Case Reports in Clinical Medicine
Vol.3 No.5(2014), Article ID:45940,5 pages DOI:10.4236/crcm.2014.35067
Barbiturate Coma: Rebound and Refractory Hyperkalemia
Bonnie C. Greenwood1, Christopher D. Adams1, Zain I. Khalpey2, Peter C. Hou3
1Department of Pharmacy, Brigham and Women’s Hospital, Boston, USA
2Department of Surgery, University of Arizona, Tucson, USA
3Department of Emergency Medicine, Brigham and Women’s Hospital, Boston, USA
Email: cdadams@partners.org
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 19 February 2014; revised 18 March 2014; accepted 17 April 2014
ABSTRACT
Purpose: To describe a traumatic brain injury patient who experienced profound dyskalemia upon the initiation and cessation of a pentobarbital infusion and propose management options for future patients receiving this intervention. Methods: Case report. Results: Case report. Conclusions: Dyskalemia has become an anticipated side effect of high dose barbiturate infusions in the setting of elevated intracranial pressure. Hypokalemia during the administration of a barbiturate infusion has been identified within this patient population and was an expected adverse event during this intervention. However, in this case we observed a significant and complicated refractory hyperkalemia upon cessation of the barbiturate infusion which required aggressive management. An objective causality assessment suggests that this adverse event was possibly related to pentobarbital. What this case documents that other cases have not is that upon re-introduction of the pentobarbital infusion, serum potassium levels did not normalize. This questions whether severe rebound hyperkalemia is a pharmacodynamic or infusion-related reaction. More data is needed to identify the mechanism of this adverse event and recommend an appropriate treatment approach.
Keywords
Barbiturate, Rebound, Refractory Hyperkalemia

1. Introduction
Barbiturates have been used for more than 70 years, generally at low doses for their anesthetic and anti-seizure properties. Higher doses of barbiturates have been shown to be effective in reducing refractory elevated intracranial pressure (ICP) and arterial vasospasms following subarachnoid hemorrhage [1] . The higher doses required to lower ICP typically result in a comatose state and increase the risk for adverse events such as: arterial hypotension, infection, immunosuppression, respiratory complications, hepatic dysfunction, renal dysfunction and suppressed cortical activity [2] [3] . Furthermore, barbiturates are known to cause profound and specific electrolyte disturbances, such as dyskalemia. Barbiturate-induced hypokalemia, initially observed in laboratory animals, was documented in humans almost 10 years later [4] . There is little data in the literature evaluating the mechanism of this life-threatening adverse event nor describing an effective intervention to safely correct this abnormality.
We report the case of a traumatic brain injury patient with refractory, elevated ICP who was initiated on a pentobarbital infusion. During therapy, the patient predictably experienced persistent hypokalemia despite aggressive, exogenous potassium supplementation. Almost immediately after discontinuation of the pentobarbital infusion, the patient experienced hyperkalemia. In addition to worsened ICP control, resistance of hyperkalemia to standard therapeutic interventions directed the team to re-start the pentobarbital infusion. Re-initiation of the infusion was found to have no impact on serum potassium levels.
2. Case Report
A 20-year-old male presented to an outside hospital with a Glasgow Coma Score (GCS) of 7 following assault with a blunt weapon. His GCS deteriorated further prompting rapid sequence induction and intubation in order to maintain airway protection. The initial head computed tomography (CT) scan revealed a diffuse right subdural hematoma with mass effect and a one millimeter (mm) midline shift. En route to our institution, the patient received a mannitol bolus per paramedic emergency protocol as the right pupil was noted to be fixed and dilated at 6 mm and the GCS deteriorated to 3T. Upon admission to our institution (approximately 2 and a half hours after the initial injury), a second head CT scan revealed an approximate 10 mm midline shift to the left (Figure 1). In the emergency department, the patient was noted to have positive gag and cough reflexes. He was transferred to the trauma intensive care unit (ICU) and an ICP monitoring device was placed by the neurosurgical team.
