Open Journal of Nephrology
Vol.3 No.1(2013), Article ID:29225,4 pages DOI:10.4236/ojneph.2013.31011
A Case of Severe Hypomagnesaemia*
1Department of Internal Medicine, Spital Zollikerberg, Zollikerberg, Switzerland
2Department of General Paediatrics, University Children’s Hospital, Münster, Germany
3Department of Nephrology and Haemodialysis, Spital Zollikerberg, Zollikerberg, Switzerland
Email: gurpreet.anand@spitalzollikerberg.ch, konradma@uni-muenster.de, ludwigtheodor.heuss@spitalzollikerberg.ch, #robert.schorn@spitalzollikerberg.ch
Received December 22, 2012; revised February 7, 2013; accepted March 7, 2013
Keywords: Hypocalcaemia; hypomagnesaemia; hypokalaemia; weakness
ABSTRACT
Hypomagnesaemia is a common finding that occurs in hospitalised patients and can be caused by renal or extrarenal wasting. Because it is not routinely tested, even severe hypomagnesaemia can be missed. In cases of extrarenal wasting, the kidney can decrease its magnesium excretion to very low levels. A systematic diagnostic approach allows the proper diagnosis and treatment of hypomagnesaemia. We report on a 72-year-old Caucasian male patient with repeated drops and amyosthenia caused by severe hypomagnesaemia. In this report, we discuss the pathophysiology, clinical signs, diagnostic investigation and treatment of magnesium depletion.
1. Introduction
Despite the fact that hypomagnesaemia is a frequent finding, ranging from 12% in all hospitalised patients to 60% in an intensive care setting, plasma magnesium is not measured routinely [1,2]. The clinical manifestations of magnesium depletion are rather nonspecific. Early signs include nausea, vomiting, fatigue, weakness, hyperreflexia, carpopedal spasm, tremor and tetany. A prolonged QT interval, ST depression, ventricular arrhythmias and the enhancement of digitalis toxicity are typical cardiac findings [3]. Symptomatic hypomagnesaemia is often associated with hypokalaemia due to urinary potassium wasting and hypocalcaemia due to lower parathyroid hormone secretions, as well as end-organ resistance to its effects. The diagnosis of magnesium depletion is challenging because the clinical manifestation may be absent; additionally, the serum magnesium concentration does not reflect the total body magnesium content.
2. Case Report
A 72-year-old male Caucasian patient presented to the emergency department with a history of progressive amyosthenia developing over eight weeks. Hypocalcaemia and 25-hydroxyvitamin D deficiency had been detected twenty-two days previously, and replacement was started without clinical improvement. The patient had a history of chronic renal failure, coronary artery disease, hypertensive cardiomyopathy and hyperlipidaemia. His medications included aspirin, bisoprolol, losartan, pravastatin, torasemid, pantoprazole, calcium and vitamin D3. There was no evidence of alcohol consumption. His blood pressure was 129/99 mmHg, and his pulse rate was 102 b.p.m. Upon examination, he demonstrated general muscle weakness, absent reflexes in the lower extremities, positive Chvostek’s sign and an otherwise normal clinical status. His laboratory analysis showed remarkable results for total serum calcium (1.79 mmol/l), potassium (3.5 mmol/l), 25-hydroxyvitamin D (<30 nmol/l), iPTH (6.3 pmol/l) and creatinine (112 µmol/l). Based on the progressive amyosthenia, positive Chvostek’s sign and hypocalcaemia despite the calcium and vitamin D substitution, a magnesium analysis was performed that revealed severe hypomagnesaemia with 0.11 mmol/l (Figure 1). Other blood values, including pH and bicarbonate, and the urinary sediment were normal. The loop diuretic and proton pump inhibitor (PPI) were ceased, and a 24-h urine (600 ml) collection yielded the following results: calcium 0.32 mmol/l, magnesium 0.17 mmol/l, phosphate 16.2 mmol/l and creatinine 14.7 mmol/l. Electrocardiography, chest X-ray, head CT-scan and abdominal sonography showed no abnormalities.
The fractional excretion of magnesium (FeMg) was 9.3%, indicating renal magnesium wasting. In the context of hypocalciuria, this raised the suspicion of Gitelman syndrome (GS) or autosomal-dominant renal hypomagnesaemia associated with hypocalciuria (IDH). Because of the reported manifestation of inherited forms of

Figure 1. Serum magnesium and calcium levels over time, following calcium and magnesium substitution.
hypomagnesaemia at older ages, despite a negative family history, we performed a genetic analysis that ruled out GS and IDH (no mutations in SLC12A3 or FXYD2). The screened siblings did not demonstrate renal magnesium wasting.
After intravenous magnesium substitution, the clinical picture improved rapidly (Figure 1). The follow-up by the family physician showed normal serum magnesium concentrations under oral magnesium substitution despite the restarted loop diuretic and PPI medication.
