Open Journal of Orthopedics
Vol.2 No.2(2012), Article ID:20120,4 pages DOI:10.4236/ojo.2012.22014
Comparison between Different Modalities of Treatment of Ewing Sarcoma
![]()
1Department of Orthopaedics, Bankura Sammilani Medical College, Bankura, India; 2Bankura Sammilani Medical College, Bankura, India.
Email: ranadeb.b@rediffmail.com, arindamm02@gmail.com, ujjal_doc@yahoo.com
Received March 10th, 2012; revised April 20th, 2012; accepted May 8th, 2012
Keywords: Ewing Sarcoma; EBRT; Chemotherapy; Surgery; Kaplan-Meier Survival Curves; Cox Proportional Hazards Model
ABSTRACT
Background: Ewing sarcoma is the most common primary malignant tumour in patients younger than 10 years of age. The incidence is less than 1 per 1 million per year. Usually it is located in the diaphysis of long bones. Prognosis of these tumours has improved dramatically since the introduction of multiagent chemotherapy, from an erstwhile 10% survival rate to the current 70% for patients with non-metastatic Ewing sarcoma. Method: A retrospective review of patients with histologically confirmed Ewing sarcoma who were treated in the Department of Orthopaedics, B.S. Medical College during the time period from April 2000 to March 2012 was performed. Patients were divided into two groups: Group A included those treated by External Beam Radiotherapy (EBRT) + chemotherapy while Group B included the patients treated with surgery + chemotherapy. Results were analysed depending on the survival rates. Kaplan-Meier survival curves were compared using log-rank test and a multivariate Cox proportional hazards model was calculated. Result: The survival curves of both the groups were not found to be significantly different. Conclusion: Treatment of Ewing tumour has multiple options. No one treatment modality is superior. Survival rates of patients treated by radiation + chemotherapy are not significantly different from those treated with surgery + chemotherapy.
1. Introduction
James Ewing (1866-1943) first described the tumour, establishing that the disease was separate from lymphoma and other types of cancer known at that time. Because a common genetic locus is responsible for a large percentage of Ewing sarcoma and primitive neuroectodermal tumors, these are sometimes grouped together in a category known as the Ewing family of tumors. The diseases are, however, considered to be different: Peripheral primitive neuroectodermal tumours are generally not associated with bones, while Ewing sarcomas are most commonly related to bone [1].
Ewing sarcoma occurs most frequently in teenagers, with a male/female ratio of 1.6:1. Although usually classified as a bone tumour, Ewing sarcoma can have characteristics of both mesodermal and ectodermal origin, making it difficult to classify. Ewing sarcoma is more common in males and usually presents in childhood or early adulthood, with a peak between 10 and 20 years of age. It can occur anywhere in the body, but most commonly in the pelvis and proximal long tubular bones, especially around the growth plates. The diaphyses of the femur are the most common sites, followed by the tibia and the humerus. Thirty percent are overtly metastatic at presentation. Patients usually experience extreme bone pain. It is positive for CD99 and negative for CD45. Other entities that may have a similar clinical presentation include osteomyelitis, osteosarcoma (especially telangiectatic osteosarcoma) and eosinophilic granuloma. Soft tissue neoplasms such as pleomorphic undifferentiated sarcoma (malignant fibrous histiocytoma) that erode into adjacent bone may also have a similar appearance [2].
Because almost all patients with apparently localized disease at diagnosis have occult metastatic disease, multidrug chemotherapy (often including ifosfamide and etoposide) as well as local disease control with surgery and/or radiation is indicated in the treatment of all patients. Treatment often consists of neo-adjuvant chemotherapy generally followed by a limb salvage or an amputation and may also include radiotherapy. Complete excision at the time of biopsy may be performed if malignancy is confirmed at the time it is examined. Treatment lengths vary depending on location and stage of the disease at diagnosis. Radical chemotherapy may be as short as 6 treatments at 3 week cycles, however most patients will undergo chemotherapy for 6 - 12 months and radiation therapy for 5 - 8 weeks [2]. Antisense oligodeoxynucleotides have been proposed as possible treatment by down-regulating the expression of the oncogenic fusion protein associated with the development of Ewing sarcoma resulting from the EWS-ETS gene translocation. In addition, the synthetic retinoid derivative fenretinide [4-hydroxy(phenyl)retinamide] has been reported to induce high levels of cell death in Ewing sarcoma cell lines in vitro, and to delay growth of Ewing sarcoma xenografts in vivo mouse models [3].
