Advances in Nanoparticles
Vol. 2 No. 4 (2013) , Article ID: 39018 , 8 pages DOI:10.4236/anp.2013.24049
Development of Cu Nanoparticle Loaded Oil Palm Fibre Reinforced Nanocomposite
1Engineering and Processing Division, Malaysian Palm Oil Board (MPOB), Kajang, Selangor, Malaysia
2Faculty of Chemical and Natural Resources Engineering, University of Malaysia Pahang (UMP), Kuantan, Malaysia
Email: *ridz@mpob.gov.my
Copyright © 2013 Ridzuan Ramli et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received September 2, 2013; revised October 9, 2013; accepted October 21, 2013
Keywords: Biodegradation; Nano-Composite; Nanoparticle; Natural Fibre; Polyester Resin
ABSTRACT
Functionalized copper nanoparticles (FCuNPs) have been synthesized by chemical reduction method and polyvinyl alcohol (PVA) performed as a stabilizer in that medium. Analysis observed that the average size of the synthesized FCuNPs was 3.5 nm. The obtained FCuNPs were loaded in the oil palm empty fruit bunch (EFB) natural fibre. Before the loading of FCuNPs in EFB fibres, the surface of the fibres is tailored by the cationic agent CHPTAC since they have a natural tendency to exhibit negatively charged surface owing to the presence of large amount of hydroxyl groups. Thereafter, different types of composite were developed and their properties were studied. The composites were developed by using the untreated empty fruit bunch (UEFB) fibres and FCuNPs loaded EFB (NP-CAEFB) fibres with commercially available unsaturated polyester resin (UPER). The synthesized composites were characterized through FTIR, FESEM, XRD, DSC, tensile strength tests, etc. The obtained biodegradation results indicated that significant weight loss was not observed for neat PER and PER/FNP-CAEFB nanocomposite, whereas, the UEFB/VUPER composite showed ca. 21.4% weight loss at 90 days, which was considered rationally due to the preferential degradation of the fibre.
1. Introduction
For a long time, fibre-reinforced plastic composites have played a potential role due to a variety of applications for their high specific strength and modulus. The traditional fibre-reinforced plastics which are usually made up of glass, carbon or aramid fibres-reinforced thermoplastic and thermoset resins are considered critically and because of their environmental difficulties the manufacture, use and removal of traditional fibre-reinforced composites are less popular. Of late, there has been an increasing interest for alternatives which are the biodegradable plastic composites by using natural fibres [1-4]. The benefits of natural fibres are: biodegradability, renewability, low density, and low cost. This low density of the natural fibres can show some superior specific stiffness and strengths which are comparable to the respective quantities of synthetic fibres like glass fibres. The promising applications of natural fibre reinforced composites: doorpackage trays, instrument panels, arm rest, seat back and glove boxes [1]. However, the natural and common drawbacks of natural fibres are the high degree of moisture absorption, poor wettability with non-polar polymers, low-adhesion of untreated fibres with polymer-matrix which facilitates the retardation of significant bonding during aging [3-5]. To overcome these drawbacks for increasing the applications of natural fibre reinforced composites, the possible recommendations are the fabriccation method of fibre, cautious choice of pair (fibre and matrix), and development procedures [6,7]. Numerous researches have been done on the potentiality of the natural fibres such as kenaf, flax, and jute reinforcements for plastic composite materials [1-5]. Amongst those, oil palm empty fruit bunch (EFB) fibres are getting popularity by researchers for using this huge biomass as an alternative reinforcing agent in composite materials. EFB fibre is mainly a cellulose enriched natural product and an available renewable resource in some countries like Malaysia, Indonesia, India, Brazil, etc. However, in spite of their great potentials, EFB fibres still remain low potentiality in the fabrication of new biomaterials even though they have environmentally friendly and biocompatible characteristics. It is known that cellulose containing materials are extensively involved in the fields of medical supplies, textiles, packaging, electronic devices, energy storage, biomedical materials and composite. Although the EFB fibres contain 42% - 65% cellulose, their poor mechanical strength and easy degradation to bacterial attack make them almost useless materials in various applications. Moreover, EFB fibres are often very prone to the growth of microorganisms when disposed to land, thereby causing various types of diseases. Consequently, improving the mechanical and antibacterial performance of cellulosic materials using facile approach attracts many researchers. Cu is known to exhibit a broad-spectrum biocide and effectively inhibit the growth of bacteria, fungi and algae. Hence, the present work has been undertaken to synthesize FCuNPs, which, if embedded in EFB fibres, is expected to improve fibres mechanical strength and antimicrobial activity so that metallic NPs reinforced fibres can be exploited in due applications especially for nanocomposite. Previous recasts on cellulosic natural fibres have suggested that many valueadded industrial products cater to the needs in automotive, agriculture and other industrial sectors [8]. Very few studies on the EFB reinforced thermosetting composites have been reported so far, hence it is the first time the attempt has been made to study the properties of NPs loaded EFB fibre-reinforced unsaturated polyester resin (UPER) composites. In this work, FCuNPs have been synthesized by chemical reduction method and polyvinyl alcohol (PVA) performing as a stabilizer in that medium. The obtained FCuNPs were loaded in the EFB natural fibre. Before the loading of FCuNPs in EFB fibres, the surface of the fibres is tailored by the cationic agent CHPTAC since they have a natural tendency to exhibit negatively charged surface owing to the presence of large amount of hydroxyl groups [9]. Thereafter, different types of composite were developed and their properties were studied. The composites were developed by using the untreated empty fruit bunch (UEFB) fibres and FCuNPs loaded EFB (NP-CAEFB) fibres with comercially available UPER. The developed materials were characterized through FTIR, FESEM, XRD, DSC, tensile strength tests, etc.