Over the ensuing 24-hour period, medical management escalated with the use of mannitol (0.5 - 0.8 g/kg) boluses, hypertonic saline (23.4%, 30 ml) boluses, norepinephrine (6 - 8 mcg/min) and sedation with propofol, midazolam, and fentanyl. Even though ICPs remained high and fluctuated significantly (15 - 41 mm Hg), the cerebral perfusion pressure (CPP) was maintained above 60 mm Hg. A subsequent interval head CT scan was largely unchanged. However, due to rising ICPs on hospital day two, the patient underwent partial right craniectomy. Initially, the patient showed clinical improvement; however, within 12 hours of the procedure, ICPs increased (range: 28 - 32 mm Hg) and CPPs trended downward (range: 55 - 60 mm Hg). Additionally, the patient began to demonstrate signs of renal dysfunction with decreased urine output and increased serum creatinine. At this time, the patient was being treated with propofol, midazolam, fentanyl, cisatracurium, norepinephrine, and phenylephrine.
On day three, pentobarbital therapy was initiated for refractory elevated ICPs with a 10 mg/kg bolus and infusion titrated between 1 - 2 mg/hr to obtain complete burst suppression on electroencephalogram. Upon initia-
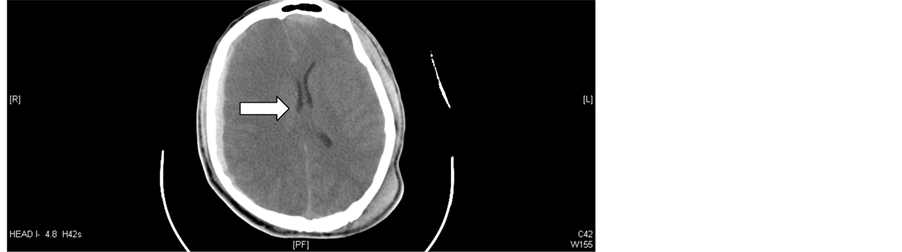
Figure 1. Head computed tomography scan on admission to our institution. Multi-compartment acute intracranial hemorrhage, as above, with significant mass effect, midline shift (arrowed), and effacement of the basal cisterns with impending herniation.
tion of the pentobarbital infusion, the potassium level was 4.1 mmol/L. It was also postulated that it may suppress the intractable rebound hyperkalemia. After 27 hours of barbiturate therapy, the potassium level trended down to 1.7 mmol/L. The patient received a total of 340 mEq of potassium over 45 hours for hypokalemia (potassium range 1.7 - 4.1 mmol/L). Throughout hospital day four, ICPs remained below 25 mm Hg; however, the CPP declined to less than 60 mm Hg. Also, the patient began to show signs of infection with increased temperature, leukocytosis, and a positive sputum culture. Vasopressin was added to norepinephrine and phenylephrine. An external ventricular drain was placed by the neurosurgical team, and following insertion, ICPs ranged between 10 and 21 mm Hg.
On hospital day five, ICP and CPP were well-controlled and vasopressors were weaned. The pentobarbital infusion was stopped in an attempt to assess the patient’s neurological response since it was expected that it would take some time for the clinical effect to wear off. One hour after pentobarbital was stopped, the potassium increased from 3.2 mmol/L to 6.2 mmol/L (Figure 2). Hyperkalemia was treated with multiple boluses of calcium gluconate, insulin, 50% dextrose and sodium bicarbonate. Despite the fluxes in potassium, every effort was made to avoid acidemia and the patient maintained a pH range between 7.29 - 7.37 prior to becoming acidotic on his last ICU day (Figure 3).

Figure 2. Creatinine and electrolytes before, during and after pentobarbital infusion.
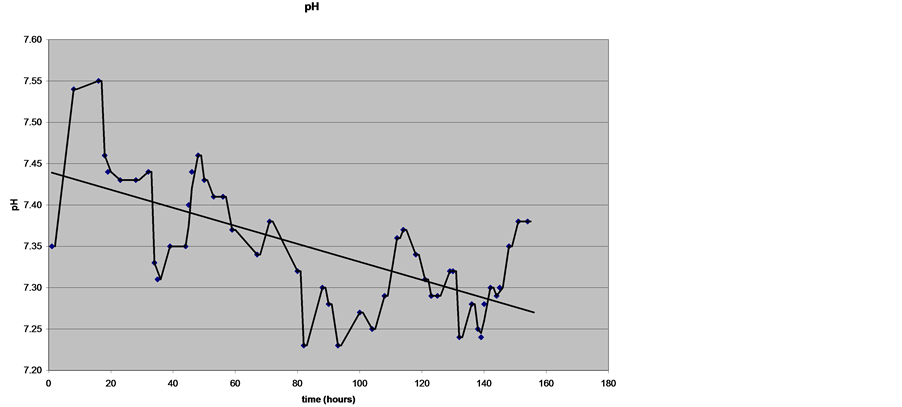
Figure 3. pH trend during admission.
