Journal of Computer and Communications
Vol.03 No.06(2015), Article ID:57254,10 pages
10.4236/jcc.2015.36011
Comparative Study of the Parallelization of the Smith-Waterman Algorithm on OpenMP and Cuda C
Amadou Chaibou1, Oumarou Sie2
1Laboratoire de Mathématiques et Informatique (LAMI), Université de Ouagadougou, Ouagadougou, Burkina Faso
2Département de Mathmatiques et Informatique, Université de Ouagadougou, Ouagadougou, Burkina Faso
Email: chaibouam@univ-ouaga.bf, chaibouam@yahoo.fr, sie@univ-ouaga.bf
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 14 April 2015; accepted 15 June 2015; published 18 June 2015
ABSTRACT
In this paper, we present parallel programming approaches to calculate the values of the cells in matrix’s scoring used in the Smith-Waterman’s algorithm for sequence alignment. This algorithm, well known in bioinformatics for its applications, is unfortunately time-consuming on a serial computer. We use formulation based on anti-diagonals structure of data. This representation focuses on parallelizable parts of the algorithm without changing the initial formulation of the algorithm. Approaching data in that way give us a formulation more flexible. To examine this approach, we encode it in OpenMP and Cuda C. The performance obtained shows the interest of our paper.
Keywords:
Cuda, GP-GPU, OpenMP, Parallel Computing, Smith-Waterman

