Advances in Microbiology
Vol.4 No.9(2014), Article ID:48204,15 pages
DOI:10.4236/aim.2014.49064
Characterization of Avirulent TnphoA Mutants in Agrobacterium tumefaciens to Enhance Transformation Efficiency
Dilip K. Das1*, Eugene W. Nester2
1P.G. Department of Biotechnology, Tilka Manjhi Bhagalpur University, Bhagalpur, India
2Department of Microbiology, School of Medicine, University of Washington, Seattle, USA
Email: *dilipdas1@live.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

![]()

Received 11 May 2014; revised 15 June 2014; accepted 20 July 2014
Protein fusion with the Escherichia coli alkaline phosphatase is used extensively for the analysis of the topology of membrane protein. Agrobacterium strain A6007 was mutagenized with E. coli strain mm294A plasmid pRK609 having TnphoA to obtain mutants defective in virulence. Because alkaline phosphatase activity is only detected when the PhoA gene product from the transposon is secreted out of the protoplasm, the virulence mutants are located in genes that code for transmembrane or periplasmic proteins. Attempts were made to obtain the sequences adjacent to the TnphoA inserts through several different approaches including Inverse PCR, Cloning, and Tail PCR. Transposon-adjacent sequence was obtained from one membrane anchor subunit in Bradyrhizobium japonicum i.e. succinate dehydrogenase which has enhanced transformation efficiency.
Keywords: Transposon, Mutagenesis, Agrobacterium tumefaciense, Bradyrhizobium japonicum

Agrobacterium tumefaciens is a Gram-negative, soil-inhabiting, pathogenic bacterium. It causes crown gall diseases in dicotyledonous plants. It harbors a big Ti plasmid, which has vir, con, origin of replication and T-DNA regions. It affects the wounded portion of the plant, which secretes a phenolic substance named as acetosyringone which activates the vir region. It has many vir genes viz vir A, B, C, D, E & G etc. which encode a variety of proteins i.e. vir A, B, C, D, E & G etc. Vir A binds with acetosyringone and activates vir genes, and consequently vir D2 proteins bind with left and right borders of T-DNA and a single stranded T-DNA comes out which binds with different vir proteins and channelizes through vir B protein made pore and goes to the plant cell and integrates into the plant genome, and consequently the integrated genes express and plant becomes transformed. It was believed that virulence genes were present on Ti plasmid only but it was found that besides Ti plasmid, vir genes were also present on chromosomal DNA viz ChvA, ChvB, and exoC etc. [1] [2] . These chromosomal virulence genes also transfer T-DNA which causes tumor in the plant. On mutation of these, genes cause avirulent strains which do not form tumor. TnphoA construct (Figure 1) which is a derivative of Tn5 encoding kanamycin resistance, is successfully used for mutagenesis [3] . This construct generates a hybrid protein which has a version of the alkaline phosphatase (EC 3.1.3.1) gene with its signal sequence and promoter deleted, which will result in a blue colony phenotype on X-phos containing media when secreted into the periplasm, if the insertion occurs in the correct orientation and reading frame. This feature has allowed TnphoA to be used in the analysis of transmembrane protein topology [3] and also to identify chromosomal genes that are secreted into the periplasm [4] . Protein fusions have played a central role in molecular genetics studies of the mechanism of protein export in bacteria [5] [6] . In most of the studies, the hybrid containing amino terminal sequences of exported protein fused with cytoplasmic protein β-galactosidase moiety, which is unable to pass through the cytoplasmic protein. To study the exported protein from transmembrane, TnphoA construction is used to locate the membrane-localized chromosomal genes, where alkaline phosphatase bound protein is secreted from the membrane. If the missing signal peptide is substituted by either own moiety or other exported protein, a TnphoA mutagenesis approach has successfully been used in Agrobacterium tumefaciens to identify additional genes critical for virulence. Before the transposon mutagenesis could begin, however, it was first necessary to derive PhoA mutants; this was because A. tumefaciens expresses endogenous alkaline phosphatase activity that would completely mask the mutant phenotype [7] . Chemical mutagenesis was used successfully to obtain multiple strains which had stable PhoA minus phenotypes and continued virulence was confirmed to ensure that virulence related genes remained functional (wood, personal communication). TnphoA was introduced into the PhoA minus, virulent strain A6007 on a plasmid (pRK 609 from E. coli strain MM294A) which was not maintained in A. tumefaciens (wood, personal communication) identified approximately 37 avirulent or weakly virulent mutants which expressed alkaline phosphatase activity (wood, personal communication).