2. Experimental
2.1. Material
Copper chloride dihydrate salt with 98% purity and analytical grade sodium hydroxide were procured from ALDRICH. Polyvinyl alcohol (PVA) of molecular weights MW = 30,000 - 70,000, sodium borohydride (NaBH4) Reagent Plus (purity 99%), ascorbic acid (purity 99.7%) and 60% solution of (3-Chloro-2-hydroxypropyl) trimethylammoniumchloride solution (CHPTAC) were purchased from ALDRICH. Raw EFB fibres were collected from the LKPP Corporation Sdn. Bhd., Kuantan, Malaysia. Unsaturated polyester resin 268BQT obtained from PERMULA Chemicals SDN BHD was used as matrix. Laboratory grade of 2-Butanone peroxide (ALDRICH) was used as hardener.
2.2. Methods
The raw fibres of golden-brown colour with an average diameter of 0.175 ± 0.071 mm were washed to remove dust particles and dried under sunlight for 3 days. The fibres thus obtained were referred to untreated EFB (UEFB) fibres and then this fibre was chopped to make on an average length of 4 mm. CuNPs were synthesized in 0.5% PVA solution with deionized (DI) water. Copper chloride (250 mg) and ascorbic acid (20 times with respect to the precursor molar ratio) were subsequently added in 100 mL PVA solution with vigorous stirring. About 7.0 mL solution of sodium borohydride (0.347 M) was injected drop-wise to the above-mentioned reaction mixture with continuous stirring for 2 h for the preparation of CuNPs sols. The UEFB fibres were dipped in 20% CHPTAC solution by maintaining a liquor ratio of 1:25 in a water bath for cationization reaction. During the cationization process, the bath was set at a constant temperature of 60˚C along with continuous agitation. Thereafter, 15% sodium hydroxide (40 g/L) with respect to the fibre-weight was added in three steps at an interval of 5 min, and the mixture was further stirred for 15 min.
2.2.1. Modifications of EFB Fibers by FCuNPs
The cationized EFB (CAEFB) fibers were removed from the bath, rinsed several times with water, neutralized and then dried at the ambient temperatures. The CAEFB fibers were introduced in the synthesized nanocopper sol of copper concentration 250 mg/L, maintaining the fiber to sol ratio of 1:25 with continuous shaking by a mechanical shaker for 12 h in order to adsorb FCuNPs. Then, the FCuNPs reinforced CAEFB (NP-CAEFB) fibers were removed from the bath, rinsed with water, and dried in the dark place at room temperature.
2.2.2. Composite Formation
Required amount of resin and hardener (1%) were mixed in a beaker. Thereafter, required amount of chopped fibres were also well-mixed with the resin mixture by had mixing technique. This fibre-resin mixture was poured in the mold and covered the mold with a stainless steel plate to fill the mold properly and then removed the composite after 12 h.
2.2.3. Fourier Transformed Infrared Spectroscopy
Fourier-transformed infrared (FTIR) spectra of liquid samples were taken with a PerkinElmer Spectrum 100 FTIR Spectrometer over the frequency range 4000 - 650 cm−1. On the other hand, FTIR spectra of different solid samples were recorded over the frequency range 4000 - 650 cm−1 using a Thermo Scientific Model Smart Performer attenuated total reflectance (ATR) accessory of Ge crystal, attached to a Thermo Scientific spectrophotometer (Model Nicolet Avatar-370) with a single bounce. The FTIR parameters were as follows: angle of incidence 45˚, sampling area 2 mm; number of background scans, 32; number of scans, 32; optical resolution, 4.00 cm−1. From the FTIR spectrum, the degree of cationization (Cd) was calculated according to the following relation [9]:
 (1)
(1)
where I1648 and I1495 are the maximum intensities of peak at 1648 and 1495 cm−1, respectively.
2.2.4. Transmission and Scanning Electron Microscopies
Transmission electron microscopy (TEM) was performed by a LEO 912 AB EFTEM operating at 120 kV to monitor the size of nanoparticles. A small drop of the liquid sol was placed over a carbon coated microscopic copper grid (200 mesh size) and dried under vacuum at room temperature before measurements. Treated and untreated EFB fiber surface were investigated by using a scanning electron microscope (SEM) of model ZEISS, EVO50, Germany. Surface morphologies of UEFB, CAEFB and NP-CAEFB fibers were also investigated by using a field emission scanning electron microscope (FE-SEM) of JEOL JSM-7600F, USA, equipped with an energy dispersive X-ray (EDX) system (OXFORD INCA. Fibers were mounted on sample holders with carbon tape and sputtered by gold.