Pentobarbital was restarted soon thereafter for worsening ICP control. Although the pentobarbital infusion remained on for the next 48 hours and the patient continued to receive treatment for hyperkalemia, potassium continued to increase. On hospital day six, potassium peaked at 8.4 mmol/L with widening QRS interval on electrocardiogram and a troponin leak (peaking at 1.32 ng/ml). Intracranial pressures again trended up, ranging between 27 and 42 mm Hg. On hospital day seven, the family made a decision to withdraw care.
3. Discussion
Although fluctuations in serum potassium concentrations have been directly associated with barbiturates, there are few published case reports describing life-threatening, rebound hyperkalemia upon the reduction or cessation of the infusion [2] [3] [5] [6] . Cairns et al., described the significance of severe, life-threatening, rebound hyperkalemia following therapeutic barbiturate coma in a case series of three neurosurgical intensive care patients; one of the three patients presented with traumatic brain injury and unlike our patient, none of the patients in this case series had documented renal dysfunction [2] . In all three patients, rebound hyperkalemia occurred within 24 hours of cessation of thiopental infusions. Schalen et al., reported that 31/38 (82%) of their patients with brain injury on thiopental infusions experienced hypokalemia despite normal potassium repletion and low to normal urinary potassium loss [3] . These investigators also found that serum potassium levels increased to normal or supra-normal levels upon reduction or cessation of a thiopental regimen.
More recently, Bouchard et al., described a case series of three traumatic brain injury patients who experienced dyskalemia after receiving thiopental therapy to lower ICP [5] . The patients received between 14 and 141 hours of thiopental infusion and experienced nadir potassium levels of between 1.7 and 2.8 mmol/L despite aggressive intravenous potassium supplementation. Within eight hours of discontinuing barbiturate therapy, all patients experienced rebound hyperkalemia (peak potassium levels 6.8 to 9.5 mmol/L). Similarly, Neil and Dale describe the case of a 14-year-old patient with severe head injury who experienced hypokalemia (2.8 mmol/L) during continuous infusion of thiopental and hyperkalemia (6.5 mmol/L) within six hours of its discontinuation [6] .
Barbiturate-induced mechanisms of dyskalemia remain unclear; however, one hypothesis suggests a concentration-dependent effect on neuronal voltage-dependant potassium channels. Thiopental reversibly inhibits potassium currents in a concentration-dependent manner resulting in hypokalemia with the assumption that upon cessation of thiopental, there would be normokalemia [7] . Another mechanism thought to contribute to barbiturate-induced dyskalemia is the inhibition of phosphofructokinase, which leads to a reduction in the intracellular production of pyruvate and lactate, culminating in a decrease in extracellular pH [8] .
Neither of these mechanisms however, explains the significant rebound hyperkalemia observed immediately after cessation of barbiturate infusion. If hypokalemia were a direct side effect of the medication, rebound hyperkalemia would be expected to occur after the drug concentration became subtherapeutic, typically three to five half-lives after discontinuation of the medication. The pharmacodynamic properties of thiopental indicate that it remains active at the cellular level long after the cessation of the infusion. The serum half-life of thiopental can range from 3 to 18 hours depending upon the patient’s age, hepatic function and duration of exposure to the medication. The serum half-life of pentobarbital can range from 15 to 48 hours.
Our experience parallels what is reported in the literature in that rebound hyperkalemia is related to either weaning or discontinuation of a barbiturate infusion. Following discontinuation of the infusion, our patient rapidly became hyperkalemic and was unresponsive to conventional corrective treatment. Upon re-initiation of the pentobarbital infusion, in the hope that it would alleviate the serum potassium load, there was no sustained lowering of serum potassium. It is important to note that this was in the setting of worsening renal function and significant exogenous potassium supplementation during the period of hypokalemia (Figure 2). During this time, the patient’s urine output was consistently above 0.5 ml/kg/hr. The fractional excretion of sodium (FeNa) on hospital day three was 0.2. On day four it was 2.6 without administration of loop diuretics. Of note, hyperkalemia only became noticeably intractable 33 hours after there was a significant worsening of creatinine, suggesting that there would be another mechanism involved in the pathogenesis of the refractory hyperkalemia rather than simply multiple organ dysfunction and acute renal impairment. Use of the Naranjo Adverse Drug Reaction Probability Scale indicated a possible relationship between rebound hyperkalemia and pentobarbital therapy in this patient [9] .
