International Journal of Clinical Medicine
Vol.1 No.2(2010), Article ID:3268,3 pages DOI:10.4236/ijcm.2010.12017
Rhizomelic Chondrodysplasia Punctata Type I
![]()
Medical Center of Central Georgia, Macon, USA.
Email: ab_mouataz@hotmail.com
Received August 8th, 2010; revised August 13rd, 2010; accepted August 17th, 2010.
Keywords: Rhizomelic Chondrodysplasia Punctata Type I, Seizure, Short Humerus
ABSTRACT
Rhizomelic Chondrodysplasia Punctata Type I is one of the rare peroxisome disorders. We report the case of a newborn white male that developed seizures and skeletal dysmorphism. The baby had short humerus bones with stippled epiphyses, consistent with the disease. He had also delay in myelinization on brain MRI with bilateral subependymal cysts over the atria and frontal horns of the lateral ventricles. Usually, infants with this disorder do not live long. This unfortunate little patient died at 5 weeks age from pneumonia. We emphasize the importance of antenatal screening for these disorders especially when a family history of dysmorphism is positive.
1. Introduction
Peroxisomes are subcellular organelles of variable diameter ranging from 0.05 to 0.5 micron that can be found in all cells except erythrocytes. The highest concentration of peroxisomes is in the liver and kidney [1]. Peroxisomal disorders are a heterogeneous group of metabolic disorders that impair peroxisome’s function. They are thought to occur in at least one in 20,000 live births [2]. In most cases, patients display neurologic dysfunction of varying extent.
2. Case Report
A white male baby born after a full-term pregnancy with skeletal dysmorphism, hypotonia, seizures, and difficulty feeding was admitted to the Neonatal Intensive Care Unit (NICU) and underwent multiple screening exams. Echocardiography showed small patent ductus arteriosus, patent foramen ovale, and mild tricuspid regurgitation. X-ray of the upper extremities showed short humerus bones with stippled epiphyses (Figure 1). The radius, ulna and hand appeared too normal. The clinical and radiologic findings were consistent with chondrodysplasia punctata type 1 (RCDP1). Brain magnetic resonance imaging (MRI) revealed delay in myelinization with bilateral subependymal cysts over the atria and frontal horns of the lateral ventricles (Figure 2). A small focal hemorrhage in the right cerebella hemisphere measuring about 7 mm in diameter was also found (Figure 2). There was a small amount of subdural blood surrounding the right cerebellum and to a lesser degree the left cerebellum (Figure 2).
The patient was stabilized and twelve days later the baby boy was discharged home after improvement in clinical condition (feeding). The possibility of having a peroxisomal disorder was discussed with the parents who denied a family history of dysmorphism. Biochemical tests of peroxisome function confirmed the diagnosis of chondrodysplasia punctata type I (red blood cell concentration of plasmalogens, phytanic acid, plasma concentration of very-long-chain-fatty acids (VLCFA). Parents were referred for genetic consultation.
After ten days, the baby was seen by the primary-care physician (PCP) for a new-onset fever and poor feeding. The patient was put on amoxicillin for otitis media but no clinical improvement ensued. Two days later, the patient was admitted to the emergency room with a swollen tongue, dehydration, poor feeding, respiratory deterioration, cyanosis and seizures. He expired shortly after (at 5 weeks age).
3. Discussion
Prenatal diagnosis of these entities is now available by peroxisomal studies of chorionic villus biopsy or cultured amniocytes. Analysis of chorion villus samples has resulted in successful diagnosis of at-risk pregnancies at as early as 10 weeks [3]. Sonography can also be used for the intrauterine diagnosis of rhizomelic chondrodysplasia punctata type I. What difference in our patient’s case was

Figure 1. X-ray of the upper extremities showing short humeri.