Various approaches were used in an attempt to obtain the sequences adjacent to the transposon insertion. This would allow for the determination of whether the avirulent phenotype was due to mutating a previously identified gene or an unknown gene; these approaches included Inverse PCR, Cloning (including southern analysis) and Tail PCR. Additionally, the mutants were screened again for both virulence and alkaline phosphatase activity.
A chromosomal virulence locus, ChvE, codes for a glucose-galactose-binding protein that interacts with the periplasmic domain of the sensory protein vir A and is involved in a synergistic induction of vir genes by plant phenolic compounds and sugars [8] -[10] . It has been reported that a succinate dehydrogenase mutant strain of Rhizobium meliloti showed delayed nodulation of Lucerne plants, and the nodules were white and ineffective [11] . It is also reported that the succinate dehydrogenase mutant has 3.5 lower rate of consumption of oxygen than the wild type [12] .
Agrobacterium tumefaciens wild strain A6007 was mutated by mixing with E. coli strain mm294A having TnphoA transposited plasmid on nitrocellulose membrane and incubated for 6 - 8 hrs on MG/L medium. Blue

Figure 1. TnphoA construct.
colonies were selected on MG/L medium supplemented with Kanamycin (950 μg/ml), Nalidixic acid (50 μg/ml), Streptomycin (100 μg/ml) and X-phos (5-bromo-4-chloro-3-indolyl phosphate toluidine salt) and screened for virulence on Kalanchoe leaves (Figure 2). Mutant strains were rescreened by streaking out from glycerol onto MG/L medium having Km/Sm/Nal/X-phos in the same concentration as stated above and each strain was tested for virulence on Kalanchoe leaves with A6007 strain as a positive control on each leaf. Chromosomal DNA was isolated by the CTAB method from 17 of the 37 mutants, including all of those which were totally avirulent on plants and expressed alkaline phosphatase on X-phos containing media. These approaches were attempted to characterize the TnphoA adjacent sequences:
1) The first approach used in the attempt to characterize TnphoA adjacent sequences was Inverse PCR. Chromosomal DNA of all 17 mutants including 45-B and A6007 were digested with SalI enzyme from BRL, which cuts at only one place in the transposon. Followed by SalI digestion an overnight self ligation was carried out at 16˚C with T4DNA ligase enzyme from BRL. Inverse PCR was then performed using two outward pointing primers (Jon 1: 5’ GCAGTAATATCGCCCTGAGCAGCC 3’, TnphoA 2: 5’ CCAGGAAACCAGCAGCGGCTATCC 3’) which amplified the intervening sequence between them containing TnphoA adjacent sequence up to the next SalI site and visible as clear bands (Figure 3). These bands were sliced from the gel and extracted the DNA through QIAgen extraction kit and re-amplified through PCR using both above mentioned primers. The amplified DNAs were PCR sequenced and sent for sequencing.
2) The second approach attempted was cloning scheme. It was done by two methods. (a) Selection by Kanamycin resistance—genomic DNAs of all mutants including 45-B and pBluescript II vector were digested with SalI enzyme from BRL. All mutants DNAs were digested and run on gel (2%) and approximately above than 4.0 kb DNA was sliced from each lane of gel mutant DNA and DNAs were extracted through QIAgen extraction kit. These extracted DNAs were treated as inserts. Both vector and inserts were ligated together in the ratio of 1:4 with T4DNA ligase from BRL. Transformation was performed both by electroporation using protocol of [7] as well as CaCl2 using protocol of [13] and plated onto MG/L Kan (50 μg/ml) plates. If fragments from the SalI digested containing the kanamycin resistance gene of TnphoA were ligated into the vector, colonies should grow up but none appeared on these plates (Figure 4). So second method was used to increase the efficiency of cloning. (b) Southern analysis—in this method six different digests and double digested were designed using SalI and BamHi (which cut only once within the transposon) and EcoRV and XbaI (which do not cut within the transposon) in combination with the single cutters. This should result in chromosomal fragments containing the TnphoA Kanamycin resistance gene and extending varying lengths beyond the transposon into the adjacent sequence depending on whether the restriction sites lie in that area of the genome. After these digestions were performed, they were run out on a 0.8% agarose gel for 2 hr at 80 V. Followed by they were transferred to a

Figure 2. Mutagenesis procedure & virulence on Kalanchoe leaves.