2.2.5. X-Ray Diffractometry
X-ray diffraction (XRD) data were collected by using a RigakuMiniFlex II (30 kV and 15 mA), Japan, equipped with computer-controlled software to set up the apparatus and analyze the data. To prepare the disc specimen of same thickness for each category of differently treated EFB fibers, 1 g of chopped fibers were compressed in a cylindrical mold with a pressure of 1 MPa. The specimens were step-wise scanned over the operational range of scattering angle (2h) between 10˚ to 50˚, with a step of 0.02˚, using the CuKa radiation of wavelength k = 1.541 Å. The data were recorded in terms of the diffracted X-ray intensity (I) versus 2h. The average size of the cellulose crystallites, Dhkl, was determined with the full width at half-maximum (FWHM) of the (002) peak by using the following Scherer Formula (2):
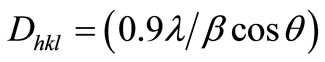 (2)
(2)
where, β is the FWHM (in radians) and θ is the diffraction angle. The β value was determined by curve fitting after subtracting the amorphous background. The Gaussian curve was fitted at the top of the peak for determining β and the position using an appropriate program. The X-ray crystallinity, XCrl, was calculated by the help of Segal’s method with the following Equation (3) [10]:
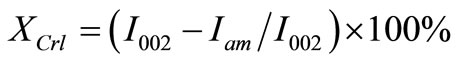 (3)
(3)
where, I002 is the maximum intensity of 002 reflections from the crystalline and amorphous components, and Iam is the minimum intensity of diffraction from the amorphous part of EFB fibers. During the calculation process, the height of the (002) peaks were used at a 2θ ≈ 22˚, while the intensity of Iam was taken from the minimum intensity between the (002) and (110) peaks at a 2θ ≈ 16˚.
2.2.6. Mechanical Testing
The single EFB fibers were subjected to the ASTM D3379-75 standard test method for tensile strength and Young’s modulus. Each UEFB fiber was separated by hand and mounted on a paper. Its two ends were strongly glued with the paper, and they were inspected under a calibrated optical microscope of 200× magnification to measure the diameter of each fiber. The single fibers were then individually placed in the grips of a DIA-STRON LTD/U.K (FDAS 765) tensile testing machine, and the supporting sides of the paper were carefully cut to maintain the gauge length of 30 mm. The fibers were then tensile tested to failure at a rate of 3 mm/min using a 500 N-load cell. Average strength and modulus were obtained using results from 50 specimens. Single fiber tensile tests were also performed for NPCAEFB fibres. The tensile tests of composites were carried out using the same machine as according to the standard methods ASTM D3039. The used wt% of fibre was 30% which was optimized and so it was used.
The crosshead speed of the tensile testing was set at 3 mm/min. The dimension for tensile specimen is 60 mm long and 13 mm wide. All the results were taken as the average value of twenty samples.
2.2.7. Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) was performed to determine the glass transition temperature (Tg), melting temperature (Tm) and crystallization temperature (Tc), using a TA/Q1000 apparatus under nitrogen atmosphere. In order to avoid moisture effect, all the test specimens were dried at 90˚C for 6 h prior to the measurements. For monitoring DSC, the samples were initially heated at 25˚C - 350˚C with a heating rate 10˚C min−1, then cooled down to 25˚C with a cooling rate 5˚C min−1 and re-heated from 25˚C -350˚C to monitor calorimetric properties.
2.2.8. Biodegradability Test
Biodegradability was determined by measuring the weight loss of the specimens which are buried in soil. The test was done by using the soil of 1:1 mixture of black soil and leaf mold for gardening. The selected each specimen was buried in the aforesaid soil in planters and incubated at room temperature (25˚C - 30˚C). At 2 days intervals, water was poured so that the soil was kept in humid condition. After burial of particular time the specimens were removed from the soil, whre the burial duration followed for 30, 60 and 90 days, respectively. Thereafter, the dugged out specimens were washed with water and dried at 40˚C in a vacuum oven up to the of achievement a constant weight. The weight loss (%) is calculated according to the following Formula (4):
 (4)
(4)
where, Wi and Wf are the dry weights of composites at the before and after the burial, respectively.
3. Results and Discussion
A change of sol colour is observed (image shown in Scheme 1) after the addition of reducing agent, where the light-blue colored aqueous solution changes to wine red, which is a convincing indication of the formation of FCuNPs in the sol.
Figure 1 illustrates the FTIR spectra of solutions before and after nanoparticle formation.
