Open Journal of Pediatrics
Vol. 2 No. 2 (2012) , Article ID: 19870 , 4 pages DOI:10.4236/ojped.2012.22026
Fatal hemophagocytic lymphohistiocytosis presenting as Reye’s syndrome: Report of two cases
![]()
Department of Pediatrics, Wakayama Medical University, Kimiidera Wakayama City, Japan
Email: *nami@mail.wakayama-med.ac.jp
Received 6 February 2012; revised 6 April 2012; accepted 26 April 2012
Keywords: Hemophagocytic Lymphohistiocytosis; Reye’s Syndrome; Cytokine; Mitochondria; Encephalopathy
ABSTRACT
We report two cases of rapidly fatal hemophagocytic lymphohistiocytosis (HLH) that presented as Reye’s syndrome (RS). The patients were referred to our hospital because of altered level of consciousness with hyperammonemia and hypoglycemia. The first patient, a 24-month-old girl (Case 1), died soon after arrival and was clinically diagnosed as having RS, but a diagnosis of HLH was established on the basis of autopsy findings. The other patient, a 4-month-old boy (Case 2), was diagnosed as having HLH in view of the bone marrow findings on admission, but immunosuppressive therapy failed to prevent a rapidly fatal course. Marked hypercytokinemia was found in both patients, and liver pathology demonstrated panlobular microvesicular steatosis of hepatocytes, resembling that in RS. Mononuclear cell infiltration with hemophagocytosis in the liver was evident in case 2, but not in case 1. In both cases, hypercytokinemia was thought to have caused mitochondrial dysfunction, resulting in RS-like microvesicular steatosis of hepatocytes. HLH is an important differential diagnosis of children presenting with RS-like picture.
1. INTRODUCTION
Reye’s syndrome (RS) is a rare non-inflammatory encephalopathy associated with hepatic dysfunction and fatty infiltration of the viscera, principally affecting children [1]. A proposed criteria for RS by the Centers for Disease Control are as follows; an acute non-inflammatory encephalopathy, characteristic liver histology or raised serum transaminases or ammonia values, and no other explanation for illness [2]. The precise pathogenesis of RS has not been completely elucidated, but mitochondrial dysfunction and consequent metabolic changes explain its clinical features [3]. Although the incidence of RS has decreased significantly because of reduced use of salicylates in children, RS is an important differential diagnosis in children with encephalopathy because it requires rapid and intensive therapy for full recovery [4]. Also, physicians must pay particular attention to any underlying congenital metabolic disorders, infectious diseases or toxic conditions that might present as RS [2,5].
Hemophagocytic lymphohistiocytosis (HLH) is a lifethreatening condition characterized by uncontrolled activation of T-cells and macrophages resulting in hypercytokinemia. The cardinal symptoms are prolonged fever, hepatosplenomegaly and central nervous system symptoms, and a diagnostic guideline of HLH was proposed by Histiocyte Society [6]. The eight criteria in the 2004 guideline are as follows; 1) fever; 2) splenomegaly; 3) cytopenias affecting at least two of three lineages in the peripheral blood; 4) hypertriglyceridemia and/or hypofibrinogenemia; 5) hemophagocytosis in bone marrow, spleen or lymphnodes; 6) low or absent NK-cell activity; 7) hyperferritinemia; and 8) high levels of soluble interleukin-2 receptor (sIL2R) [7]. Altogether five of the eight criteria must be fulfilled except for patients with a molecular diagnosis consistent with HLH. Clinical features of HLH can largely be explained by high serum concentrations of inflammatory cytokines such as tumor necrosis factor alpha (TNF-α). Although involvement of TNF-α in the pathogenesis of RS has been reported [8], only one case with an underlying relationship between RS and HLH has been reported previously [9].
Here we report two fatal cases of HLH that presented as RS, including details of the serum cytokines levels and findings of liver pathology.