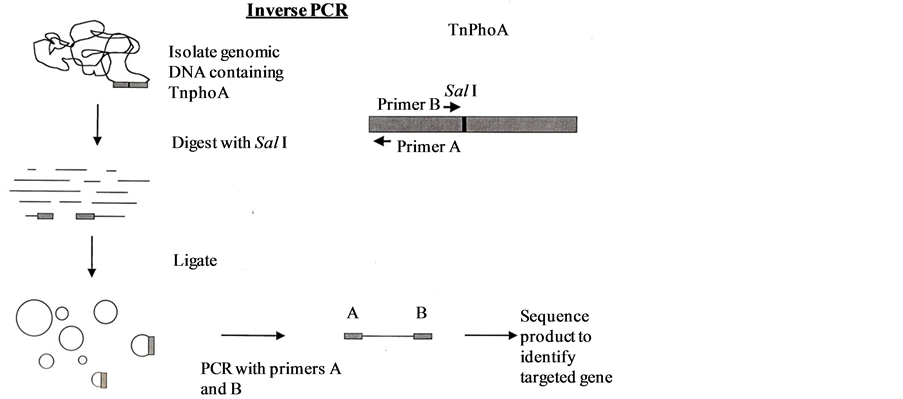
Figure 3. Invese PCR.
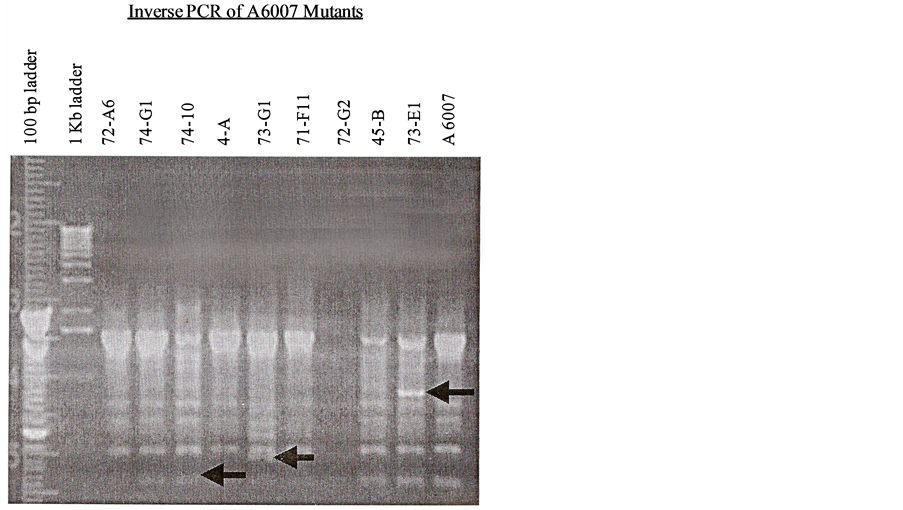
Figure 4. Invers PCR of A6007 mutants.
nylon (Hybond, Amersham) membrane and PhoA gene with primers TnphoA 5’: 5’ CCGCTCGAGGATCCTGTTCTGGAAAACC 3’ and TnphoA 3’: 5’ GGCTCTAGATTATTTCAGCCCCAGAGC 3’ (from Lishan Chen). This probe was gel extracted with QIAgen kit and labeled with a kit from Amersham. Wash buffers and hybridization buffers were also made according to the Amersham kits protocol. Hybridization was carried out at 55˚C overnight with four washes the following day. ECF (Electro Chemical Front) substrate was applied to blots which were then incubated and scanned (Figure 5 and Figure 6). Genomic DNAs were partially digested with various restriction enzymes though some of them were Cs (Cesium chloride) purified was big hurdle to take Southern results of all mutants, so third approach Tail PCR was used.