Abroad absorption band of hydroxyl (O-H) groups of PVA chains appear at 3359 cm−1 and 3341 cm−1 before and after NPs formation, respectively, showing 18 units red shift of this polar group. This shifting of wavenumber demonstrates that PVA molecules facilitate coordination with Cu2+ species. Such coordination through the ester bond of PVA to Cu species due to the electrostatic attractions was described elsewhere [11]. The ester bond is located at 1047 cm−1 before NP formation and shifted to 1039 cm−1 after NP formation, indicating the coordination of polar -O-H groups with Cu species. The morphologies and sizes of CuNPs in the sol were monitored by TEM as shown in Figure 2. TEM image clarifies that the particles developed in the sol are in sizes in the order of a few nanometers with spherical shape. Analysis from 100 to 500 particles of diameters 1.2 - 10.5 nm shows the average size of 3.5 ± 1.1 nm.
The morphologies and sizes of FCuNPs were also observed by FESEM as shown in Figure 3, which clearly demonstrated the bonding between NPs and fibre.
The adsorption mechanism is shown in Scheme 2. The adsorptions of copper on CAEFB fibres from sols measured by EDX also (shown in EDX spectrum of Figure 4). According to Scheme 2, when CAEFB fibres are immersed in a nanocopper sol, FCuNPs stabilized by nega-

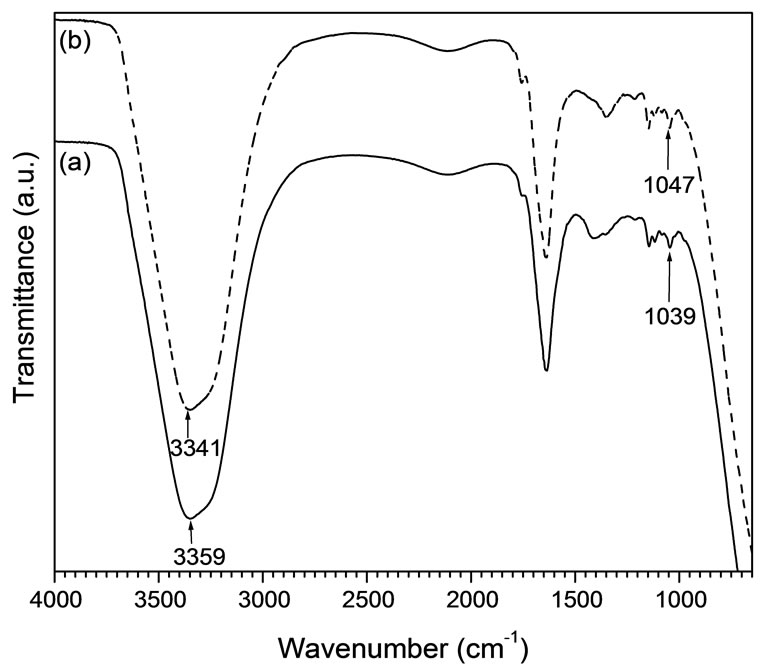
Figure 1. FTIR spectra of copper sols: (a) before and (b) after NPs formation.

Figure 2. TEM image of synthesized FCuNPs.

Scheme 2. Mechanism of cationization of UEFB fibres by CHPTAC.

Figure 3. FESEM image of loaded FCuNPs in CAEFB fibres.
tively charged PVA spheres can readily be adsorbed by the positive sites of cationized fibres.
A large amount of positive charges may cause the charge reduction at the fibre surface because of cationiczation of EFB fibres, thereby increasing the adsorption capability of functionalized copper nanoparticles due to an attractive interaction between fibres and FCuNPs.
The X-ray diffractograms of different fibres are illustrated in Figure 5. The peak appearing at 2θ = 42.3˚ for NP-CAEFB fibre has been reportedly attributed to the 111 reflection from the cubic copper crystal [12]. This finding demonstrates the CuNPs impregnation in the fibres. The average size of FCuNPs as analyzed from (1 1 1) peak is ~17 nm.
Figure 6 depicts the TS of different fibres before burial (BB) and after burial (AB) under soil. Analyses show that the increase in TS of NP-CAEFB fibre from UEFB fibre before burial is 25%. This increase in TS is rational, because the functionalized CuNPs can enter into fibre matrices and act as filler or plasticizer, which contribute to load sharing during stretching the fibres.
On the other hand, the TS of UEFB and CuNPsCAEFB fibres after soil burial decreases by 32% and 25% from their respective TS before burial, resulting in 7% antifungal activity. However, the TS-decrease of UEFB fibres is more than NP-CAEFB fibres, demonstrating that CuNPs reinforced fibres are more durable than the control ones.