1. Introduction
In this paper, we discuss the parallelization of the Smith-Waterman algorithm [1] -[3] on the proteins sequences alignment. This algorithm permits to compare protein sequence of large sizes. The sequence alignment analyses sequences of amino acids to extract similar subsequences. The results of such analysis answer questions such as:
• Is that a new sequence fully or partially in the database?
• Does this sequence contain a given gene?
• How a gene can migrate from other previously identified genes?
• etc.
Answers to these questions can help to simulate changes or mutations used in medicine, the recognition of body (from the classification of individuals based on genetic maps), phylogeny (comparing very similar sequences for inferring evolutionary relationships of proteins within families), etc.
Many algorithms are used in sequence alignment. They can be classified into two types:
• Approach gives rigorous results but is extremely slow. The algorithm of Needleman and Wusch [4] for global search and the Smith-Waterman’s algorithm for local search belong to the algorithms of this category.
• Very fast approach but with results less satisfactory for very large databases. This is a compromise between speed and sensitivity. BLAST1 [5] and FASTA2 [6] are two algorithm of this category. BLAST algorithm uses a heuristic to detect the anchor points to locate areas of identical sequences. FASTA is used for a quick comparison of protein or nucleotide.
Our works focuses on the search for satisfactory solutions with reduced execution time.
2. Preliminaries
The Smith-Waterman algorithm is used to find the large alignment between two sequences based on the substitution matrix and the fixed penalty. It allows to extract the longest similar segments in the two aligned sequences.
2.1. Principle of the Algorithm
To determine similarity between two nucleotide or protein, the Smith-Waterman algorithm compares all possible segments and assigns a score. It returns segment with the highest score. For example, consider s and t two sequences to be compared. The algorithm begins by creating a matrix M of dimensions equal to the lengths of the sequences s and t. Then the cell values of matrix M are calculated starting from the cell in the upper left corner to the cell at the bottom right corner. Formula (1) gives the expression of 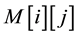
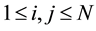 .
.
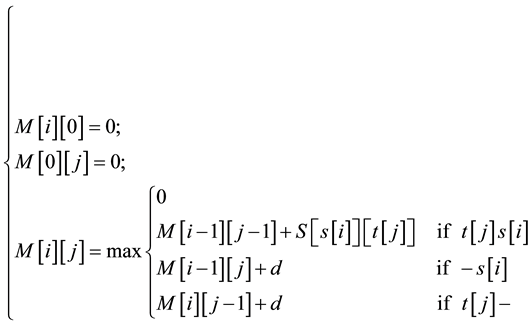 (1)
(1)
Where:
-S is a “Blosum Scoring matrix”3.
-d is a fixed constant corresponding to the alignment of a letter and an empty score (-).
-t [j] s [i] means that t [j] and s [i] are animo acide.
-s[i] and t[j]―correspond respectively to the alignement of the―a animo acide (animo acide with-).
-M [i] [j] is intuitively the score of an alignment ending with t [i] s [j]. At each step, when a maximum value is calculated, it is stored along the direction in which it is obtained: on the diagonal (i − 1, j − 1), just above (i − 1, j) or to the left (i, j − 1). Computing the matrix M and the information regarding the directions in which the highest values are obtained, require much time and memory space.
To restore the best alignment, we proceed as follows:
• Find the maximum value in the matrix M. This is the end of the local alignment with the best score.
• Go to (the) cell (s) adjacent(s) Maximum score:
-movement on the diagonal shows an alignment of the letters t [i] and s [j];
-an horizontal movement means of a bias t [i] and a blank (−) between s [j − 1] and s [j];
-an vertical movement is analogous to the horizontal displacement;
-when the maximum score is 0, it means that the optimal local alignment starts at M [i + 1] [j + 1].
• The optimum score is given by the equation:
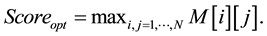
2.2. Application Example
Here are two sequences to be compared:
t: CGGGTATC
s: CCCTAGGT
Figure 1 shows the values in the matrix of scores at the end of the execution of the Smith Waterman algorithm:
Once all the cells of the matrix scores are calculated to find the best local alignment, we start with the cell where the maximum score has been identified, then back to the cell that was used to determine the score and so on. And the optimal local alignment in the Smith-Waterman algorithm is:
C G G G --A T
---C T A G G T
2.3. Highlighting Parallelizable Calculations
As shown in Figure 2, calculations are done in parallel according to the anti-diagonal.
At time T1, a single cell is calculated, at time T2, two cells are calculated, at time T3, three cells are calculated, etc.
Generally, the cell M(i,j) is computed at time Tij = i + j ? 1.
2.4. Linear Representation of Cells
The number of cells calculated at each iteration Ti is given as above.
T1 at first, then T2 and T3, and so on. We obtain the representation in Figure 3.
![]()
Figure 1. Example of sequence alignment.
![]()
Figure 2. Cases calculable at the same time Ti.
![]()
Figure 3. Linear representation of the parallelizable boxes.
This representation of the scoring matrix clearly shows the tasks that can be performed simultaneously.
2.5. Evolution of the Number of Cells at the Same Step
Suppose we have two sequences of identical size N. The number of computable Nbi cells at the same time Ti is given by the Formula (2).
![]() (2)
(2)
We assume Nb the sum of Nbi. Formula (3) permits to verify that the new approach takes into account the N2 cells of the scoring matrix.
![]() (3)
(3)
Thus without taking into account the dependancies between the cells during the computation, the number of iterations to calculate the N2 cells is 2N − 1.
So, if ![]() represents the average number of cells computable per passage we have in (4),
represents the average number of cells computable per passage we have in (4), ![]()
![]() (4)
(4)
3. Proposed Models and Materials
To evaluate the matrix scoring, most of the existing approaches use directly the matrix in Figure 2 through a double iteration on the rows and columns.
These approaches have the merit of simplicity. However, given the dependencies between the cells, the value of a cell may not be calculated during the first passage so that the matrix scoring requires more than ![]() iterations.
iterations.
To remedy this situation, we use a approach that consists of a transformation of the initial matrix [7] -[10] , which doesn’t change its essential properties but rather optimizes the calculation order of cells.
3.1. Transformation of the Matrix of Scores
M represents the matrix of scores, i the line number and j the column number.
We define an application ![]() as follows:
as follows:
For each cell referenced (i, j):
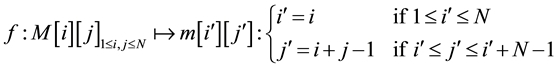
 is an bijective application. It transforms the matrix in Figure 2 to the matrix in Figure 3.
is an bijective application. It transforms the matrix in Figure 2 to the matrix in Figure 3.
In fact  is a change of reference from (i, j) ® (i, i + j − 1).
is a change of reference from (i, j) ® (i, i + j − 1).
The function f represents a rotation of the scoring matrix cells which transforms antidiagonals to columns. However, we must keep in mind the treatments which must be applied on sequences to be aligned.
3.2. Dependency of the Calculations in the New Representation
In the original representation, M [i] [j] depends on the values of cells M [i] [j − 1], M [i − 1] [j] and M [i − 1] [j − 1], as shown in Figure 4.
In the new representation, computing m [i] [j] depends on m [i-1] [j − 2], m [i − 1] [j − 1] and m [i] [j − 1] as shown in Figure 5.
The change of representation permits to calculate values of cells on the same column. So, the genomic sequence located on the column reference changes as we have shown in Figure 5. It follows a refitting of the value S [s [i]] [t [j]] used in the research of the value of m [i] [j] which becomes S [s [i’]] [t [j’ − i’]] for m [i’] [j’].
3.3. Reconstitution of the Solution
Once all the values of the matrix scores are calculated, we must produce the results. The score is obtained in the same way as in the original form, i.e. the maximum value of cells, but acids are obtained using function .
.