3) The third approach was Tail PCR: Tail (Thermal asymmetric interlaced) PCR was used with some modification of protocol of [14] . In this method four primers were used (Figure 7). Primer 1 (TnphoA II: 5’ GTGCAGTAATATCGCCCTGAGCA 3’) anneals to the 5’ end of TnphoA and is used in combination with the partially degenerate primers (a), (b), or (c) that all contain the following sequence: 5’ GGCCACGCGTCGTC GACTAGTACNNNNNNNNNN 3’ followed by AGAG (a), ACGCC (b), or GATAT (c), for two rounds of PCR at very low annealing temperatures (30˚C and 43˚C sequentially). A 1:1 dilution of this PCR product is made and 1 μl of this dilution is used to do another PCR reaction for the normal 30 cycles with a 60˚C annealing temperature. This second PCR reaction uses primer 2 (Hah-1: 5’ GTTTTCCAGAACCAGGGCAAAACGG 3’) which anneals to the complement of the tail end of the TnphoA sequence and primer D (CEKG4: 5’ GGCCACGCGTCGACTAGTAC 3’) which anneals to the complement of the tail end of primer (a), (b), (c). In this way unknown sequences adjacent to a known sequence can be amplified by PCR which can then be followed by a sequencing PCR reaction [14] . Resulting bands (Figure 7 and Figure 8) were gel extracted, re-amplified with another round of PCR, and sequenced.
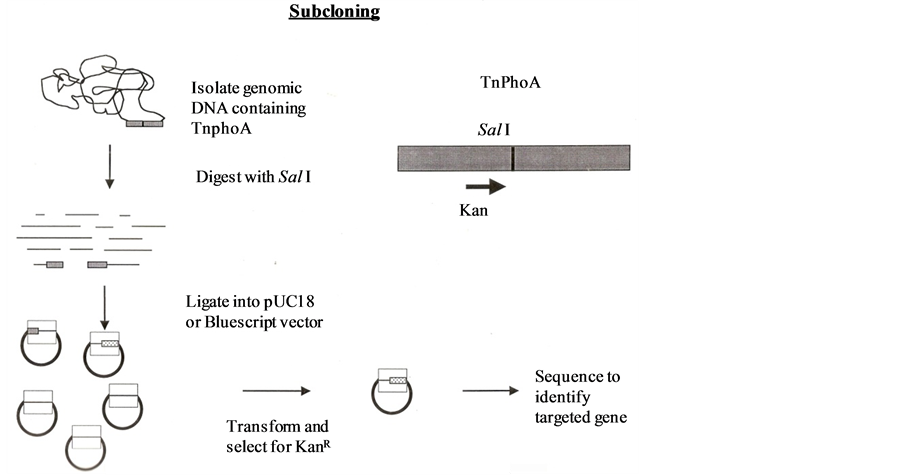
Figure 5. Cloning & subcloning of DNA containing TnphoA.
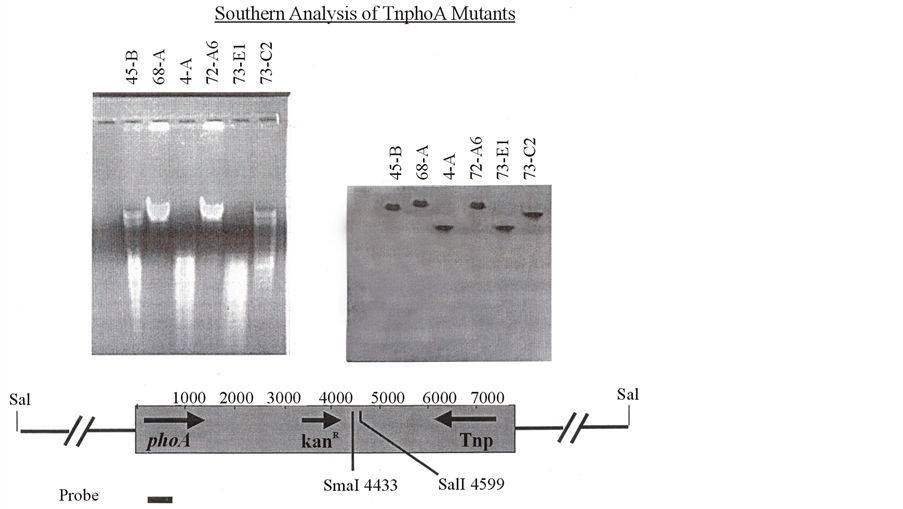
Figure 6. Southern analysis of TnphoA mutants.