2. Materials and Methods
A retrospective review of patients younger than 21 years (median age, 16 years) was performed on the patients admitted in the Department of Orthopaedics of B.S. Medical College during the study period was of about 12 years from April 2000 to March 2012. Eligibility criteria for the patients included in the study were as follows:
1) Patients who were younger than 21 years age;
2) Had typical radiographic and histological features of Ewing sarcoma;
3) Tumour was located in extremity;
4) No evidence of metastasis.
A review of 17 patients, who were diagnosed as Ewing sarcoma by histology during the study period of 12 years, were included in the study. The patients were divided into two groups. Patients who had been treated by External beam radiotherapy (EBRT) followed by chemotherapy were placed in Group A, while those treated by surgery (amputations or limb salvage) followed by chemotherapy were kept in Group B. All patients in both the groups had received six cycles of chemotherapy. The chemotherapy regime consisted of Inj. Actinomycin D (500 mcg Day 1 to 3), Inj. Vincristine (2 g on Day 1), Inj. Doxorubicin (80 mg on Day 1), Inj. Cyclophosphamide (1000 mg on Day 1). The cycles had been repeated every three weeks. Patients in Group A had received EBRT followed by chemotherapy while those in Group B had undergone surgery followed by chemotherapy.
The patients were analysed statistically based on the survival rates. Survival was denoted as “1” if the event i.e., failure (in our study which is death of the patient) occurred. This means if the patient entered the study at a certain point of time and died after x yrs within the study period, the survival was marked as 1 at the end of study and his time of survival was x yrs. While, “0” was used when the data are censored, i.e., we don’t know what exactly happened to the patient after that time or can’t use the information anymore. Thus if the patient could be followed up for z yrs during the study period and no further information was available at the end of the study or if he was still alive at the end of study, his survival was denoted as “0”.
Kaplan-Meier survival curves were compared using log-rank test and a multivariate Cox proportional hazards model. P ≤ 0.05 was regarded as significant.
3. Results
The patients under the study were divided into 2 groups.
Group A: EBRT + chemotherapy;
Group B: Surgery (amputation/limb salvage) + chemotherapy.
Ewing Sarcoma involving the bones of the extremities i.e., humerus, femur or tibia were studied. Size of the tumor was not considered even though it has a definite prognostic significance, as specific parameters are not available in this regards. Also it is difficult to differentiate the effect of location and size as most proximally located tumors are larger at presentation than distally located tumors.
The results were statistically analysed and compared by Kaplan-Meier survival curves using log-rank test and a multivariate Cox proportional hazards model. P ≤ 0.05 was regarded as significant.
During the study, it was found that of the 17 patients, 10 patients had been treated by EBRT + chemotherapy (Group A), while 7 patients had been treated by surgery + chemotherapy (Group B).
Log-rank (Mantel-Cox) Test performed showed a Chi square value of 1.521, P-value 0.2175 which was not significant. The survival curves of both the groups were not found to be significantly different (Table 1, Figure 1).
The Hazard ratio was calculated to be 0.3692 and 95% confidence interval (CI) of the ratio was 0.07577 to 1.799.
4. Discussion
Ewing Sarcoma is the fourth most common primary malignant tumour of bone, but is the second most common in patients younger than 30 years and the most common in patients younger than 10 years of age. The incidence is less than 1 per 1 million per year. The most common location include metaphysis of long bones and the flat bones of the shoulder and pelvic girdles.
Pain is almost universal complaint of patient with Ewing sarcoma. It is usually insidious in onset. The pain may be only mild and intermittent initially and may respond to initial conservative treatment. In addition to pain, patients may also present with erythema, fever, swelling suggesting osteomyelitis. Classically, Ewing sarcoma appear radiographically as a destructive lesion in the diaphysis of long bone with an onion skin periosteal reaction [4]. Regardless of the location, MRI of the full bone should be ordered to evaluate the full extent of the lesion.