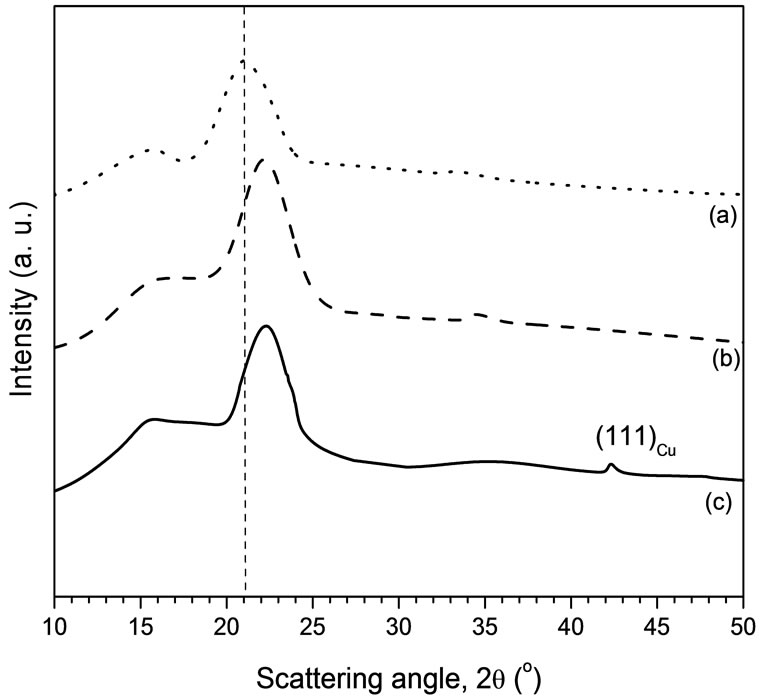
Figure 5. XRD patterns of different fibres: (a) UEFB; (b) CAEFB and (c) NP-CAEFB.
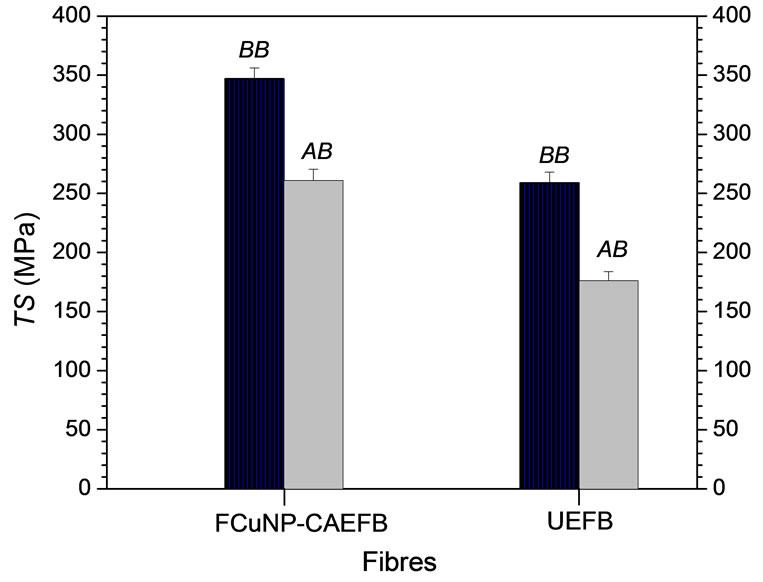
Figure 6. Mechanical strength results of different fibres in different conditions: where BB (before soil burial) and AB (after soil burial).
This result may be explained by the facts that the metallic nanoparticles can show a certain degree of sterilization, because the catalytic properties of metallic species partly help to create active oxygen in water, which dissolve the organic substances to maintain sterilizing effect [13].
Figure 7 represents the FTIR spectra of (a) VUPER, (b) NP-CAEFB-VUPERC. The major absorbance peaks obtained for the VUPER are: 1) the peak around 3450 cm−1, which may be attributed to the OH stretching, 2) the strong peaks at 3089 and 2976 cm−1 that can be assigned to the C-H stretching in aromatic and aliphatic moieties, respectively, 3) the C=O stretching (from ester linkage) observed at 1732 cm−1, also observed at 1955 cm−1 [14] (iv) the CH2 deformation appearing at 1450 cm−1, and (v) the C-O asymmetric mode of the ester groups observed at 1073 cm−1. The FTIR spectrum of NP-CAEFB-VUPERC also indicates the presence of aforesaid functional groups absorption frequencies with some shifting in some important positions. The crucial changes of peak at 1955 cm−1 is almost invisible in the spectrum of Figure 7(b) as well as the significant red shift occurred for C=O and C-O groups from ester moiety. These observed findings are indicating the significant degree of bonding among the components of composites.
DSC thermograms of a) VUPER, (b) NP-CAEFBVUPERC are shown in Figure 8. On heating, the pure VUPER displays three main successive transitions: a glass transition, a cold crystallisation exotherm and a melting endotherm, which are respectively characterized by temperatures Tg, Tc and Tm.
