapid, simple and inexpensive preparation of high-purity samples that are ready for chemical analysis. It may very well be that further improvement in the sol-gel IAP method will eliminate the need for the SPE step, as in the case of consecutive ELISA analysis.
4. Summary and Conclusion
At present, immunochemical assays such as ELISA can be developed for almost any compound—artificial or natural, and they offer many advantages for quick, inexpensive, and efficient analysis of large numbers of samples. However, the ability to use an ELISA in a reliable manner requires sample purification which eliminates matrix interference with the assay. The IAP sol-gel based method developed in this study overcomes most of the difficulties associated with residue analysis in biological samples, e.g., serum, and offers many advantages over the commonly used IAP methods based on Abs that are entrapped in, adsorbed by, or covalently bound to various matrixes. The sol-gel method meets the IAP requirements for purification of analytes from crude samples, without the need for any preliminary treatment prior to the IAP step; it simplifies examination of the samples, and decreases the analysis time and cost by enabling sample analysis—by either immunochemical or instrumental chemical analytical methods—after only a single purification step. A combination of both approaches could provide a basis for analysis of MPA in biological samples in order to monitor their pharmacokinetic properties, and implementation of these approaches could be extended to studying population exposure to MPA and also to monitoring occurrence of MPA contamination in food, soil, and other environmental samples.
5. Acknowledgements
This research was supported by the European Commission STREP contract FOOD-CT-2004-513953 F & F on: “Pharmaceutical products in the environment: Development and employment of novel methods for assessing their origin, fate and effects on human fecundity”. The funding source had no role in the study design, in collection or analysis of the data, in writing the report, or in deciding whether and where to submit the paper for publication. This paper is contribution no. 505/11 from the Agricultural Research Organization, the Volcani Center, Bet Dagan 50250, Israel.
REFERENCES
- M. Crane, C. Watts and T. Boucard, “Chronic Aquatic Environmental Risks from Exposure to Human Pharmaceuticals,” Science of the Total Environment, Vol. 367, No. 1, 2006, pp. 23-41. doi:10.1016/j.scitotenv.2006.04.010
- K. Fent, A. A. Weston and D. Caminada, “Ecotoxicology of Human Pharmaceuticals,” Aquatic Toxicology, Vol. 76, No. 2, 2006, pp. 122-159. doi:10.1016/j.aquatox.2005.09.009
- I. Figa-Talamanca, M. E. Traina and E. Urbani, “Occupational Exposures to Metals, Solvents and Pesticides: Recent Evidence on Male Reproductive Effects and Biological Markers,” Occupational Medicine, Vol. 51, No. 3, 2001, pp. 174-188. doi:10.1093/occmed/51.3.174
- M. Yang, M. S. Park and H. S. Lee, “Endocrine Disrupting Chemicals: Human Exposure and Health Risks,” Journal of Environmental Science and Health, Vol. 24, No. 2, 2006, pp. 183-224. doi:10.1080/10590500600936474
- P. K. Jjemba, “Excretion and Ecotoxicity of Pharmaceutical and Personal Care Products in the Environment,” Ecotoxicology and Environmental Safety, Vol. 63, No. 1, 2006, pp. 113-130. doi:10.1016/j.ecoenv.2004.11.011
- O. A. H. Jones, N. Voulvoulis and J. N. Lester, “Human Pharmaceuticals in Wastewater Treatment Processes,” Critical Reviews in Environmental Science and Technology, Vol. 35, No. 4, 2005, pp. 401-427. doi:10.1080/10643380590956966
- A. Nikolaou, S. Meric and D. Fatta, “Occurrence Patterns of Pharmaceuticals in Water and Wastewater Environments,” Analytical and Bioanalytical Chemistry, Vol. 387, No. 4, 2007, pp. 1225-1234. doi:10.1007/s00216-006-1035-8
- A. H. Piersma, M. Luijten, V. Popov, V. Tomenk, M. Altstein, F. Kagampang and H. Schlesinger, “Pharmaceuticals,” In: I. Shaw, Ed., Endocrine-Disrupting Chemicals in Food, Woodhead Publishing Limited, Cambridge, 2009, pp. 459-518. doi:10.1533/9781845695743.4.459
- L. Sun, W. Yong, X. Chu and J. M. Lin, “Simultaneous Determination of 15 Steroidal Oral Contraceptives in Water Using Solid-Phase Disk Extraction Followed by High Performance Liquid Chromatography-Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 1216, No. 28, 2009, pp. 5416-5423. doi:10.1016/j.chroma.2009.05.041
- M. Velicu and R. Suri, “Presence of Steroid Hormones and Antibiotics in Surface Water of Agricultural, Suburban and Mixed-Use Areas,” Environmental Monitoring and Assessment, Vol. 154, No. 1-4, 2009, pp. 349-359. doi:10.1007/s10661-008-0402-7
- M. Shalev, M. Bardugo, A. Nudelman, A. Krol, H. Schlesinger, A. Bronshtein and M. Altstein, “Monitoring of Progestins: Development of Immunochemical Methods for Purification and Detection of Levonorgestrel,” Analytica Chimica Acta, Vol. 665, No. 2, 2010, pp. 176-184. doi:10.1016/j.aca.2010.03.029
- M. Shalev and M. Altstein, “Sol-Gel Entrapped Levonorgestrel Antibodies: Activity and Structural Changes as a Function of Different Polymer Formats,” Materials, Vol. 4, No. 3, 2011, pp. 469-486.