3.4. Materials
3.4.1. Dataset
To examine the acceleration rate, we use the Smith-Waterman algorithm on the alignment of genomic sequences. This algorithm has been subject of several parallel implementations [2] [11] . For illustration, we will consider the computation of cell values of the dynamic matrix used in this algorithm. The sequences we use have been downloaded from the existing genomic databases. The substitution matrix used is BLOSUM [62] with penalty-2.
3.4.2. Specifications of the Sequential Computer Used
It has the following features:
Processor: Pentium (R) Dual-Core CPU E5500 @ 2.80 Ghz 2.80 Ghz;

Figure 4. Calculating M[i][j] dependency.

Figure 5. New dependencies computing m[i][j].
Ram: 3.00 Go HD: 500 Go
Operating System: Ubuntu 11.10.
C compiler used: gcc version 4.4.6.
3.4.3. GP-GPU Specifications
The graphic’s card used is a NVIDIA GeForce GTX 670, which consists of seven multiprocessors equivalent to 1344 CUDA cores, clocked at 1.4 GHz, two (2) gigabytes of memory shared between cores hearts, 64 KB constant memory and 64 KB of shared memory per CPU.
4. Experiments and Results
4.1. Classical Approach versus Our Approach in Sequential Mode
In Figure 6, we have compared the sequential computation time of the matrix’s scoring in its initial representation and the new reorganization. We note that the new representation has almost the same performance as the initial representation for sequences of length less to fourteen thousand (14,000) nucleotides. This experiment aims to show that the two representations of the matrix’s scoring are equivalent, sequentially in the range of sequences that we study: no spare time. This enables us to guarantee for the continuation of the study that considering the new representation of the matrix’s coring did not induce additional execution time. For the rest, we will use the same interval of sequences.
Note that, we have been limited in trying to go beyond this size of sequence due to our calculation capabilities.
4.2. OpenMP Implementation of Our Method
OpenMP is based on the principle of shared memory [12] -[15] . The computation to be performed is decomposed into multiple tasks. Tasks are performed by the available computational units. The treatment to be performed, and data variables can be stored in a location accessible to all processing units. Shared variables are declared in the Shared() option while thread-specific variables are in the Private() optional list().
Experiments were performed on DNA sequences of various lengths using OpenMP. As we shall see, the optimum dosages of the block size depend mainly on the size N of the sequences to align.
We propose two opportunities for parallelization of the calculation of the values of cells in the matrix scoring by the Smith-Waterman algorithm.
The scoring matrix is reorganized: all cells in a column can be calculated simultaneously. Thus at each iteration, the cells of a single column are calculated. Each thread will calculate elements of one or more cells.
A thread can read the updated content elements from another thread to calculate its own elements.

