Journal of Cancer Therapy
Vol.2 No.3(2011), Article ID:6661,10 pages DOI:10.4236/jct.2011.23049
Retinoblastoma as an Epigenetic Disease: A Proposal
![]()
Department of Biomedical Sciences, University of Siena, Siena, Italy.
Email: mastrangelo@unisi.it
Received May 6th, 2011; revised July 15th, 2011; accepted July 25th, 2011.
Keywords: Retinoblastoma, Epigenetics, Genome Imprinting, DNA Methylation, Histone Acetylation/Deacetylation
ABSTRACT
The aim of the present review is to give new insights into the pathogenesis of retinoblastoma, by applying the principles of Epigenetics to the analysis of clinical, epidemiological, and biological data concerning the disease. As an emerging new scientific approach linking the genome to the environment, Epigenetics, as applied to the interpretation of clinical, epidemiological and biological data in retinoblastoma, can not only explain the inconsistencies of the mutational (“two hit”) model, but also opens new outstanding scenarios in the fields of diagnosis, treatment and prevention of this eye tumour. This review is both a collection of literature data arguing against the role of the mutational (“two hit”) model in the genesis of retinoblastoma, and a documented evaluation of how the Epigenetic, rather than the genetic model fit the variegated phenotypic expression of the disease. The epigenetic model in the genesis of retinoblastoma, proposed herein, emphasizes the role of environment and the interaction of the environment with the genome, in generating retinoblastoma in young children. Environmental toxicants, including radiations, wrong diets, and infectious diseases, among others, all play a major role in conditioning the degree of DNA methylation in embryos and foetuses during pregnancy, thus leading to stable, functional alterations of the genome, which, on the other hand, can be also transmitted from generation to generation, thus mimicking a hereditary disease. An accurate analysis of the currently available literature on both retinoblastoma and Epigenetics, coupled with the knowledge of the variegated phenotypic expression of the disease, can easily lead to the conclusion that retinoblastoma is an epigenetic, rather than a genetic disease.
1. Introduction
Although rare, retinoblastoma is the most common eye tumour affecting children under the age of five years [1]. In 1971, Alfred Knudson [2], by reviewing a series of 48 cases, formulated a hypothesis according to which this tumour may be determined by the loss or inactivation of both copies of a single gene. The presumptive gene responsible for tumour development in retinoblastoma was later identified and indicated as “Rb1”, and its complete DNA sequence fully described and characterized [3]. Since the beginning, retinoblastoma has been considered a hereditary tumour, and this view has been further reinforced by DNA investigations (polymorphism and conformational DNA analysis followed by DNA sequencing) demonstrating that retinal tumours usually bear mutations on both copies of the Rb1 gene [4], thus apparently confirming the mechanisms hypothesized by Knudson in 1971, and allowing the identification a new class of “cancer genes” defined “tumour suppressor genes” [5]. Knudson’s mutational model, maintains that two sequential mutations of the Rb1 gene are necessary to develop a retinoblastoma, and the timing and target of these two mutational events determines the clinical phenotype of the disease. Namely, when both the first and second mutations involve the somatic cells, the individual will develop a tumour affecting only one eye (unilateral retinoblastoma), but when the first mutation occurs in the germinal cells of one parent, and the second involves the individual’s somatic retinal cells, the disease will affect both eyes (bilateral retinoblastoma) [6,7]. This fundamental diversity in the pathogenesis of the tumour represents the basic distinction between two different clinical retinoblastoma phenotypes; e.g.:
1) unilateral retinoblastoma (65% - 70% of all cases), which is sporadic (i.e. non hereditary), occurs at a later age, and usually presents with a single tumour focus on the retina of the affected eye, and;
2) bilateral retinoblastoma (30% - 35% of all cases), which is hereditary, occurs at an earlier age, involves both eyes, and commonly presents with multiple tumour foci in the retina of at least one eye [2].
The theoretical model proposed by Knudson was accepted and used worldwide to explain some of the most important features concerning the different genetic, clinical, and epidemiological aspects of retinoblastoma, but the model itself has been more recently challenged by evidences showing that both aneuploidy and genetic instability play an essential role in the genesis of cancer [8-10]. Nevertheless, neither the mutational nor the aneuploidy model seem to be able to explain the variegated phenotypic expression of retinoblastoma.
We propose herein, that the pathogenesis and clinical expression of retinoblastoma can be better understood and explained if the principles of Epigenetics are applied to the study of this eye tumour affecting young children.