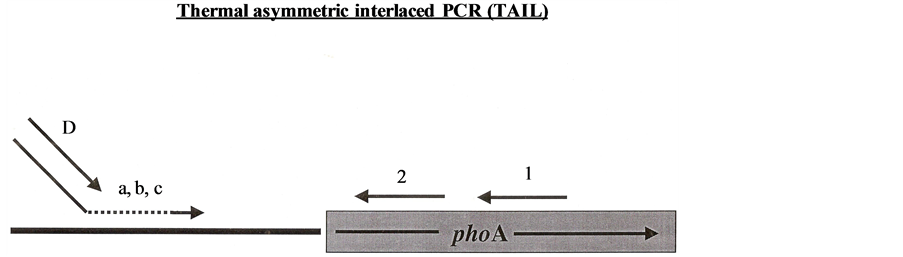
Figure 7. Tail PCR.
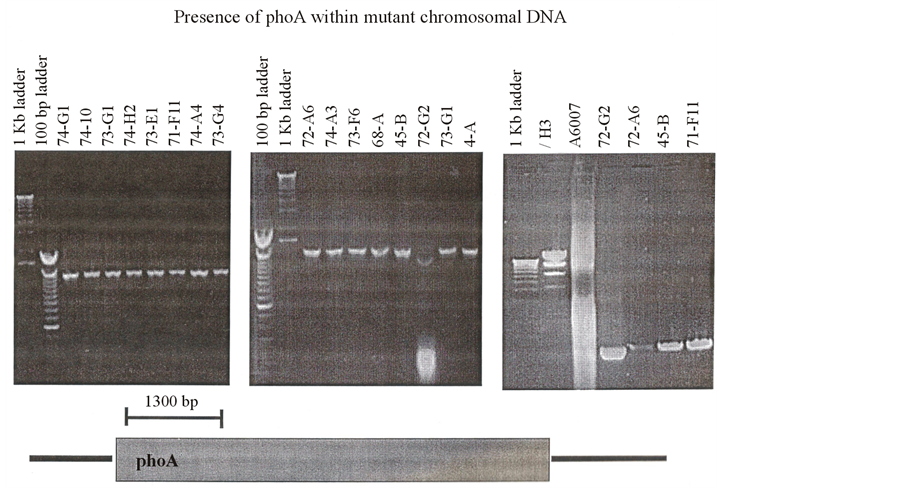
Figure 8. Presence of PhoA within mutant chromosomal DNA.
The results of the alkaline phosphatase activity assayed of all mutants were shown in Table 1, Table 2, Table 3, Table 4 and Table 5 along with the results of re-screening the mutants for virulence on Kalanchoe leaves. In Inverse PCR the shelf ligated DNAs were not amplified with the use of Taq DNA polymerase so that no bands were visible but with the use of Pfu enzyme which is a proof reading DNA polymerase and high fidelity in DNA synthesis, bands were appeared (Figure 4). In cloning approach followed by ligation and transformation, no colonies were grown up on MG/L Kan (50 μl/ml) selection plates but in southern analysis discreet bands of TnphoA adjacent sequences of different mutants digested with SalI enzyme, were visible (Figure 5 and Figure 6). In Tail PCR the presence of PhoA was screened by using primers TnphoA 5’ and TnphoA 3’ with the results being positive in each mutant except 74-G2 showed somewhat ambiguous. In all mutants one TnphoA band was seen in comparison to A6007 (control, no TnphoA band) (Figure 7 and Figure 8). In the Tail and Inverse PCR many bands were sequenced but except 45-B mutant band sequence (Figure 9) no other mutant’s band sequence gave any promising result. Its (45-B) band sequence showed a high degree of homology 984% similar to a succinate dehydrogenase membrane anchor subunit in Bradyrhizobium japonicum (Figure 9(a) and Figure 9(b)). Two distinct regions of homology were present, the first being 26 amino acids in length and the second being 13 amino acids in length. The E-value for this homology was 5 × 10−11, showing that the protein coded by the 45-B gene and the polypeptide in B. japonicum are almost certainly homologues (Figure 10 and Figure 11) and confirming that the succinate dehydrogenase gene is adjacent to the transposon (Figures 11(a)-(d)).

Table 1. Expression of TnphoA mutant’s alkaline phosphatase activity on X-phos & virulence on Kalanchoe leaves.
Attempts were made to characterize transposon adjacent sequences in Agrobacterium tumefaciens mutants defective in tumor-forming ability using a variety of approaches. One likely reason for the lack of success with the Inverse PCR approach was possibility that the size of DNA that was to be amplified was too large. Taq polymerase can’t reliably amplify fragments larger than 3 kb and if the closest SalI or BamHI site was farther away than that from the 5’-end of TnphoA, then successful Inverse PCR would have been difficult. Similarly, size may have also been a factor in the attempt to clone part of the adjacent gene, since the transposon fragment remaining after digestion with SalI or BamHI would have been at least 4 kb. It is right because large fragments can be cloned and make many copies in the vector like the PCR; if the next SalI or BamHI occurred at a great distance from the 5’ end of the transposon, size would have likely been prohibitive.