2. CASE REPORT
Case 1. A 24-month-old girl with normal development for age was referred to our hospital because of severe encephalopathy. The patient’s medical and family history was unremarkable. She had become febrile two days earlier, and had been diagnosed as having viral gastroenteritis because of vomiting and watery diarrhea. Acetaminophen and the anti-emetic agent metoclopramide had been administered. On the third day of illness, she had developed recurrent episodes of generalized convulsions with altered level of consciousness and was admitted to a nearby hospital, but was soon transferred to our hospital for intensive care. On arrival, her consciousness level was Glasgow Coma Scale E1V1M1. Anisocoria was evident, and the papillary light reflex was sluggish. She had ataxic respiration and generalized cyanosis. Severe dehydration was suggested by reduced skin turgor, dry oral cavity and prolonged capillary refilling time. No hepatosplenomegaly and icterus were noticed. She was clinically diagnosed as having RS on the basis of the clinical and laboratory findings (Table 1). Soon after arrival, she developed sudden ventricular tachycardia and died before any therapy could be started. Rotavirus and adenovirus antigen in stools were negative, as were throat, stool and blood cultures for pathogen. Analysis of serum amino acids and urine organic acids did not show any specific metabolic diseases. Autopsy performed two hours after death, revealed prominent hemophagocytosis in the bone marrow, small and large intestine, and mesenteric lymph nodes (Figure 1(A)). The weight of the brain was elevated at 1400 g, reflecting edema, but there was no evident meningeal inflammation or infiltration with inflammatory cells. There were also no vascular lesions suggestive of influenza encephalopathy [10]. Liver pathology revealed panlobular microvesicular lipid droplets in the hepatocytes, but no infiltration with mononuclear cells and hemophagocytosis in serial sections (Figure 2). Because the patient fulfilled 5 of the 8 diagnostic criteria for HLH: fever, hemophagocytosis, elevated levels of serum ferritin (14,778 ng/ml) and sIL2R (2618 IU/ml) and decreased level of plasma fibrinogen (30 mg/dl), a diagnosis of HLH was established. Neither genetic analyses of primary HLH nor electron microscopy studies of the liver were conducted.
Case 2. A 4-month-old boy was referred to our hospital because of 9 days of fever, altered level of consciousness and hepatic derangement. The patient’s medical and family history was unremarkable. He was admitted to a nearby hospital on the fourth day of illness. Although no focus of infection was evident, cefpirome sulphate was administered intravenously because of elevated levels of serum C-reactive protein at 5 mg/dl and leucocytosis. As fever persisted and in view of red lips and mild liver derangement, aspirin (30 mg/kg/day) was started on the 7th day of illness with the suspicion of Kawasaki disease, but with no response. On the 8th day of illness, altered level of consciousness was noticed. On admission to our hospital on the 9th day of illness, RS was suspected because of hyperammonemia, hypoglycemia (Table 1), and also a history of aspirin use. The patient appeared pale and his consciousness level was Glasgow Coma Scale E2V1M4. A computed tomography scan of the head
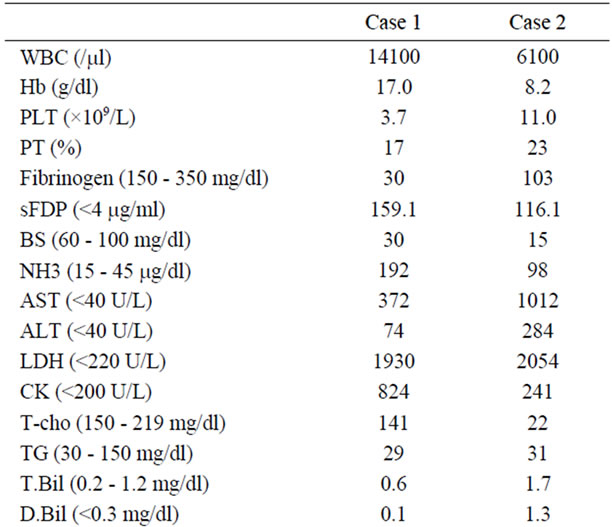

Table 1. Laboratory and cytokine data.