Our experience differs from published literature in that rebound hyperkalemia was associated with halting the infusion completely; others tend to wean the infusion rate of the barbiturate. Schalen et al. weaned the barbiturate infusion; however in doing so, it was noted that over the next 24 hours the ICP began to rise again, as did the serum potassium [3] . This was treated with a high-concentration bolus of thiopental w wh which successfully corrected the rebound hyperkalemia. We did not give a barbiturate bolus in our patient, but instead tried to reverse the hyperkalemia by reintroducing the barbiturate infusion. Differences in the response of serum potassium to bolus administration compared to continuous infusion suggest that this may be an infusion-related effect.
Furthermore, there are other factors that may have influenced the non-responsiveness to the re-introduction of the barbiturate infusion. These include: acute brain injury, hypothermia, endogenous and exogenous catecholamines, diuretic therapy, insulin, acid base balance and acute renal dysfunction. However, by plotting each of the administered medications as well as other factors that may influence serum potassium levels versus time, we ruled out all other confounders.
4. Conclusion
Clinicians should be cognizant of all adverse effects of barbiturate therapy for treatment of elevated ICPs. Refractory and severe hyperkalemia can be life-threatening and may occur following cessation of a barbiturate infusion. One should be aware of this phenomenon during therapy and should establish a threshold for treating hypokalemia during the duration of the infusion in order to avoid potential rebound hyperkalemia while optimizing the function of multiple organ systems. A protocolized approach should be tailored for each patient including recommendations for specific doses, monitoring and weaning of barbiturate therapy during the ICU stay.
References
- Roberts, I. (1999) Barbiturates for Acute Traumatic Brain Injury. Cochrane Database of Systematic Reviews, 3, CD000033. http://dx.doi.org/10.1002/14651858.CD000033
- Cairns, C.J.S., Thomas, B., Fletcher, S., Parr, M.J. and Finfer, S.R. (2002) Life-Threatening Hyperkalemia Following Therapeutic Barbiturate Coma. Intensive Care Medicine, 28, 1357-1360. http://dx.doi.org/10.1007/s00134-002-1399-y
- Schalen, W., Messeter, K. and Nordstrom, C.H. (1992) Complications and Side Effects during Thiopental Therapy in Patients with Severe Head Injuries. Acta Anaesthesiologica Scandinavica, 36, 369-377. http://dx.doi.org/10.1111/j.1399-6576.1992.tb03483.x
- Bali, I.M., Dundee, J.W. and Assaf, R.A.E. (1973) Proceedings: Immediate Changes in Plasma Potassium Induced by Intravenous Induction Agents. British Journal of Anaesthesia, 45, 1238. http://dx.doi.org/10.1093/bja/45.12.1238
- Bouchard, P.M., Frenette, A.J., Williamson, D.R. and Perreault, M.M. (2008) Thiopental-Associated Dyskalemia in Severe Head Trauma. Journal of Trauma, 64, 838-842. http://dx.doi.org/10.1097/TA.0b013e3180341f65
- Neil, M.J.E. and Dale, M.C. (2009) Hypokalemia with Severe Rebound Hyperkalemia after Therapeutic Barbiturate Coma. Anesthesia & Analgesia, 108, 1867-1869. http://dx.doi.org/10.1213/ane.0b013e3181a16418
- Friederich, P. and Urban, B.W. (1999) Interaction of Intravenous Anesthetics with Human Neuronal Potassium Currents in Relation to Clinical Concentrations. Anesthesiology, 91, 1853-1860. http://dx.doi.org/10.1097/00000542-199912000-00040
- Carlsson, C., Nordstrom, C.-H. and Siesjo, B.K. (1979) Metabolic Changes in the Cerebral Cortex of the Rat Induced by Intravenous Pentothal Sodium. Acta Anaesthesiologica Scandinavica, 57, 7-17.
- Naranjo, C.A., Busto, U., Sellers, E.M., Sandor, P., Ruiz, I., Roberst, E.A., et al. (1981) A Method for Estimating the Probability of Adverse Drug Reactions. Clinical Pharmacology & Therapeutics, 30, 239-245. http://dx.doi.org/10.1038/clpt.1981.154