- N. Skalka, A. Krol, H. Schlesinger and M. Altstein, “Monitoring of the Non-Steroid Anti-Inflammatory Drug Indomethacin: Development of Immunochemical Methods for Its Purification and Detection,” Analytical and Bioanalytical Chemistry, Vol. 400, No. 10, 2011, pp. 3491-3504. doi:10.1007/s00216-011-5027-y
- A. Black, D. Francoeur, T. Rowe, J. Collins, D. Miller, T. Brown, M. David, S. Dunn, W. A. Fisher, N. Fleming, C. A. Fortin, E. Guilbert, L. Hanvey, A. Lalonde, R. Miller, M. Morris, T. O’Grady, H. Pymar, T. Smith and E. Henneberg, “Canadian Contraception Consensus,” Journal of Obstetrics and Gynaecology Canada, Vol. 26, No. 2, 2004, pp. 347-436.
- A. M. Kaunitz, “Long-Acting Injectable Contraception with Depot Medroxyprogesterone Acetate,” American Journal of Obstetrics and Gynecology, Vol. 170, No. 5, 1994, pp. 1543-1549.
- S. Kennedy, A. Bergqvist, C. Chapron, T. D’Hooghe, G. Dunselman, R. Greb, L. Hummelshoj, A. Prentice and E. Saridogan, “ESHRE Guideline for the Diagnosis and Treatment of Endometriosis,” Human Reproduction, Vol. 20, No. 10, 2005, pp. 2698-2704. doi:10.1093/humrep/dei135
- E. R. Guilbert, J. P. Brown, A. M. Kaunitz, M. S. Wagner, J. Berube, L. Charbonneau, D. Francoeur, A. Gilbert, F. Gilbert, G. Roy, V. Senikas, R. Jacob and R. Morin, “The Use of Depot-Medroxyprogesterone Acetate in Contraception and Its Potential Impact on Skeletal Health,” Contraception, Vol. 79, No. 3, 2009, pp. 167-177. doi:10.1016/j.contraception.2008.10.016
- C. Choksuchat, S. Zhao, T. D. Deutch, T. D. Kimble and D. F. Archer,” Effects of Progesterone, Levonorgestrel and Medroxyprogesterone Acetate on Apoptosis in Human Endometrial Endothelial Cells,” Contraception, Vol. 79, No. 2, 2009, pp. 139-145. doi:10.1016/j.contraception.2008.08.008
- C. Lanari, C. A. Lamb, V. T. Fabris, L. A. Helguero, R. Soldati, M. C. Bottino, S. Giulianelli, J. P. Cerliani, V. Wargon and A. Molinolo, “The MPA Mouse Breast Cancer Model: Evidence for a Role of Progesterone Receptors in Breast Cancer,” Endocrine-Related Cancer, Vol. 16, 2009, pp. 333-350. doi:10.1677/ERC-08-0244
- H. Chang, S. Wu, J. Hu, M. Asami and S. Kunikane, “Trace Analysis of Androgens and Progestogens in Environmental Waters by Ultra-Performance Liquid Chromatography-Electrospray Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 1195, No. 1-2, 2008, pp. 44-51. doi:10.1016/j.chroma.2008.04.055
- G. Kaklamanos, G. Theodoridis and T. Dabalis, “Gel Permeation Chromatography Clean-up for the Determination of Gestagens in Kidney Fat by Liquid Chromatography-Tandem Mass Spectrometry and Validation According to 2002/657/EC,” Journal of Chromatography A, Vol. 1216, No. 46, 2009, pp. 8067-8071. doi:10.1016/j.chroma.2009.04.050
- G. Kaklamanos, G. Theodoridis and T. Dabalis, “Determination of Anabolic Steroids in Muscle Tissue by Liquid Chromatography-Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 1216, No. 46, 2009, pp. 8072-8079. doi:10.1016/j.chroma.2009.04.051