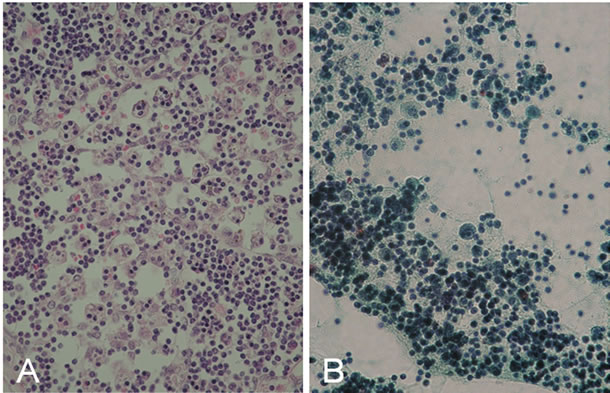
Figure 1. Hemophagocytic features in two cases. (A) Prominent hemophagocytic features in mesenteric lymph node in Case 1. (B) Papanicolou stain of ascites in Case 2. Numerous mononuclear cells with hemophagocytosis were seen.
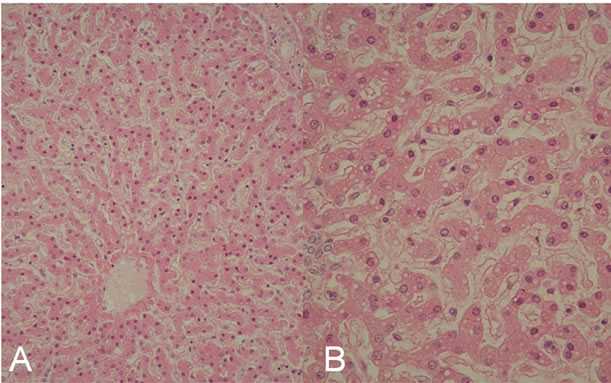
Figure 2. The liver pathology in Case 1. Microvesicular steatosis of hepatocytes was seen. (A) ×100; (B) ×400.

Figure 3. The liver pathology in Case 2. (A) Microvesicular steatosis of hepatocytes similar to Case 1 was seen ×400. (B) Lymphohistiocytic cells with hemophagocytosis in portal areas was seen ×400.
demonstrated no apparent brain edema. The abdomen was distended due to ascites, and moderate hepatosplenomegaly was also noticed at 3 cm and 1 cm below right and left costal margins, respectively. Because of mild pancytopenia and hepatosplenomegaly, a bone marrow examination was performed, and this revealed apparent hemophagocytosis. Because the patient fulfilled 6 of the 8 diagnostic criteria for HLH: fever, splenomegaly, hemophagocytosis in bone marrow, elevated levels of serum ferritin (62,426 ng/ml) and sIL2R (2618 IU/ml), and decreased level of plasma fibrinogen (103 mg/dl), a diagnosis of HLH was established. Methylprednisolone pulse therapy was immediately started in addition to glucose and mannitol infusion, but his general condition deteriorated rapidly. Intractable hypoglycemia and hyperanmonemia persisted with a maximum blood ammonia level of 199 μg/dl. Mechanical ventilation and vigorous circulatory support with inotropic agents were also started, but the patient died 36 hours after admission. Cerebrospinal fluid examination could not be performed. Antibody tests for Epstein-Barr virus, cytomegalovirus and human herpes virus type-1 were all negative. Adenovirus was not detected in throat swabs and stool samples. A stool test for rotavirus antigen was also negative. No pathogen was detected in blood, stool, urine and throat cultures. Postmortem needle liver biopsy showed not only panlobular microvesicular steatosis in the hepatocytes, but also mild mononuclear cell infiltration with hemophagocytosis into portal areas (Figure 3). Numerous mononuclear cells with hemophagocytosis were present in the ascites (Figure 1(B)). Neither genetic analyses of primary HLH nor electron microscopy studies of the liver were conducted.