2. Purpose
While the literature concerning the genetic origin of retinoblastoma has flourished in the last four decades, and the idea that this tumour is determined by two sequential mutations of the Rb1 gene still persists among geneticists and ophthalmologists, evidence is cumulating which clearly argues against the role of DNA mutations in cancer in general [8-10] and in retinoblastoma in particular (see below). The most relevant arguments raised against the mutational model are reported in Table 1 together with the related references, and although they have been abundantly discussed elsewhere (see. References 8 - 32, in Table 1), and the role of aneuploidy itself, in the genesis of cancer, is now well established, no clear cut “alternative” has been proposed, so far, which consistently explains how and why retinoblastoma develops in human beings.
The main purpose of the present review is to show that according to the currently available evidence, the concept of “epigenetic” gene regulation offers a totally new and consistent explanation of both aetiology and pathogenesis of retinoblastoma, by taking into consideration the complex gene/environment interactions which account for the variable and variegated phenotypic expression of the disease.
3. What Is Epigenetics
“Epigenetics” is a term coined in 1940 by the developmental biologist Conrad Waddington to define “…the interactions of genes with their environment, which bring the phenotype into being”. Literally, “epi” —genetics means “above genetics” and the term properly designates events which modify gene expression without modifying the structure of the genes themselves .
Although epigenetic regulation of gene expression is the basic mechanism through which billions of specialized cells belonging to an organism differentiate, starting from a single embryonic ancestor and one and the same DNA, the idea that gene expression can be stably modified in the absence of structural alterations of the DNA sequence, has never been taken into serious consideration, in the pathogenesis of cancer.
After Waddington, Hollyday and Plough proposed, among others, the methylation of Cytosine—Guanine (CpG) dinucleotide rich regions of the DNA , as the biochemical basis of epigenetic regulation of gene expression, showing that gene expression can be either totally stopped or increased in total absence of evident or detectable changes (mutations) of the basic DNA structure of the genes.
Other mechanisms of epigenetic gene regulation do exist, such as histone acetylation/deacetylation, and micro RNAs, but a detailed analysis of all the possible mechanisms involved, is beyond the scope of the present review.
However, the discovery that epigenetic (or functional) modulation of gene expression is dependent on the environment, is “stable”, and can be transmitted from one generation to the next, has opened a completely new perspective in the study of the interactions between environment and human genome and will, most probably, ultimately clarify how these interactions lead to the development of many different human diseases, including cancer. This is why one of the most recently reported definitions of “epigenetics” is: “an emerging branch of investigation in cancer research (but also in other fields of clinical pathology) which studies the interactions between environment and genome in determining disease” .
Epigenomics has shown that environmental exposure to nutritional, chemical, and physical factors may stably modify gene expression through, among others, methylation of CpG rich DNA portions, such as the promoter regions of some “housekeeping” genes, transposable elements adjacent to genes with metastable epialleles, and regulatory elements of imprinted genes. In other words, the methylation state of different regions of the genome, determines whether a gene is expressed or not, within a cell.
The purpose of the next paragraphs is to show that the epigenetic control mechanisms of gene expression are active in retinoblastoma and therefore retinoblastoma can be viewed as an epigenetic rather than a “genetic” disease [35,36].
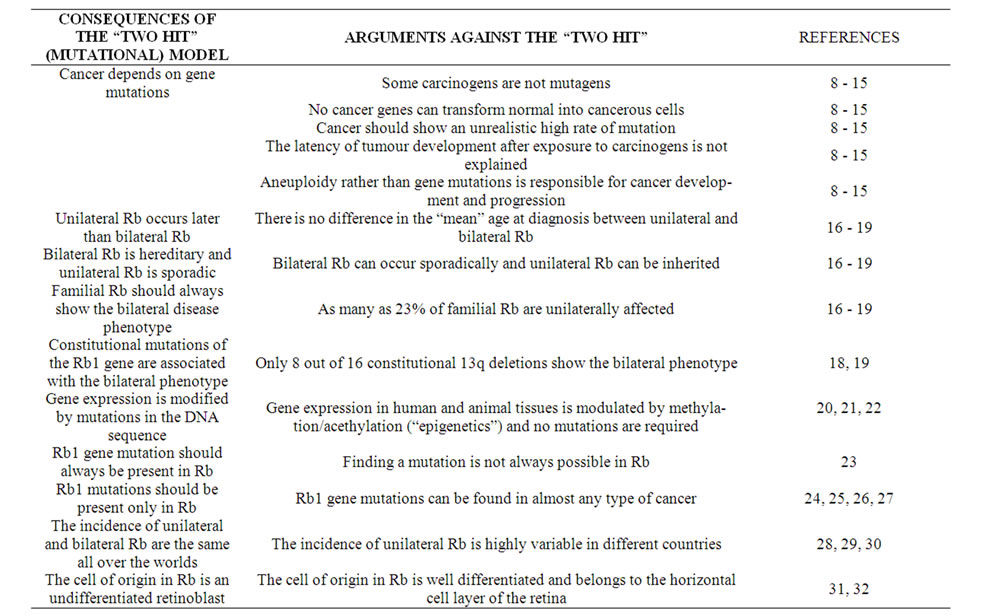
Table 1. Arguments against the mutational (”two hit”) model in the genesis of retinoblastoma (and related references).