The presence of single melting peak of the composites is due to the existence of single phase of composite, developed during the controlled cooling process in the
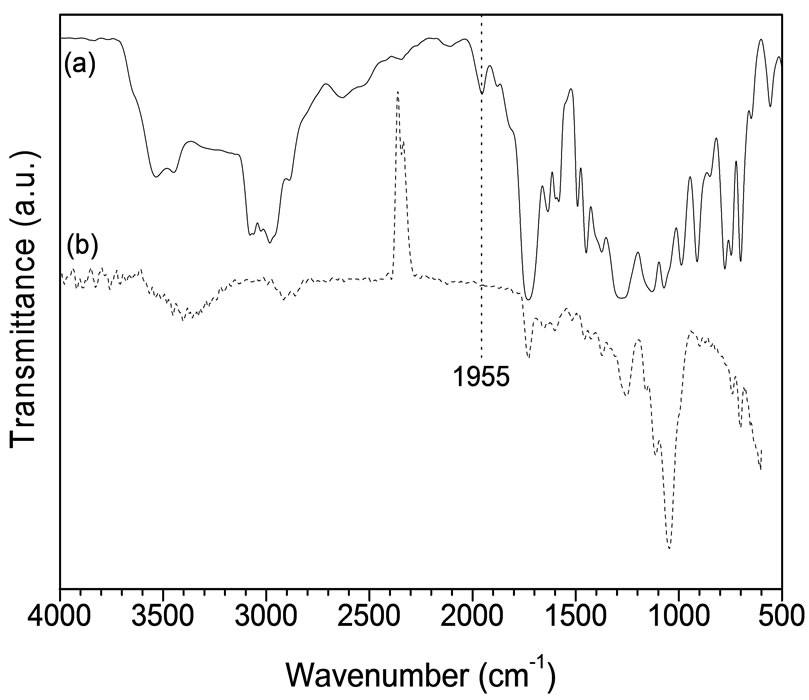
Figure 7. FTIR spectra of (a) VUPER and (b) FNP-CAEFBVUPERC.
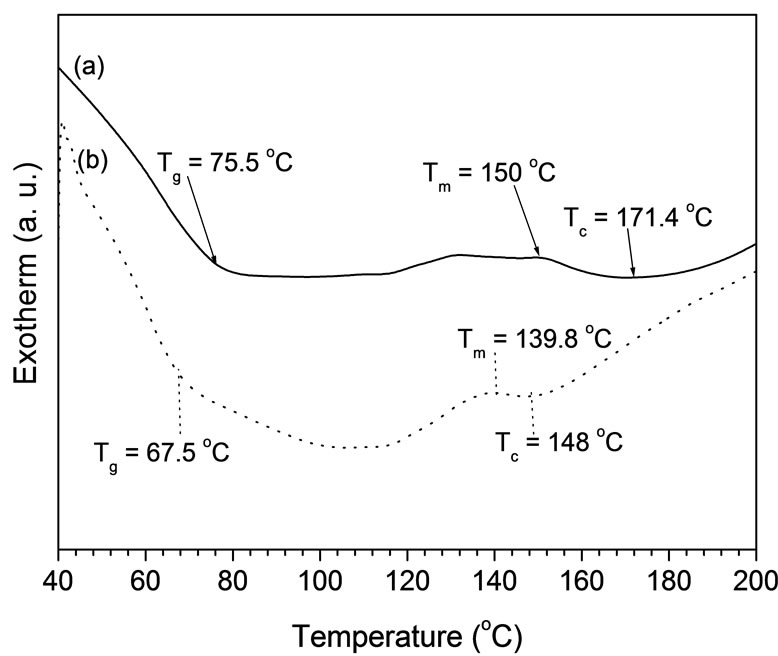
Figure 8. DSC thermograms of (a) VUPER and (b) NPEFB-VUPERC.
presence of NP-CAEFB fibers [15]. The Tg, Tc and Tm values for all samples are introduced in Figure 7. While the Tg value of VUPER in the NP-CAEFB-VUPERC decreases. This fact can be explained on the basis of the polymer chain mobility, which is easier in VUPER as compared to NP-CAEFB-VUPERC, because the higher crystallinity for the latter, as obvious from the XRD study, is a strong evidence for more restrictions of chain movement. The Tc decreases significantly for composites, as an VUPER > NP-CAEFB-VUPERC. Interestingly, changes of melting endotherm of composites are also shoing the same pattern.
The most important factor to obtain a good fiber reinforced composites is the adhesion between matrix polymers or the fiber and interphase properties. Due to the presence of hydroxyl group in natural fibers, the moisture absorption is high which leads to poor wettability and weak interfacial bonding between fibers and hydrophobic matrices. Therefore, in order to develop composites with better mechanical properties, it is necessary to impart hydrophobicity to the fibers by suitable treatments [16]. The surface modification of UEFB would not only decrease the moisture absorption, but would improve the wettability of fiber and matrix at interphase region hence subsequently improve the mechanical properties of the composites.
The tensile properties of the resin at 0 wt% of fiber are used as control. It is observed that the use of NP treated CAEFB fibre of 30 wt% performed the improved tensile properties of the VUPER composites. The superior mechanical properties of NP treated CAEFB based composites as compared to UEFB composites are attributed to the fact that treatment improves the fiber surface adhesives characteristics by fibrillation process [17].
Table 1, represents the TS of VUPER, UEFB-VUPERC, and NP-CAEFB-VUPERC. Clearly, NP-CAEFBVU-PERC showed the highest TS, which are immediately followed by UEFB-VUPERC. The respective increase of TS of NP-CAEFB-VUPERC from UEFBVUPERC is about 27%, and from VUPER is 58%, denoting a significant figure to enhance materials properties. This better performance of the resulting composites can be attributed to the increased compatibility between NPCAEFB fibers and VUPER because of the modification of UEFB fibers surface by both CHPTAC and FCuNPs treatments. On the other hand, the increase of TS in the composites merely for the inclusion of UEFB fibers content may be assigned to the robust TS value of the individual EFB fiber [18]. However, when UEFB fiber content is greater than 30 wt%, the composites become inhomogeneous because of the fiber-fiber interaction whose effect may cause to develop fiber-agglomeration in the composites, as reported elsewhere [19].