3. DISCUSSION
Our two reported patients both presented with suspected RS because they exhibited acute consciousness disturbance and liver derangement with hyperammonemia and hypoglycemia. However, as hemophagocytic features and elevated levels of serum ferritin and sIL2R were found, and they were finally diagnosed as having HLH. Inherited metabolic disorders, infections and toxic conditions have been reported to present with a clinical and pathological picture resembling RS, but HLH has not been listed as a differential diagnosis of RS [2,5]. The present cases underline the need to include HLH in the differential diagnosis of patients with encephalopathy suspected to have RS on the basis of the serum levels of ferritin and sIL2R, and hemophagocytic features in the bone marrow, regardless of the peripheral full blood count.
Panlobular microvesicular steatosis has been reported to be a histological hallmark of RS [1], although it is not specific to RS [2]. Interestingly, the liver pathology in both patients showed microvesicular steatosis similar to that in RS. Microvesicular steatosis is a result of mitochondrial dysfunction, and in RS, mitochondrial failure is thought to result from inhibition of oxidative phosphorylation and fatty acid β oxidation of long-chain fatty acids [4,11,12]. Although the precise pathogenesis of mitochondrial dysfunction in RS has not been completely characterized, cytokines such as TNF-α have been reportedly involved [3,13]. In the present cases, severe hypercytokinemia including TNF-α was also evident, and it could have caused RS-like mitochondrial dysfunction resulting in microvesicular steatosis of hepatocytes.
HLH often shows hepatic involvement. Hasegawa et al. reported that the level of serum Fas ligand was elevated in HLH, and speculated that the Fas-Fas ligand system was involved in the hepatic derangement [14]. Pathologically, chronic hepatitis-like mononuclear cell infiltration with hemophagocytic features has been reported to be characteristic of HLH, and the severity of mononuclear cell infiltration is reportedly correlated with the hepatic derangement [15,16]. Although hepatic steatosis has been described previously [17,18], RS-like microvesicular steatosis has not been emphasized. Our reported two cases both exhibited RS-like hepatic derangement and microvesicular steatosis of hepatocytes. Our experience suggests that RS-like hepatic dysfunction might be one of the pathogenesis of hepatic injury in HLH.
The brain pathology in Case 1 was non-specific, except for RS-like brain swelling. Hypercytokinemia has been reported to be an important etiological factor for encephalopathy, especially that associated with influenza virus infection [10]. Ioi et al. reported a case of rotavirus infection complicated by RS and accompanied by hypercytokinemia [19]. Our two patients also exhibited prominent hypercytokinemia, which was assumed to be related to their encephalopathy, although we were unable to examine the cerebrospinal fluid and brain pathology in Case 2.
In conclusion, HLH should be included in the differential diagnosis of children presenting with RS-like picture.
REFERENCES
- Reye, R.D., Morgan, G. and Baral, J. (1963) Encephalopathy and fatty degeneration of the viscera. A disease entity in childhood. Lancet, 2, 749-752. doi:10.1016/S0140-6736(63)90554-3
- Casteels-Van, D.M., Van Geet, C., Wouters, C. and Eggermont, E. (2000) Reye syndrome revisited: A descriptive term covering a group of heterogeneous disorders. European Journal of Pediatrics, 159, 641-648. doi:10.1007/PL00008399
- Glasgow, J.F.T. and Middleton, B. (2001) Reye syndrome-insights on causation and prognosis. Archives of Disease in Childhood, 85, 351-353. doi:10.1136/adc.85.5.351
- Hardie, R.M., Newton, L.H., Bruce, J.C., Glasgow, J.F.T., Mowat, A.P., Stephenson, J.B.P. and Hall, S.M. (1996) The changing clinical pattern of Reye’s syndrome 1982- 1990. Archives of Disease in Childhood, 74, 400-405. doi:10.1136/adc.74.5.400
- Pugliese, A., Beltramo, T. and Torre, D. (2008) Reye’s and Reye’s-like syndromes. Cell Biochemistry & Function, 26, 741-746. doi:10.1002/cbf.1465
- Gupta, S. and Weitzman, S. (2010) Primary and seconddary hemophagocytic lymphohistiocytosis: Clinical features, pathogenesis and therapy. Expert Review of Clinical Immunology, 6, 137-154. doi:10.1586/eci.09.58
- Henter, J.I., Horne, A., Arico, M., Egeler, R.M., Filipovich, A.H., Imashuku, S., Ladisch, S., McClain, K., Webb, D., Winiarski, J., Janka, G., for the Histiocyte Society (2007) HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer, 48, 124-131. doi:10.1002/pbc.21039
- Odeh, M. (1995) A possible role for antibodies to tumor necrosis factor α and to endotoxin in the treatment of Reye’s syndrome. Gut, 37, 441-443. doi:10.1136/gut.37.3.441
- Caldarella, A., Gori, F., Bartoloni Saint-Omer, F. and Sarti, A. (1994) Reye’s syndrome and hemophagocytic histiocytosis in a 16-month-old boy: Is there a possible common etiopathogenetic basis? Pathologica, 86, 329-332.