Table 2. Expression of TnphoA mutant’s alkaline phosphatase activity on X-phos.
Table 3. Expression of TnphoA mutant’s alkaline phosphatase activity on X-phos.
Table 4. Expression of TnphoA mutant’s alkaline phosphatase activity on X-phos.
Table 5. Expression of TnphoA mutant’s alkaline phosphatase activity on X-phos.
From the southern analysis which was intended to determine the size of the fragment, it was seen that all mutants have a single TnphoA insert and its size is about 23 kb; we were unable to get the size standard included in the Amersham kit to light up on the blot so we had to develop our own ladder to know the size of fragment. Southern results couldn’t get with all mutants due to incomplete digestion of chromosomal DNA though we used Cs (Cesium chloride) purified DNA and used highly concentrated BamHi. Rather than continue working to figure out and solve this problem, we chose to attempt the Tail PCR approach.
The Tail PCR approach has been used successfully in a number of bacterial species by Manoil (wood, personal communication) and although our attempts have not yielded instant and easy success in the case of each mutant, the results we have obtained from 45-B are very promising; we cannot be sure that whether the succinate dehydrogenase homologue in Agrobacterium tumefaciens is critical for virulence at this time. It has been seen that succinate dehydrogenase mutant strain of Rhizobium meliloti showed delayed nodulation of Lucerne plants and the formed nodules were white and ineffective.
Revertant strain induced red and effective nodules. Succinate dehydrogense mutant rate of oxygen consumption also goes down due to choking of respiratory pathway for the liberation of kinetic energy. Sugar and plant phenolic compound-acetosyringone activates the vir A protein which induces vir genes and consequently transfers the T-DNA from agrobacterium to the plant. Succinate dehydrogenase which catalyses the conversion of
 (a)
(a) (b)
(b)
Figure 9. (a) Blast search of 45-B lower band sequence producing significant alignment: score and E-value; (b) Succinate dehydrogenase membrane anchor subunit.

Figure 10. Blast search of 45-B lower band sequence.
 (a)
(a)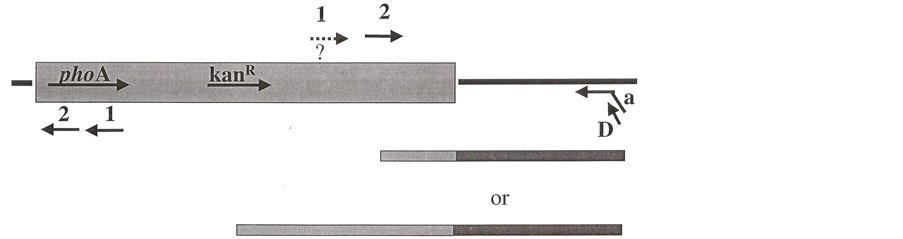 (b)
(b) (c)
(c)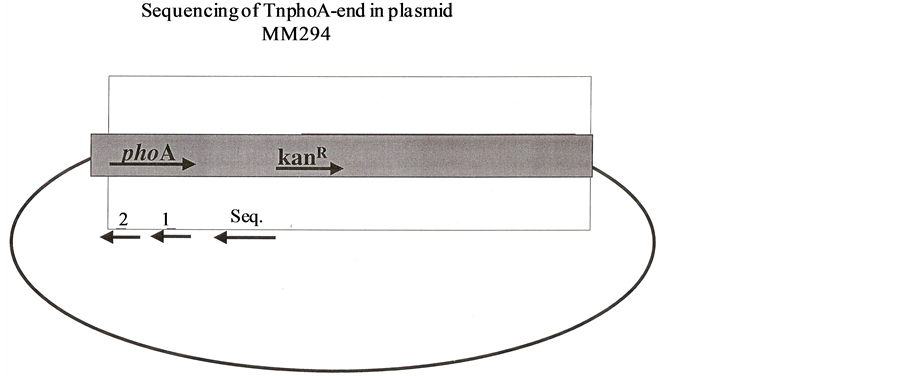 (d)
(d)
Figure 11. (a) Binding of primer & with TnphoA and adjaunt sequence; (b) Tail PCR shows the action of primer and (a) (b) or (c) & (d) on right side of TnphiA adjacent sequence; (c) TnphoA adjacent sequence of Tail PCR; (d) Sequencing of TnphoA-end in plasmid MM294.
succinate to fumarate and goes in further step of the production of energy, to convert fumarate to phosphoenolpyruvate which later on by the reversal of glycolysis changes into sugar which activates vir genes. It seems that succinate dehydrogenase may be involved in virulence and T-DNA transfer from agrobacterium to the plant with high efficiency for transformation.