4. Retinoblastoma and Methylation of the Promoter Region of Housekeeping Genes
A “Housekeeping” gene is a gene that is expressed at a fairly consistent level throughout the cell cycle and from tissue to tissue, because it is usually involved in routine cellular metabolism, (i.e. basic cell functions which are common to all different cell types). Moreover, gene expression is regulated by a given DNA region called “promoter”, which, therefore, can be defined as a sequence of DNA needed to turn a gene “on” or “off”.
Given their functions, “housekeeping” genes are usually expressed in almost any kind of human cells. The Rb1 gene is one of such genes and its function is to regulate cell growth by preventing cells from dividing too fast or in an uncontrolled way [37]. Its key role in the development of cancer has been highlighted by different Authors [24-27,38,39].
As it has been shown, in vitro methylation of the promoter region of the Rb1 gene, dramatically reduces pRb expression [40] particularly in sporadic retinoblastoma [41] which, on the other hand, is the most commonly accepted form of non hereditary disease.
Moreover, methylation of the promoter regions of housekeeping genes is a common mechanism that contributes to inactivating cell cycle control related genes (Rb1, among others) in the early stages of development of glial tumours [42]. Interestingly, as a key gene in cell cycle control, Rb1 has been found aberrantly methylated, alone or together with other cell cycle regulating genes, in different types of cancer; among others: epithelial odontogenic tumours [43], bladder cancer [44], radon induced rat lung tumors [45], follicular lymphoma [46], SV40 associated B cell lymphoma [47], gastric carcinoma [48], neuroblastoma [49], and pituitary adenoma [50].
Furthermore, aberrant methylation of the Rb1 gene promoter region has been found in 20% of embryos with chromosomal mosaicism that die in utero, thus suggesting that aberrant epigenetic genomic modifications at early stages of human embryonic development, can be one of the mechanisms promoting genomic instability (realized in the form of mosaic abnormalities of the karyotype), that are incompatible with the normal course of embryogenesis [51].
Finally, retinoblastoma frequently shows aberrant methylation of other genes such as HIN-1 [52], HIC-1 [53], Caspase 8 and 10 [54], and RASSF1A [55], all of which are commonly considered key genes in the development of cancer in young children.
Current evidence, therefore, suggests that at least DNA methylation, which is a fundamental mechanism in epigenetic regulation of gene expression, plays a major role in all Rb1-dependent cancers investigated so far, including retinoblastoma.
5. Retinoblastoma, Metastable Epialleles, and Transposable Genetic Elements
Metastable epialleles are defined as gene loci that can be epigenetically modified (i.e. modified by the environment) in a variable and reversible manner, such that a distribution of phenotypes can occur from genetically identical cells.
Currently, only a few genes with metastable epialleles have been identified, but experiments with these genes have produced very interesting results. For example, Jirtle and coll. have shown that in the “Agouti” mice, maternal dietary exposure to phytoestrogen genistein during gestation shifts the coat-colour distribution of viable yellow offspring towards brown, and that the genistein-induced hypermethylation protects the offspring from obesity in adulthood.
Moreover, genistein, when given at a level that is comparable to that consumed by humans with high soy diets, increases DNA methylation even though it is not a methyl-donating compound; the mechanism through which this is accomplished is still unknown.
Taken together, these results suggest the interesting possibility that hypermethylating dietary supplements could reduce the effect of environmental toxicants that cause DNA hypomethylation, thereby protecting the epigenome from their deleterious effects [56].
Furthermore, talking about genes that can be epigenetically modified in a variable and reversible manner (i.e. genes with no structural DNA alterations), it is of interest to note that the phenotypic expression of retinoblastoma is not only highly variable, encompassing clinical entities such as, for example, “retinoma”, which is considered a precancerous lesion [57], but can also be “modulated” as if they would depend on variable environmental exposures. At this regard it must be outlined that the literature refers of cases of spontaneously regressed retinoblastoma [58,59], which underwent a new malignant transformation [60].