- Togashi, T., Matsuzono, Y., Narita, M. and Morishima, T. (2004) Influenza-associated acute encephalopathy in Japanese children in 1994-2002. Virus Research, 103, 75-78. doi:10.1016/j.virusres.2004.02.016
- Jaeschke, H., Gores, G.J., Cederbaum, A.I., Hinson, J.A., Pessayre, D. and Lemasters, J.J. (2002) Mechanisms of Hepatotoxicity. Toxicological Sciences, 65, 166-176. doi:10.1093/toxsci/65.2.166
- Trost, L.C. and Lemasters, J.J. (1996) The mitochondrial permeability transition: A new pathophysiological mechanism for Reye’s syndrome and toxic liver injury. Journal of Pharmacology and Experimental, 278, 1000-1005.
- Corkey, B.E., Geschwind, J.F., Deeney, J.T., Hale, D.E., Douglas, S.D. and Kilpatrick, L. (1991) Ca2++ response to interleukin 1 and tumor necrosis factor in cultured human skin fibroblasts. Journal of Clinical Investigation, 87, 778-786. doi:10.1172/JCI115081
- Hasegawa, D., Kojima, S., Tatsumi, E., Hayakawa, A., Kosaka, Y., Nakamura, H., Sako, M., Osugi, Y., Nagata, S. and Sano, K. (1998) Elevation of the serum Fas ligand in patients with hemophagocytic syndrome and DiamondBlackfan anemia. Blood, 91, 2793-2799.
- De Kerguence, C., Hillaire, S., Molinié, V., Gardin, C., Degott, C., Erlinger, S. and Valla, D. (2001) Hepatic manifestations of hemophagocytic syndrome: A study of 30 cases. American Journal of Gastroenterology, 96, 852-857.
- Chen, J.H., Fleming, M., Pinkus, G., Pinkus, J.L., Nichols, K.E., Mo, J.Q. and Perez-Atayde, A.R. (2010) Pathology of the liver in familial hemophagocytic lymphohistiocytosis. American Journal of Surgical Pathology, 34, 852-867. doi:10.1097/PAS.0b013e3181dbbb17
- Tsui, W.M.S., Wong, K.F. and Tse, C.C.H. (1992) Liver changes in reactive haemophagocytic syndrome. Liver, 12, 363-367. doi:10.1111/j.1600-0676.1992.tb00590.x
- Jaffe, R. (2004) Liver involvement in the histiocytic disorders of childhood. Pediatric and Development Pathology, 7, 214-225. doi:10.1007/s10024-003-9876-z
- Ioi, H., Kawashima, H., Nishimata, S., Watanabe, Y., Yamanaka, G., Kashiwagi, Y., Yamada, N., Tsuyuki, K., Takekuma, K., Hoshika, A. and Kage, M. (2006) A case of Reye syndrome with rotavirus infection accompanied with high cytokines. Journal of Infection, 52, e124-e128. doi:10.1016/j.jinf.2005.07.028
NOTES
*Corresponding author.