Figure 6. Original version versus the new version.
This solution has the advantage of providing the list of computable elements in an iteration but has the disadvantage of combining expectations.
It should also be noted that at this level, it is possible to calculate in multiple loops. At each loop,  threads are created.
threads are created.
As there’s no extra time outside access for reading or writing to the different cells of the matrix, the runtime in both cases are the same. So, we will treat one case.
4.2.1. Mathematical Modeling of the OpenMP Runtime
We assume:
N: length of the sequences to be aligned;
 : time performance of each iteration (i, j). It is also the time to treat; the value of a cell of the matrix;
: time performance of each iteration (i, j). It is also the time to treat; the value of a cell of the matrix;
 : initialization time before starting calculations;
: initialization time before starting calculations;
 : latency time: wait time for all threads to finish their tasks in iterated;
: latency time: wait time for all threads to finish their tasks in iterated;
E(x): denotes the integer portion of x.
From these assumptions:
The sequential computational time of the cell values of the scoring matrix is given in (5).
 (5)
(5)
As we shall see, the optimum dosages of the block size depend mainly on the size of the sequences to align N. So that, we propose two (2) opportunities for the parallelization of calculation of the scoring matrix cells using the Smith-Waterman algorithm.
During the kth executing of the loop,  , there are exactly k cells to calculate.
, there are exactly k cells to calculate.
If  there are N-(k-N) (or 2N-k) cells to calculate. At each iteration, each thread is responsible for
there are N-(k-N) (or 2N-k) cells to calculate. At each iteration, each thread is responsible for
computing  cells (first phase) and then
cells (first phase) and then .
.
We deduce T the total time calculation using OpenMP as follows:

Hence T is given in Formula (6)
 (6)
(6)
4.2.2. Determining the Optimum Value of nb-Threads

Differentiating the expression of T relative to nb-threads, we obtain:


No value of  cancels the derivative.
cancels the derivative.
4.2.3. Estimation of the Theoretical Acceleration
Acceleration is calculated in Formula (7).
 ;
;
 (7)
(7)
4.2.4. Measured Accelerations
We distinguish two cases:
Case 1: one calculation for each thread per iteration
As shown in Figure 7, the parallelization with OpenMP does not significantly affect system performance. For sequences of 2500 and 5000 nuclides, peak is reached with two threads and acceleration is 1.5. Sequences of 10,000 nuclides, optimum acceleration is 1.5 and is obtained with 8 threads. The sequence of 14,000 nuclides gives maximum acceleration of 1.13 with 2 threads. In summary, with OpenMP, the best results are obtained with two threads.
Case 2: several calculations for each thread by iteration
We also tested if a thread performs a set of calculations rather than one. The results we have in this implementation are very similar to those obtained in the previous implementation. There is a very sleazy performance improvement of the order of a few hundredths of a second in some cases. Figure 8 recapitulates the results obtained for two (2) threads by varying the cell size to calculate each thread, per iteration.
4.3. GP-GPU Implementation of Our Method
Initial form of the Smith Waterman algorithm has many implementation on GP-GPU as in [16] -[20] . To perform the calculation on GP-GPU, the scoring matrix is represented in vector form. Each ring launched calculates the elements of a column of the new representation. These elements are identified from the parameters (i, j). In total 2N iterations are launched.
4.3.1. Mathematical Modeling of the GP-GPU’s Runtime
A GP-GPU implementation starts on a CPU then uses a kernel (program running on GP-GPU). So there is cooperation between the CPU and the GPU cores. Communications between GPU and CPU are simulated out through the GP-GPU memory. The CPU copies data to be used by the GP-GPU there and CPU also reads the contents of the GP-GPU memory for reuse in sequential calculations or simply confirm.

Figure 7. Performance based on the number of threads.