- J. C. Cornette, K. T. Kirton and G. W. Duncan, “Measurement of Medroxyprogesterone Acetate (Provera) by Radioimmunoassay,” Journal of Clinical Endocrinology & Metabolism, Vol. 33, No. 3, 1971, pp. 459-466. doi:10.1210/jcem-33-3-459
- M. Hiroi, F. Z. Stanczyk, U. Goebelsmann, P. F. Brenner, M. E. Lumkin and D. R. Mishell, “Radioimmunoassay of Serum Medroxyprogesterone Acetate (Provera) in Women Following Oral and Intravaginal Administration,” Steroids, Vol. 26, No. 3, 1975, pp. 373-386. doi:10.1016/0039-128X(75)90082-3
- G. Cui, H. Q. Chu, Y. M. Hu and C. L. Xu, “Ultrasensitive Signal Amplified Immunoassay of Medroxyprogesterone Acetate (MPA) Using the Atomic Absorption of Silver Deposited on the Surface of Gold Nanoparticles,” Food and Agricultural Immunology, Vol. 21, No. 2, 2010, pp. 165-173. doi:10.1080/09540100903556021
- L. K. Lewis, P. A. Elder and G. K. Barrell, “An Enzyme-Linked-Immunosorbent-Assay (Elisa) for Plasma Medroxyprogesterone Acetate (Mpa),” Journal of Steroid Biochemistry and Molecular Biology, Vol. 42, No. 2, 1992, pp. 179-183. doi:10.1016/0960-0760(92)90026-F
- C. F. Peng, Y. W. Chen, W. Chen, C. L. Xu, J. M. Kim and Z. Y. Jin, “Development of a Sensitive Heterologous Elisa Method for Analysis of Acetylgestagen Residues in Animal Fat,” Food Chemistry, Vol. 109, No. 3, 2008, pp. 647-653. doi:10.1016/j.foodchem.2007.12.072
- H. L. Qi, R. R. Wang, G. Yang and J. L. Liu, “An Enzyme-Linked Immunoassay for the Detection of Medroxyprogesterone Acetate in Intestines Based on Monoclonal Antibody,” Food and Agricultural Immunology, Vol. 22, 2011, pp. 125-134. doi:10.1080/09540105.2010.549208
- H. Tieming, P. Chifang, C. Xiaogang and X. Chuanlai, “Rapid Determination of Time-Resolved Fluoroimmunoassay for Medroxyprogesterone Acetate Residues in Pork Tissues and Comparison with Liquid Chromatography and Tandem Mass Spectrometry,” Journal of Fluorescence, Vol. 16, No. 6, 2006, pp. 743-747. doi:10.1007/s10895-006-0132-6
- C. F. Peng, T. M. Huo, L. Q. Liu, X. G. Chu and C. L. Xu, “Determination of Medroxyprogesterone Acetate Residues by Ce Immunoassay with Chemiluminescence Detection,” Electrophoresis, Vol. 28, No. 6, 2007, pp. 970- 974. doi:10.1002/elps.200600290
- C. Peng, Z. Li, Y. Zhu, W. Chen, Y. Yuan, L. Liu, Q. Li, D. Xu, R. Qiao, L. Wang, S. Zhu, Z. Jin and C. Xu, “Simultaneous and Sensitive Determination of Multiplex Chemical Residues Based on Multicolor Quantum Dot Probes,” Biosensors and Bioelectronics, Vol. 24, No. 12, 2009, pp. 3657-3662. doi:10.1016/j.bios.2009.05.031
- M. Altstein and A. Bronshtein, “Sol Gel Immunoassays and Immunoaffinity Chromatography,” In: J. M. Van Emon, Ed., Immunoassay and Other Bioanalytical Techniques, CRC press, 2007, pp. 357-383.