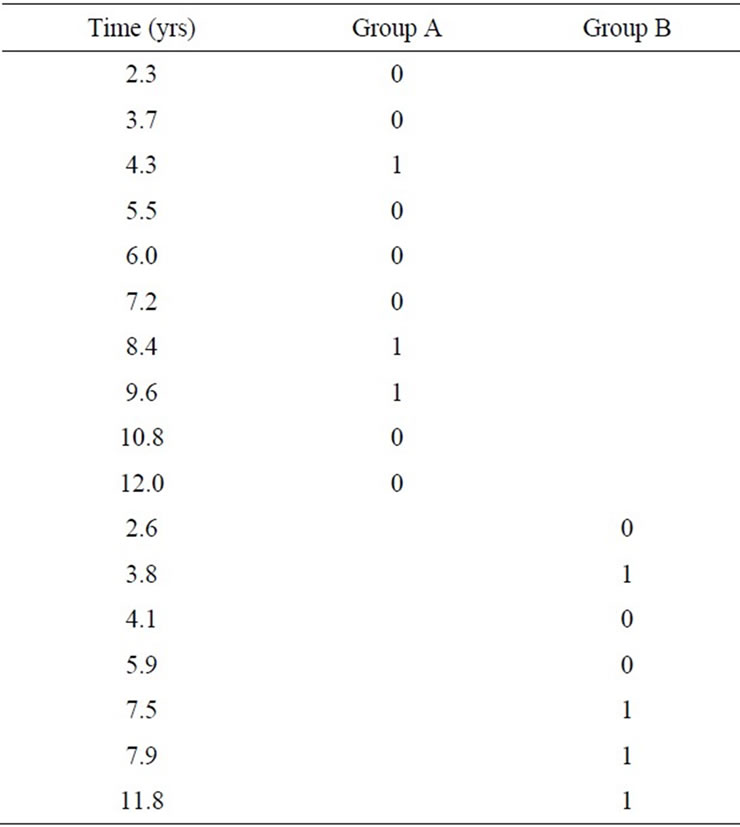
Table 1. Survival rates.

Figure 1. Survival curves comparing Group A (EBRT + chemotherapy) and Group B (surgery + chemotherapy).
Radiograph and CT scan of the chest is done as lungs are the most common site of metastasis. Histologically, Ewing sarcoma appear as small blue cells with very little intercellular matrix. Immunohistochemical studies show that t (11; 22) (q24; q12) is the most common translocation diagnostic of Ewing sarcoma and is present in 90% patients. Prognosis depends on numerous factors the worst being presence of distant metastasis. Older age, male, presence of fever, anaemia, elevated laboratory values, aberration of p53 expression and poor histological response to neo-adjuvant chemotherapy are prognostically worse [5].
A treatment program for Ewing sarcoma may include several approaches such as surgery, radiation and chemotherapy. Surgery is used to remove a tumor or remove any tumor left after chemotherapy, which typically lasts about nine weeks. Surgery is performed if complete removal of the tumor is possible without damage to vital tissue or organs. Radiation therapy is used in combination with chemotherapy and sometimes surgery. Radiation for these tumors usually is external radiation from outside the body. Studies are evaluating the effectiveness of radiation implanted in the body during surgery. For treating the Ewing’s family of tumors, surgery or radiation often is used to remove the local tumor and chemotherapy is used to kill any cancer cells that remain in the body. A supplement to the treatment options listed above is myeloablative therapy with stem cell support. This usually is reserved for patients who have resistant disease, recurrence of disease or widely disseminated disease. Myeloablative therapy is a very intense regimen of chemotherapy to destroy all cells that divide rapidly. These cells include some blood cells and hair cells, as well as cancer cells [6]. Stem cells are self-renewing cells that create all of the other various types of blood cells. Stem cell support involves enriching the stem cells to increase the number of these important cells circulating in the blood after the chemotherapy has been given to kill the remaining tumor cells. Treatment for the Ewing tumors depends on where the cancer is located, how far the cancer has spread, the stage of the disease, and the age and general health of the patient. For localized tumors treatment may be combination chemotherapy followed by surgery, radiation therapy or surgery in combination with radiation therapy, a clinical trial of intensified chemotherapy, chemotherapy after surgery with or without stem cell transplant. For metastatic tumors, treatment options include combination chemotherapy followed by radiation therapy or surgery, high-dose chemotherapy with or without radiation therapy plus additional stem cell support, a clinical trial of intensive chemotherapy with multiple chemotherapy drug combinations [7]. For recurrent tumors, treatment depends on where the cancer has recurred, how the cancer was treated before and the specific medical condition of each child. Chemotherapy may be used for children who did not previously receive chemotherapy. Radiation treatment may be given to reduce symptoms. Surgery may be used to remove tumors that have spread to the lungs or other organs. Clinical trials are testing new treatments [8].