Figure 8. Performance with two threads, varying chunk size per iteration.
The Kernel
We assume:
 : kernel initialization time;
: kernel initialization time;
NB: number of blocks per multiprocessor;
NWP: number of warps;
NTW: number tasks per warp;
 : iteration’s execution time per warp;
: iteration’s execution time per warp;
The theoretical time of calculating the scoring matrix is given in (8)
 (8)
(8)
To speed up the calculations, we transferred the data in memory GP-GPU and selected a grid of 512 ´ 512 ´ 512. This setting is sufficient for processing sequences that we handle.
4.3.2. Performance on GP-GPU
Figure 9 presents the results obtained with GP-GPU.
We note that the acceleration increases with the size of the sequences examined.
4.4. Comparison of the Implementations on OpenMP and GP-GPU
Figure 10 represents the results obtained.
For OpenMP we examine three (3) cases. The first case uses two (2) threads.
The second uses also two (2) threads with a chunk of fifty (50) and the last one two (2) threads with a chunk of two hundred and fifty (250).
The best case is the second one. We notice also that beyond 14,000 acids per sequence, the three cases have equivalent results.
The implementation on GP-GPU gives better acceleration compared to OpenMP. For sequences used, the performance is improved more than 25 times.
5. Conclusions
In this paper, we present a method based on the rotation of the scores matrix in order to improve the implementation of the Smith Waterman algorithm.
This transformation explicits the parallelism contained in this algorithm and facilitates its exploitation across different platforms of parallelization.
We validate the application of this method with OpenMP and Cuda C. For each representation, we also measure the performance while executing the loop of the Smith-Waterman algorithm. It appears that the number

Figure 9. Performance on GP-GPU.