- D. Avnir, T. Coradin, O. Lev and J. Livage, “Recent Bio-Applications of Sol-Gel Materials,” Journal of Materials Chemistry, Vol. 16, 2006, pp. 1013-1030. doi:10.1039/b512706h
- M. Cichna-Markl, “Selective Sample Preparation with Bioaffinity Columns Prepared by the Sol-Gel Method,” Journal of Chromatography A, Vol. 1124, No. 1-2, 2006, pp. 167-180. doi:10.1016/j.chroma.2006.06.077
- V. B. Kandimalla, V. S. Tripathi and H. X. Ju, “Immobilization of Biomolecules in Sol-Gels: Biological AND Analytical Applications,” Critical Reviews in Analytical Chemistry, Vol. 36, No. 2, 2006, pp. 73-106. doi:10.1080/10408340600713652
- M. Alstein, O. Ben-Aziz, N. Skalka, A. Bronshtein, J. C. Chuang and J. M. Van Emon, “Development of an Immunoassay and a Sol Gel Based Imuunoaffinity Cleanup Method for Coplanar Pcb’s from Soil and Sediment Samples,” Analytica Chimica Acta, Vol. 675, No. 2, 2010, pp. 138-147.
- P. Degelmann, S. Egger, H. Jurling, J. Muller, R. Niessner and D. Knopp, “Determination of Sulfonylurea Herbicides in Water and Food Samples Using Sol-Gel GlassBased Immunoaffinity Extraction and Liquid Chromatography-Ultraviolet/Diode Array Detection or Liquid Chromatography-Tandem Mass Spectrometry,” Journal of Agricultural and Food Chemistry, Vol. 54, No. 6, 2006, pp. 2003-2011. doi:10.1021/jf052718+
- R. J. Hodgson, M. A. Brook and J. D. Brennan, “Capillary-Scale Monolithic Immunoaffinity Columns for Immunoextraction with in-Line Laser-Induced Fluorescence Detection,” Analytical Chemistry, Vol. 77, No. 14, 2005, pp. 4404-4412. doi:10.1021/ac048142p
- M. Kaware, A. Bronshtein, J. Safi, J. M. Van Emon, J. C. Chuang, B. Hock, K. Kramer and M. Altstein, “Enzyme-Linked Immunosorbent Assay (ELISA) and SolGel-Based Immunoaffinity Purification (IAP) of the Pyrethroid Bioallethrin in Food and Environmental Samples,” Journal of Agricultural and Food Chemistry, Vol. 54, No. 18, 2006, pp. 6482-6492. doi:10.1021/jf0607415
- P. Pulido-Tofino, J. M. Barrero-Moreno and M. C. Perez-Conde, “Analysis of Isoproturon at Trace Level by Solid Phase Competitive Fluoroimmunosensing after Enrichment in a Sol-Gel Immunosorbent,” Analytica Chimica Acta, Vol. 562, No. 1, 2006, pp. 122-127. doi:10.1016/j.aca.2005.12.072
- F. Tsagkogeorgas, M. Ochsenkuhn-Petropoulou, R. Niessner and D. Knopp, “Encapsulation of Biomolecules for Bioanalytical Purposes: Preparation of Diclofenac Antibody-Doped Nanometer-Sized Silica Particles by Reverse Micelle and Sol-Gel Processing,” Analytica Chimica Acta, Vol. 573-574, 2006, pp. 133-137. doi:10.1016/j.aca.2006.03.006
- L. E. Vera-Avila, J. C. Vazquez-Lira, L. M. Garcia and R. Covarrubias, “Sol-Gel Immunosorbents Doped with Polyclonal Antibodies for the Selective Extraction of Malathion and Triazines from Aqueous Samples,” Environmental Science & Technology, Vol. 39, No. 14, 2005, pp. 5421-5426. doi:10.1021/es048000c
- X. Zhang, D. Martens, P. M. Kramer, A. A. Kettrup and X. Liang, “Development and Application of a Sol-Gel Immunosorbent-Based Method for the Determination of Isoproturon in Surface Water,” Journal of Chromatography A, Vol. 1102, No. 1-2, 2006, pp. 84-90. doi:10.1016/j.chroma.2005.10.072
- X. Zhang, D. Martens, P. M. Kramer, A. A. Kettrup and X. Liang, “On-Line Immunoaffinity Column-Liquid Chromatography-Tandem Mass Spectrometry Method for Trace Analysis of Diuron in Wastewater Treatment Plant Effluent Sample,” Journal of Chromatography A, Vol. 1133, No. 1-2, 2006, pp. 112-118. doi:10.1016/j.chroma.2006.08.007
- R. R. Sokal and J. F. Rohlf, “Biometry,” W. H. Freeman and Co., New York, 1995.