The weight loss increased with burial-period for all the samples and the results are shown in Table 2. There was no significant difference between VUPER and NPCAEFB-VUPERC samples until 90 days. The UEFBVUPERC specimen buried for more than 60 days could not be fully recovered because of a considerable fragmentation. Apparent decay of neat VUPER and NPCAEFB-VUPERC is slower than that of UEFB-VUPER, indicating that UEFB has a higher biodegradability than NP-CAEFB. Careful observation reveals that some hollows generated by the biodegradation of matrix VUPER are appeared for all the VUPER samples after 30 days. The biodegradability of matrix polymer is thought to be almost the same for all the samples, as far as judging from the growth of hollows of the matrix VUPER. It is thought that the faster fragmentation of UEFB-
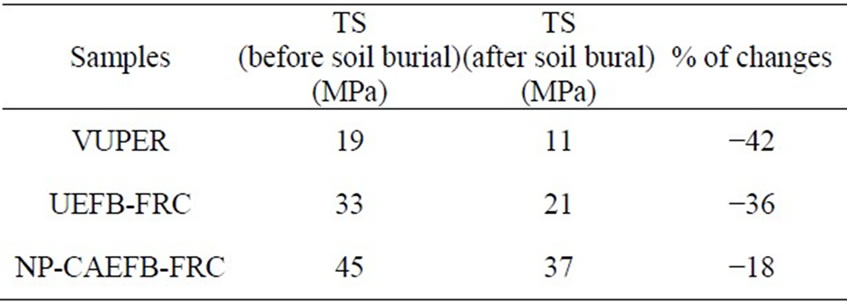
Table 1. Tensile strength results of different fibre reinforced composites (FRC) and virgin unsaturated polyester resin (VPER) in different conditions.
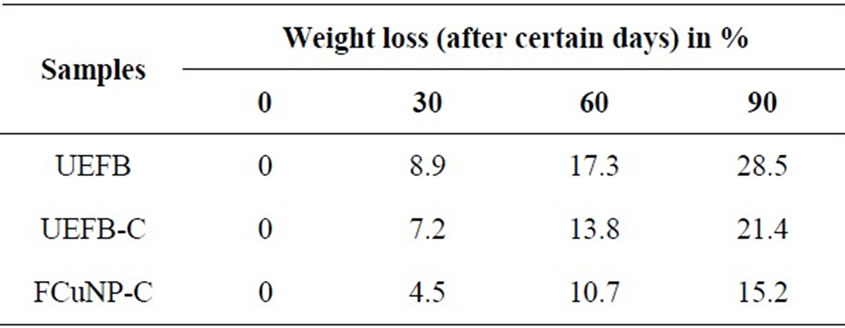
Table 2. Biodegradability test results as weight loss (%) due to the soil burial up to 90 days.
VUPERC is caused by a rapid biodegradation of UEFB exposed on the surface by the biodegradation of the matrix VUPER.
4. Conclusions
Nanocopper sol has been synthesized at atmospheric conditions and FCuNPs exist there. The synthesized FCuNPs are incorporated in UEFB fibres via cationization by CHPTAC. FTIR result indicated the 36% cationization. The morphological and XRD analysis of CuNPs impregnated fibre shows that CuNPs are impregnated in the fibre which can improve the strength and durability of the fibres. The developed NP-CAEFBVUPERC has potential mechanical strength and controlled biodegradability.
The NP-CAEFB-VUPERC shows the Tg decrease of 12%, which indicates a considerable enhancement of materials properties by NPs treated EFB fibers. Fourier transforms infrared spectroscopy has provided strong evidence of bond formation between them. Moreover, analysis observed by DSC and X-ray diffraction has been found to be consistent. Finally, obtained material and its properties are suggesting the potential usage of it as the usable materials like different types of boards.
5. Acknowledgements
The authors would like to thank the Director General of MPOB for permission to publish this paper. The authors also wish to gratefully acknowledge the funding from MPOB to this study.
REFERENCES
- D. B. Hall, P. Underhill and J. M. Torkelson, “Spin Coating of Thin and Ultrathin Polymer Films,” Polymer Engineering and Science, Vol. 38, No. 12, 1998, pp. 2039- 2045.