Figure 10. Performance based on the number of threads.
of threads used with OpenMP increases performance but depends on the size of the sequences to be compared. Similarly, on GP-GPU the choice of grid dimensions is an essential element of improving performance. We note a little performance with OpenMP and performance increases with the size of the sequences on the GP-GPU. At the end, GP-GPU improves performance of computing the Smith-Waterman algorithm. For the sequences used, the performance is improved more than 25 times compared with OpenMP. Ultimately, this study allows the following conclusions:
• Expanding the use of GP-GPU to parallel computing in addition to graphics for which they are at the basis created. The relatively low cost of GP-GPU will make parallel computing more accessible to the public.
• In the case of the Smith-Waterman algorithm, we conclude that the GP-GPU accelerates it more than OpenMP.
• In general, it is recommended to use GP-GPU than OpenMP for massively parallel and long calculations.
• We propose a mathematical modeling of time calculating of the matrix’s scoring on the OpenMP and GP-GPU. This equation setting allows us to make wise choices in the number of thread (OpenMP) and the size of the grid computing (GP-GPUs).
References
- Smith, T.F. and Waterman, M.S. (1981) Identification of Common Molecular Subsequences. Journal of Molecular Biology, 147, 195-197. http://dx.doi.org/10.1016/0022-2836(81)90087-5
- Boukerche, A., Melo, A.C.M.A., Ayala-Rincon, M. and Santana, T.M. (2005) Parallel Smith-Waterman Algorithm for Local Dna Comparison in a Cluster of Workstations. Experimental and Efficient Algorithms, 3503, 464-475. www.springerlink.com/content/xwn2q2qfm4hgvr3t
- Nguyen, V.-H. and Lavenier, D. (2009) PLAST: Parallel Local Alignment Search Tool for Database Comparison. BMC Bioinformatics, 10, 329.
- Needleman, S.B. and Wunsch, C.D. (1970) A General Method Applicable to the Search for Similarities in the Amino Acid Sequence of Two Proteins. Journal of Molecular Biology, 48, 443-453. http://dx.doi.org/10.1016/0022-2836(70)90057-4
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic Local Alignment Search Tool. Journal of Molecular Biology, 215, 403-410.
- Pearson, W.R. and Lipman, D.J. (1988) Improved Tools for Biological Sequence Comparison. Proceedings of the National Academy of Sciences of the United States of America, 85, 2444-2448. http://dx.doi.org/10.1073/pnas.85.8.2444
- Aluru, S., Futamura, N. and Mehrotra, K. (2003) Parallel Biological Sequence Comparison Using Prefix Computations. Journal of Parallel and Distributed Computing, 63, 264-272.
- Edmiston, E.W., Core, N.G., Saltz, J.H. and Smith, R.M. (1988) Parallel Processing of Biological Sequence Comparison Algorithms. International Journal of Parallel Programming, 17, 259-275. http://dx.doi.org/10.1007/BF02427852
- Rajko, S. and Aluru, S. (2004) Space and Time Optimal Parallel Sequence Alignments. IEEE Transactions on Parallel Distributed Systems, 15, 1070-1081. http://dx.doi.org/10.1109/TPDS.2004.86
- Sarje, A. and Aluru, S. (2009) Parallel Genomic Alignments on the Cell Broadband Engine. IEEE Transactions on Parallel and Distributed Systems, 20, 1600-1610.
- Lander, E., Mesirov, J.P. and Taylor, W. (1988) Protein Sequence Comparison on a Data Parallel Computer. Proceedings of the International Conference on Parallel Processing, 3, 257-263.
- Eigenmann, R. and Voss, M. (2001) OpenMP Shared Memory Parallel Programming. Lecture Notes in Computer Sci- ence 2104. Springer-Verlag, Heidelberg.
- Ferrer, M.M.R., Gajinov, V., Unsal, O.S., Cristal, A., Ayguad, E. and Valero, M. (2008) Nebelung: Execution Envi- ronment for Transactional OpenMP. International Journal of Parallel Programming, 36, 326-346.
- Chapman, B., Jost, G. and van der Pas, R. (2008) Using OpenMP Portable Shared Memory Parallel Programming. The MIT Press, Cambridge.
- Gonzalez, M., Ayguad, E., Martorell, X. and Labarta, J. (2001) Defining and Supporting Pipelined Executions in OpenMP. Proceedings of the 2nd International Workshop on OpenMP Applications and Tools, Lafayette, IN, 30-31 July 2001, 155-169. http://dx.doi.org/10.1007/3-540-44587-0_14
- Ligowski, L. and Rudnicki, W. (2009) An Efficient Implementation of Smith Waterman Algorithm on GPU Using CUDA, for Massively Parallel Scanning of Sequence Databases. Proceedings of the 2009 IEEE International Symposium on Parallel & Distributed Processing, Rome, 23-29 May 2009, 1-8. http://dx.doi.org/10.1109/IPDPS.2009.5160931
- Liu, Y., Huang, W., Johnson, J. and Vaidya, S. (2006) GPU Accelerated Smith-Waterman. Proceedings of the International Conference on Computational Science, Reading, UK, 28-31 May 2006, 188-195. http://dx.doi.org/10.1007/11758549_29
- Liu, Y., Schmidt, B. and Maskell, D.L. (2009) MSA-CUDA: Multiple Sequence Alignment on Graphics Processing Units with CUDA. Proceedings of the 20th IEEE International Conference on Application-Specific Systems, Architectures and Processors, Boston, 7-9 July 2009, 121-128.
- Voss, G., Muller-Wittig, W. and Schmidt, B. (2005) Using Graphics Hardware to Accelerate Biological Sequence Database Scanning. Proceedings of the TENCON 2005―2005 IEEE Region 10 Conference, Melbourne, 21-24 November 2005, 1-6.
- Liu, W.G., Schmidt, B., Voss, G., Schroder, A. and Muller-Wittig, W. (2006) Bio-Sequence Database Scanning on a GPU. Proceedings of the 20th International Parallel and Distributed Processing Symposium, Rhodes Island, 25-29 April 2006, 8.
NOTES

1Basic Local Alignment Search Tool.
2FAST-ALL.
3A Blosum (BLOcks SUbstitution Matrix) matrix is a substitution matrix for sequence alignment of proteins. It is used to score alignments between evolutionarily divergent protein sequences.