- M. Altstein, A. Bronshtein, B. Glattstein, A. Zeichner, T. Tamiri and J. Almog, “Immunochemical Approaches for Purification and Detection of TNT Traces by Antibodies Entrapped in a Sol-Gel Matrix,” Analytical Chemistry, Vol. 73, No. 11, 2001, pp. 2461-2467. doi:10.1021/ac001376y
- J. C. Chuang, J. M. Van Emon, R. Jones, J. Durnford and R. A. Lordo, “Development and Application of Immunoaffinity Column Chromatography for Atrazine in Complex Sample Media,” Analytica Chimica Acta, Vol. 583, No. 1, 2007, pp. 32-39. doi:10.1016/j.aca.2006.09.060
- J. D. Groopman and A. Zarba, “Immunoassays for Trace Chemical Analysis.” In: M. Vanderlan, L. H. Stanker, B. E. Watkins, D. W. Roberts, Eds., ACS Symposium Series, American Chemical Society, Washington DC, 1991, pp. 207-214.
- J. F. Lawrence, C. Menard, M. C. Hennion, V. Pichon, F. Le Goffic and N. Durand, “Use of Immunoaffinity Chromatography as a Simplified Cleanup Technique for the Liquid Chromatographic Determination of Phenylurea Herbicides in Plant Material,” Journal of Chromatography A, Vol. 732, No. 2, 1996, pp. 277-281. doi:10.1016/0021-9673(95)01274-5
- J. F. Lawrence, C. Menard, M. C. Hennion, V. Pichon, F. Le Goffic and N. Durand, “Evaluation of Immunoaffinity Chromatography as a Replacement for Organic Solvent Clean-up of Plant Extracts for the Determination of Triazine Herbicides by Liquid Chromatography,” Journal of Chromatography A, Vol. 752, No. 1-2, 1996, pp. 147-154. doi:10.1016/S0021-9673(96)00486-4
- J. F. Lawrence and C. Menard, “Determination of Clenbuterol in Beef Liver and Muscle Tissue Using Immunoaffinity Chromatographic Cleanup and Liquid Chromatography with Ultraviolet Absorbance Detection,” Journal of Chromatography B, Vol. 696, No. 2, 1997, pp. 291-297. doi:10.1016/S0378-4347(97)00240-5
- S. Perez, I. Ferrer, M. C. Hennion and D. Barcelo, “Isolation of Priority Polycyclic Aromatic Hydrocarbons from Natural Sediments and Sludge Reference Materials by an Anti-Fluorene Immunosorbent Followed by Liquid Chromatography and Diode Array Detection,” Analytical Chemistry, Vol. 70, No. 23, 1998, pp. 4996-5001. doi:10.1021/ac980533e
- W. Jin and J. D. Brennan, “Properties and Applications of Proteins Encapsulated within Sol-Gel Derived Materials,” Analytica Chimica Acta, Vol. 461, No. 1, 2002, pp. 1-36. doi:10.1016/S0003-2670(02)00229-5
- H. Saito, K. Kanamori, K. Nakanish and K. Hirao, “Real Space Observation of Silica Monoliths in the Formation Process,” Journal of Separation Science, Vol. 30, No. 17, 2007, pp. 2881-2887. doi:10.1002/jssc.200700360
- Y. W. Sun, Y. J. Wang, W. Guo, T. Wang and G. S. Luo, “Triblock Copolymer and Poly (Ethylene Glycol) as Templates for Monolithic Silica Material with Bimodal Pore Structure,” Microporous and Mesoporous Materials, Vol. 88, No. 1-3, 2006, pp. 31-37. doi:10.1016/j.micromeso.2005.08.018
- A. Bronshtein, N. Aharonson, A. Turniansky and M. Altstein, “Sol-Gel-Based Immunoaffinity Chromatography: Application to Nitroaromatic Compounds,” Chemistry of Materials, Vol. 12, No. 7, 2000, pp. 2050-2058. doi:10.1021/cm990780e
- S. Shtelzer, S. Rappoport, D. Avnir, M. Ottolenghi and S. Braun, “Properties of Trypsin and of Acid-Phosphatase Immobilized in Sol-Gel Glass Matrices,” Biotechnology and Applied Biochemistry, Vol. 15, No. 3, 1992, pp. 227- 235.
- H. H. Yang, Q. Z. Zhu, H. Y. Qu, X. L. Chen, M. T. Ding and J. G. Xu, “Flow Injection Flurescence Immunoassay for Gentamicin Using Sol-Gel-Derived Mesoporous Biomaterial,” Analytical Biochemistry, Vol. 308, No. 1, 2002, pp. 71-76. doi:10.1016/S0003-2697(02)00222-1