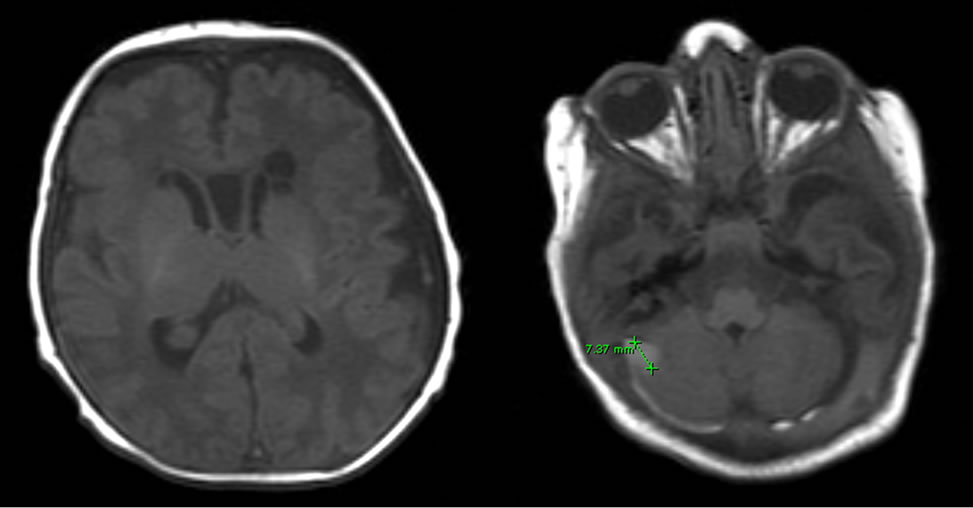
Figure 2. Brain MRI showing delayed myelinization and a focal hemorrhage in the cerebellum.
the normal images of the vertebral column and femurs. Normal spine and femurs, in addition to negative family history of dysmorphism, could have played a role in not drawing attention to this disorder during pregnancy.
The frequency of chondrodysplasia punctata is estimated at 1 per 4000 births [4]. Type I of rhizomelic chondrodysplasia punctata is more common than type II and III and is estimated at less than 0.1 per 10000 births [5]. It is thought to be an autosomal recessive disorder. Most affected infants die in the first few weeks of life from recurrent infections and that is what happened in the above case. Rare patients who survive beyond the first year are usually severely retarded and debilitated [6].
REFERENCES
- J. A. Thomas, C. L. Greene and R. M. Cohn, “Avery’s Diseases of the Newborn,” 7th Edition; WB Saunders, Philadelphia, 1998.
- J. M. Powers, R. C. Tummons, V. S. Caviness Jr., A. B. Moser and H. W. Moser, “Structural and Chemical alterations in the cerebral maldevelopment of fetal cerebro-hepato-renal (Zellweger) syndrome,” Journal of Neuropathology & Experimental Neurology, Vol. 48, No. 3, 1989, pp. 270-289.
- P. W. Gendall, C. E. Baird and D. M. O. Becroft, “Rhizomelic chondrodysplasia punctata: Early recognition with antenatal ultrasonography,” Journal of Clinical Ultrasound, Vol. 22, No. 4, 1994, pp. 271-274.
- S. H. Sastrowijoto, K. Vandenberghe, P. Moerman, J. M. Lauweryns and J. P. Fryns, “Prenatal ultrasound diagnosis of rhizomelic chondrodysplasia punctata in a primigravida,” Prenatal Diagnosis, Vol. 14, No. 8, 1994, pp. 770-776.
- N. E. Braverman, A. B. Moser and S. J. Steinberg, “Rhizomelic Chondrodysplasia Punctata Type 1,” 2010. http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=rcdp
- R. G. F. Gray, A. Green, R. B. H Schutgens, R. J. A. Wanders, P. A. Farndon and C. R. Kennedy, “Antenatal diagnosis of rhizomelic chondrodysplasia punctata in the second trimester,” Journal of Inherited Metabolic Disease, Vol. 13, No. 3, 1990, pp. 380-382.