In our retrospective review, it was found that of the 17 patients, 10 patients had been treated by EBRT + chemotherapy (Group A), while 7 patients had been treated by surgery + chemotherapy (Group B). Statistical analyses showed that the differences in treatment outcomes of both the groups were not significant. The survival curves of both the groups were not found to be significantly different (Table 1, Figure 1).
Treatment of Ewing tumour encompasses a multitude of options. Treatment depends on the tumour location, the extent of tumour spread, stage of the disease, age and general condition of the patient. From our study it is evident that survival rates are not significantly different between the patients treated with radiotherapy + chemotherapy compared with those treated by surgery (amputation/limb salvage ) + chemotherapy [9,10].
However, our study had some limitations. As Ewing Sarcoma is a rare disease, the number of patients in the study was also small. As such any definite inference could not be drawn from the study.
REFERENCES
- J. Potratz, H. Jürgens, A. Craft and U. Dirksen, “Ewing Sarcoma: Biology-Based Therapeutic Perspectives,” Pediatric Hematology-Oncology, Vol. 29, No. 1, 2012, pp. 12-27. doi:10.3109/08880018.2011.627582
- J. Potratz, U. Dirksen, H. Jürgens and A. Craft, “Ewing Sarcoma: Clinical State-of-the-Art,” Pediatric Hematology-Oncology, Vol. 29, No. 1, 2012, pp. 1-11. doi:10.3109/08880018.2011.622034
- D. Gulati, A. N. Aggarwal, S. Kumar and S. Chaturvedi, “Primary Ewing’s Sarcoma of the Second Cervical Vertebra: A Rare Entity,” Journal of Pediatric Orthopaedics B, Vol. 20, No. 6, 2011, pp. 408-412. doi:10.1097/BPB.0b013e328345d78a
- J. Schrager, R. E. Patzer, P. J. Mink, K. C. Ward and M. Goodman, “Survival Outcomes of Pediatric Osteosarcoma and Ewing’s Sarcoma: A Comparison of Surgery Type within the SEER Database, 1988-2007,” Journal of Registry Management, Vol. 38, No. 3, 2011, pp. 153-161.
- S. T. Jung, H. W. Park and J. Y. Chung, “Treatment of a Severe Neglected Valgus Deformity after Excision of the Distal Fibula for Ewing’s Sarcoma,” Journal of Bone and Joint Surgery, Vol. 94, No. 1, 2012, pp. 138-140. doi:10.1302/0301-620X.94B1.27784
- C. Müller, C. C. Winter, D. Rosenbaum, J. Boos, G. Gosheger, J. Hardes, et al., “Early Decrements in Bone Density after Completion of Neoadjuvant Chemotherapy in Pediatric Bone Sarcoma Patients,” BMC Musculoskeletal Disorders, Vol. 11, 2010, p. 287.
- T. P. Cripe, “Ewing Sarcoma: An Eponym Window to History,” Sarcoma, Vol. 2011, 2011, Article ID 457532.
- M. Huang and K. Lucas, “Current Therapeutic Approaches in Metastatic and Recurrent Ewing Sarcoma,” Sarcoma, Vol. 2011, 2011, Article ID 863210.
- Y. Funakoshi, T. Mukohara, T. Kataoka, H. Tomioka, N. Chayahara, Y. Fujiwara, et al., “Left Atrial Extension of Metastatic Lung Tumor via Pulmonary Vein: Report on the First Case of Ewing Sarcoma,” Rare Tumors, Vol. 2, No. 3, 2010, p. e53.
- S. Hafezi, R. R. Seethala, E. B. Stelow, S. E. Mills, I. T. Leong, E. MacDuff, et al., “Ewing’s Family of Tumors of the Sinonasal Tract and Maxillary Bone,” Head and Neck Pathology, Vol. 5, No. 1, 2011, pp. 8-16. doi:10.1007/s12105-010-0227-x