Concerning the underlying aetiology of the severe magnesium depletion, in this case, we hypothesised a multifactorial aetiology. The degree of hypomagnesaemia induced by loop diuretics is generally mild and associated with hypercalciuria. In theory, increasing calcium reabsorption after stopping the loop diuretic could provoke hypocalciuria. In this case, the concomitant use of a PPI could have induced extrarenal magnesium wasting through decreased intestinal absorption, and the initiation of vitamin D supplementation may have induced a calcium shift into the bones, each of which may have contributed to the development of severe magnesium deficiency.
3. Discussion
Magnesium is the second most common intracellular cation and plays an important role as a cofactor in more than 300 enzymatic reactions. It is also regarded as a natural calcium antagonist. The total magnesium content of an average adult is 24 g. Approximately 60% of the body’s magnesium is present in bone, 20% in the muscle and another 20% in the soft tissue and liver. Approximately 99% of total body magnesium is intracellular, with only 1% present in the extracellular space (blood serum or red blood cells (RBC)). Extracellular magnesium exists in three fractions: free/ionised 55% - 70%, protein-bound 20% - 30% and complexed with anions such as phosphate, bicarbonate, citrate or sulphate. Of these fractions, the ionised form has the greatest biological activity and can be measured. Total serum magnesium levels normally range from 0.65 to 1.05 mmol/l [3,4].
Magnesium homeostasis is tightly regulated by the intestine, bone and kidneys. Magnesium is absorbed in the gut and stored in bone minerals, and the excess magnesium is excreted by the kidneys and faeces. The minimal daily intake should be 350 mg for men and 430 mg for women. Magnesium is mainly (80% - 90%) absorbed in the small intestine by a passive transcellular mechanism driven by an electrochemical gradient and solvent drag. A minor fraction is transported via the transcellular transient receptor potential channel melastatin members (TRPM) 6 and 7 [3,4]. The absorption rate of dietary magnesium varies from 24% to 76% and mainly depends on the body magnesium status [5]. The kidney plays a crucial role in magnesium homeostasis, as of the filtered magnesium load (2400 mg daily), 95% is reabsorbed, and only 3% - 5% is excreted. The highest level of reabsorption occurs in the thick ascending limb of the loop of Henle (TAL) and in the proximal tubules in a passive paracellular manner (60% - 70% and 10% - 20%, respectively). In the TAL, the tight junction proteins claudin-16 and claudin-19 are responsible for this transport [4,6]. The final 5% - 10% of the filtered magnesium is reabsorbed in the distal tubule (DCT), where the transient receptor potential melastatin (TRPM) 6 channel plays a pivotal role [6]. The epidermal growth factor (EGF) and oestrogen regulate TRPM 6 activity [4]. The renal excretion of the filtered load can vary from 0.5% to 70%, allowing conservation during states of magnesium deprivation and rapid excretion in cases of excess intake [3].
The measurement of serum magnesium is relatively easy and has become the method of choice to estimate the magnesium content, although it does not reflect the total body stores (only 0.3% of the total body content is present in blood serum). However, magnesium depletion can occur in the setting of a normal serum magnesium concentration, so-called normomagnesaemic magnesium depletion [7]. Haemolysis leads to misinterpretation because the magnesium concentration in RBCs is higher than in serum [3]. The common causes of acquired hypomagnesaemia are listed in Table 1 [3,8]. A distinction between gastrointestinal and renal losses can be made by measuring the 24-hour FEMg (preferred because of the circadian variation in renal magnesium excretion) or by fractional excretion on a random urine specimen [3,4,9]. FEMg can be calculated from the following formula:
FEMg = [(UMg × PCr)/(PMg × UCr × 0.7)] × 100. In the above equation, U and P refer to the urine and plasma concentrations of magnesium (Mg) and creatinine (Cr). The plasma magnesium concentration is multiplied by 0.7 because only approximately 70% of the circulating magnesium is free (not bound to albumin) and therefore
Table 1. Causes of acquired hypomagnesaemia [3,8].
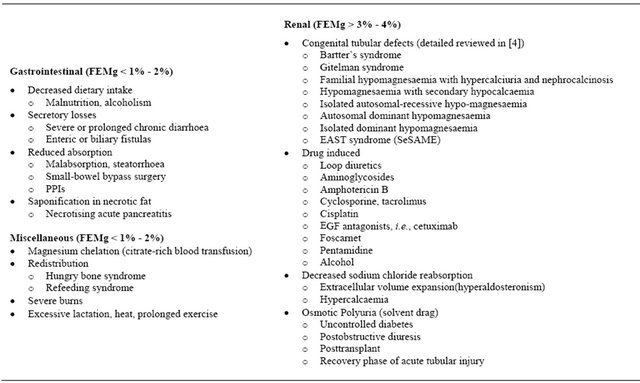
Abbreviations: EGF: Epidermal growth factor; FEMg: fractional excretion of magnesium.
capable of being filtered across the glomerulus. The normal renal response to extrarenal magnesium depletion is to restrict magnesium excretion to very low levels (typically, to <1%) [10]. Thus, the daily excretion of more than 1 mmol or a calculated FEMg above 3% to 4% in a subject with normal renal function indicates renal magnesium wasting [3,10].