- Cangelosi, G.A., Huang, L., Puvanesarajah, V., Stacey, G., Ozga, D.A., Leigh, J.A. and Nester, E.W. (1987) Common Loci for Agrobacterium tumefaciens and Rhizobium meliloti Exoplysaccharide Synthesis and Their Roles in Plant Interactions. Journal of Bacteriology, 169, 2086-2091.
- Douglas, C.J., Halperin, W. and Nester, E.W. (1982) Agrobacterium tumefaciens Mutants Affected in Attachment in Plant Cells. Journal of Bacteriology, 152, 1265-1275.
- Manoil, C. and Beckwith, J. (1985) A Transposon Probe for Protein Export Signals. Proceedings of the National Academy of Sciences USA, 87, 6684-6688.
- Long, S., McCune, S. and Walker, G.C. (1988) Symbiotic Loci of Rhizobium meliloti Identified by Random TnphoA Mutagenesis. Journal of Bacteriology, 170, 4257-4265.
- Sarthy, A., Michaelis, S. and Beckwith, J. (1981) Deletion Map of the Escherichia coli Structural Gene for Alkaline Phosphatase, PhoA. Journal of Bacteriology, 145, 288-292.
- Backwith, J. and Silhavy, T.J. (1983) Genetic Analysis of Protein Export in Escherichia coli. Methods in Enzymology, 97, 3-11. http://dx.doi.org/10.1016/0076-6879(83)97114-8
- Cangelosi, G.A., Best, E.A., Martinetti, G. and Nester, E.W. (1991) Genetic Analysis of Agrobacterium. Methods in Enzymology, 204, 584-397.
- Cangelosi, G.A., Ankenbauer, R.G. and Nester, E.W. (1990) Sugars Induce the Agrobacterium tumefaciens Genes through a Periplasmic Binding Protein and a Transmembrane Signal Protein. Proceedings of the National Academy of Sciences USA, 87, 6708-6712. http://dx.doi.org/10.1073/pnas.87.17.6708
- Huang, M.L., Cangelosi, G., Halperin, W. and Nester, E.W. (1990) A Chromosomal Agrobacterium tumefaciens Gene Required for Effective Plant Signal Transduction. Journal of Bacteriology, 172, 1814-1822.
- Shimoda, N., Toyoda-Yamamoto, A., Aoki, S. and Machinda, Y. (1990) Genetic Evidence for an Interaction between the VirA Sensor Protein and the ChvE Sugar Binding Protein of Agrobacterium tumefaciens. Journal of Biological Chemistry, 268, 26552-26558.
- Gardiol, A., Arias, A., Cervenansky, C. and Martinez-Drets, G. (1982) Succinate Dehydrogenase Mutant of Rhizobium meliloti. Journal of Bacteriology, 151, 1621-1623.
- Gardiol, A. Georges, E., Truchet, L. and Dazzo, F.B. (1987) Requirement of Succinate Dehydrogenase Activity for Symbiotic Differentiation of Rhizobium meliloti in Alfalfa Nodules. Applied and Environmental Microbiology, 53, 1947-1950.
- Cohen, S.N., Chang, A.C.Y. and Hsu, L. (1972) Non Chromosomal Antibiotic Resistance in Bacteria: Genetic Transformation of E. coli by R-Factor DNA. Proceedings of the National Academy of Sciences USA, 69, 2110. http://dx.doi.org/10.1073/pnas.69.8.2110
- Chun, K.T., Edenberg, H.J., Kelley, M.R. and Goebl, M.G. (1997) Rapid Amplification of Uncharacterized Transposon-Tagged DNA Sequence from Genomic DNA. Yeast, 13, 233-240. http://dx.doi.org/10.1002/(SICI)1097-0061(19970315)13:3<233::AID-YEA88>3.0.CO;2-E

*Corresponding author.