- A. F. Miller, “Exploiting Wrinkle Formation,” Science, Vol. 317, No. 5838, 2007, pp. 605-606. http://dx.doi.org/10.1126/science.1146680
- C. Rotella, S. Napolitano and M. Wulbbenhorst, “Segmental Mobility and Glass Transition Temperature of Freely Suspended Ultrathin Polymer Membranes,” Macromolecules, Vol. 42, No. 5, 2009, pp. 1415-1417. http://dx.doi.org/10.1021/ma8027968
- V. V. Tsukruk, “Assembly of Supramolecular Polymers in Ultrathin Films,” Progress in Polymer Science, Vol. 22, No. 2, 1997, pp. 247-311. http://dx.doi.org/10.1016/S0079-6700(96)00005-6
- L. D. Pérez, J. F. López, V. H. Orozco, T. Kyu and B. L. López, “Effect of the Chemical Characteristics of Mesoporous Silica MCM-41 on Morphological, Thermal, and Rheological Properties of Composites Based on Polystyrene,” Journal of Applied Polymer Science, Vol. 111, No. 5, 2009, pp. 2229-2237. http://dx.doi.org/10.1002/app.29245
- A. F. Vargas, W. Brostow, H. E. Hagg Lobland, B. L. Lopez and O. Olea-Mejia, “Reinforcement of Polymeric Latexes by in Situ Polymerization,” Journal of Nanoscience and Nanotechnology, Vol. 9, No. 11, 2009, pp. 6661-6667. http://dx.doi.org/10.1166/jnn.2009.1329
- H. Zou, S. Wu and J. Shen, “Polymer/Silica Nanocomposites: Preparation, Characterization, Properties, and Applications,” Chemical Reviews, Vol. 108, No. 9, 2008, pp. 3893-3957.
- C. Jiang, S. Markutsya, Y. Pikus and V. V. Tsukruk, “Freely Suspended Nanocomposite Membranes as Highly Sensitive Sensors,” Nature Materials, Vol. 3, 2004, pp. 721-728. http://dx.doi.org/10.1038/nmat1212
- M. N. K. Chowdhury, M. D. H. Beg, M. R. Khan and M. F. Mina, “Modification of Oil Palm Empty Fruit Bunch Fibers by Nanoparticle Impregnation and Alkali Treatment,” Cellulose, Vol. 20, No. 3, 2013, pp. 1477-1490. http://dx.doi.org/10.1007/s10570-013-9921-7
- T. Inagaki, H. W. Siesler, K. Mitsui and S. Tsuchikawa, “Difference of the Crystal Structure of Cellulose in Wood after Hydrothermal and Aging Degradation: A NIR Spectroscopy and XRD Study,” Biomacromolecules, Vol. 11, No. 3, 2010, pp. 2300-2305. http://dx.doi.org/10.1021/bm100403y
- H. X. Zhang, U. Siegert, R. Liu and W. B. Cai, “Facile Fabrication of Ultrafine Copper Nanoparticles in Organic Solvent,” Nanoscale Research Letters, Vol. 4, 2009, pp. 705-708. http://dx.doi.org/10.1007/s11671-009-9301-2
- W. Yu, H. Q. Xie, L. F. Chen, Y. Li and C. Zhang, “Synthesis and Characterization of Monodispersed Copper Colloids in Polar Solvents,” Nanoscale Research Letters, Vol. 4, 2009, pp. 465-470. http://dx.doi.org/10.1007/s11671-009-9264-3
- M. N. K. Chowdhury, M. D. H. Beg, Maksudur R. Khan and M. F. Mina, “Synthesis of Copper Nanoparticles and Their Antimicrobial Performances in Natural Fibres,” Materials Letters, Vol. 98, 2013, pp. 26-29. http://dx.doi.org/10.1016/j.matlet.2013.02.024
- A. Dutta, G. A. Hamilton, H. E. Hartnett and A. K. Jones, “Construction of Heterometallic Clusters in a Small Peptide Scaffold as [NiFe]-Hydrogenase Models: Development of a Synthetic Methodology,” Inorganic Chemistry, Vol. 51, No. 18, 2012, pp. 9580-9588. http://dx.doi.org/10.1021/ic2026818
- M. Pluta and A. Galeski, “Crystalline and Supermolecular Structure of Polylactide in Relation to the Crystallization Method,” Journal of Applied Polymer Science, Vol. 86, No. 6, 2002, pp. 1386-1395.
- V. Vilay, M. Mariatti, R. M. Taib and M. Todo, “Effect of Fiber Surface Treatment and Fiber Loading on the Properties of Bagasse Fiber-Reinforced Unsaturated Polyester Composites,” Composites Science and Technology, Vol. 68, No. 3-4, 2008, pp. 631-638. http://dx.doi.org/10.1016/j.compscitech.2007.10.005
- A. Vazquez and V. A. Dominguez, “Bagasse FiberPolypropylene Based Composites,” Journal of Thermoplastic Composite Materials, Vol. 12, No. 6, 1999, pp. 477-497.
- S. Ochi, “Mechanical Properties of Kenaf Fibers and Kenaf/PLA Composites,” Mechanics of Materials, Vol. 40, No. 4-5, 2008, pp. 446-452. http://dx.doi.org/10.1016/j.mechmat.2007.10.006
- S. Joseph, K. Joseph and S. Thomas, “Green Composites from Natural Rubber and Oil Palm Fiber: Physical and Mechanical Properties,” International Journal of Polymeric Materials and Polymeric Biomaterials, Vol. 55, No. 11, 2006, pp. 925-945. http://dx.doi.org/10.1080/00914030600550505
NOTES
*Corresponding author.