A magnesium retention test or loading test can differentiate among patients with hypomagnesaemic and normomagnesaemic deficiencies. After oral loading, the changes in serum magnesium and urinary excretion reflect the intestinal magnesium reabsorption [4]. Parenteral magnesium administration can also identify a functional magnesium deficiency [11]. After a magnesium loading, a urinary excretion of >60% - 70% rules out any relevant magnesium depletion. However, the standardisation and utility of this test are lacking [3,7].
Hypokalaemia, which accompanies 60% of the cases, can be caused by extrarenal (e.g., diarrhoea) and renal loss. Diuretics and a loss of the inhibitory function of magnesium on the ROMK channel promote urinary potassium wasting [7]. Another classical sign of severe hypomagnesaemia is hypocalcaemia, which is induced by the suppression of PTH release, as well as skeletal resistance to PTH [7]. Low calcitriol levels contribute to a fall in calcium concentration as well. Chronic hypomagnesaemia is associated with chondrocalcinosis (magnesium is a co-factor of various pyrophosphatases) and joint swelling.
The method of magnesium repletion varies with the severity of the clinical setting. Severe hypomagnesaemia with tetany or ventricular arrhythmia requires a prompt intravenous substitution followed by oral substitution. Mild or asymptomatic hypomagnesaemia can be substituted orally. The underlying disorder should also be corrected [3,7].
4. Conclusion
The impact of hypomagnesaemia is underestimated because clinicians often fail to measure magnesium [12]. Unexplained muscle weakness, hypocalcaemia and refractory hypokalaemia point to underlying hypomagnesaemia, despite serum magnesium values within the normal limits (normomagnesaemic magnesium depletion). Magnesium depletion can be induced by either renal or extrarenal losses. A 24-hour urinary FEMg can discriminate between these two major mechanisms. Severe hypomagnesaemia requires an intravenous replacement.
REFERENCES
- E. T. Wong, R. K. Rude, F. R. Singer, S. T. Shaw Jr., “A High Prevalence of Hypomagnesemia and Hypermagnesemia in Hospitalized Patients,” American Journal of Clinical Pathology, Vol. 79, No. 3, 1983, pp. 348-352.
- G. M. Tong and R. K. Rude, “Magnesium Deficiency in Critical Illness,” Journal of Intensive Care Medicine, Vol. 20, No. 1, 2005, 20, pp. 3-17.
- W. Jahnen-Dechent and M. Ketteler, “Magnesium Basics,” Clinical Kidney Journal, Vol. 5, Suppl. 1, 2012, pp. i3-i14. doi:10.1093/ndtplus/sfr163
- J. H. F. De Baaij, J. G. J. Hoendderop and R. J. M. Bindels, “Regulation of Magnesium Balance: Lessons Learned from Human Genetic Disease,” Clinical Kidney Journal, Vol. 5, Suppl. 1, 2012, pp. i15-i24.
- L. A. Graham, J. J. Caesar and A. S. Burgen, “Gastrointestinal Absorption and Excretion of Mg28 in Man,” Metabolism, No. 9, 1960, pp. 646-659.
- B. Glaudemans, N. V. Knoers, J. G. Hoenderop and R. J. M. Bindels, “New Molecular Players Facilitating Mg2+ Reabsorption in the Distal Convoluted Tubule,” Kidney International, Vol. 77, No. 1, 2010, pp. 17-22. doi:10.1038/ki.2009.358
- Z. S. Agus, “Hypomagnesemia,” Journal of the American Society of Nephrology, Vol. 10, No. 7, 1999, pp. 168-174.
- G. Regolisti, A. Cabassi, E. Parenti, U. Maggiore and E. Fiaccadori, “Severe Hypomagnesemia during Long-Term Treatment with a Proton Pump Inhibitor,” American Journal of Kidney Diseases, Vol. 56, No. 1, 2010, pp. 168-174. doi:10.1053/j.ajkd.2010.03.013
- N. V. Knoers, “Inherited Forms of Renal Hypomagnesemia: An Update,” Pediatric Nephrology, Vol. 24, No. 4, 2009, pp. 695-705. doi:10.1007/s00467-008-0968-x
- M. Elisaf, K. Panteli, J. Theodorou and K. C. Siamopoulos, “Fractional Excretion of Magnesium in Normal Subjects and in Patients with Hypomagnesemia,” Magnesium Research, Vol. 10, No. 4, 1997, pp. 315-320.
- P. Hébert, N. Mehta, J. Wang, T. Hindmarsh, G. Jones and P. Cardinal, “Functional Magnesium Deficiency in Critically Ill Patients Identified Using a MagnesiumLoading Test,” Critical Care Medicine, Vol. 25, No. 5, 1997, pp. 749-755.
- R. Whang and K. W. Ryder, “Frequency of Hypomagnesemia and Hypermagnesemia. Requested vs Routine,” Journal of the American Medical Association, Vol. 263, No. 22, 1990, pp. 3063-3064.
NOTES
*Transparency declarations: None to declare.
#Corresponding author.