Interestingly, the observation of spontaneously regressed retinoblastoma, dates back to 1956 [61], more than a decade before the formulation of the mutational, “two hit” model, and it still represents a theoretical challenge to it. As a matter of fact, the mutational model has no clear cut explanation of how a structurally modified DNA leading to a cancer phenotype may spontaneously revert to normality, although the concept of “penetrance” has been largely abused, for this purpose, by the supporters of the mutational model. “Penetrance” is, in fact, a rather fuzzy concept, which does not correspond to any known biochemical/molecular mechanisms, and is presently viewed as a pure stochastic (but still unexplained!) fluctuation in gene expression [62].
Epigenetics, on the contrary, by looking at gene expression as the result of a functional interaction between genes and the environment (through gene methylation and other mechanisms), acknowledges the possibility that the resulting phenotype could be modulated and consequently exhibit different degrees of variability and “plasticity”.
Variations in phenotypic expression, on the other hand, can be also explained, according to Epigenetics, by the presence of “transposable” elements (“Transposons”) within the genomic DNA.
Transposons are parasitic, repetitive mobile elements dispersed throughout the mammalian genome. They are remnants of ancestral infections which became fixed in the germline DNA, and subsequently increased in copy number [63]. The sequencing of the human genome has shown that Transposons comprise roughly 45% of our genome [64], and most transposable elements are silenced by CpG methylation [65], the same biochemical process involved in epigenetic gene regulation. The epigenetic state of a subset of transposable elements is metastable; in other words, these “mobile” elements are variably expressed in genetically identical individuals due to epigenetic modifications occurred during the early development [66]. In contrast with other regions of the human mammalian genome, the epigenetic changes occurring at the insertion site of transposable elements are a stochastic event which not only causes individual variation, but also accounts for epigenetic cellular mosaicism. Therefore, given their role in silencing genes and their variability, within the same individual, Transposons are responsible of both inter-individual and intra-individual variations in phenotypic expression of the same genes within different cells of the same organism, thus leading to mosaicism.
Retinoblastoma is a cancer showing phenotypic variation, and a number of “variant” phenotypes of the disease are currently known; among others: retinoma (a form of pre-retinoblastoma) [57,67], spontaneously regressed retinoblastoma [58-61], diffuse infiltrating retinoblastoma [68,69], unilateral and bilateral retionoblastoma [2, 4], and also “trilateral retinoblastoma” [70,71].
Moreover, somatic mosaicism for Rb1 gene mutations:
1) is common in retinoblastoma, in which a high proportion of cases represent de novo mutations [72];
2) can be found in either affected patients and their unaffected parents [73], and
3) can involves both the paternal and maternal germline [74].
Both phenotypic variation and cellular mosaicism, although quite common in retinoblastoma, are unexplainable in the light of the mutational model which assumes that when the first mutation is inherited through the germline, all the somatic and germ cells of the individual will carry that mutation [2,6,7].
On the contrary, by using the epigenetic model, phenotypic variation in the clinical expression of the disease is easily explained by the variable exposure of the foetus to environmental toxicants which, in turn, will determine the degree of hypomethylation of different key genes. Within this conceptual framework, mosaicism can be viewed as the result of the interaction between the environment and the transposable elements of the genome.
6. Retinoblastoma and Imprinting
“Imprinting” is defined as “…a non-Mendelian, germline inherited epigenetic form of gene regulation involving heritable DNA methylation and histone modification”.
The human genome is subject to imprinting which represents the consequence of epigenetic inactivation (through methylation) of different genes in either the male or female gametes, so that in the resulting zygote, they complement each other, and the normal embryo development proceeds. On the contrary, two male derived or female derived genomes are incompatible with a normal growth of the embryo or foetus [22].
Since imprinted genes are epigenetically modified in both the male and female gametes, the expression of different genes in the zygote, embryo and foetus, derived from the fusion of the two, will depend on the parental environment in which both gametes (male and female) have grown and differentiate [34].
A parentally imprinted gene in one of the gametes, is not expressed; therefore the resulting zygote will be functionally haploid, (i.e. only one copy of the gene is functioning) and the consequences may be disastrous. In Knudson’s hypothesis, inheriting an imprinted Rb1 gene means that one copy of the gene is already functionally inactivated (first “hit”) [35] and only a single event is further requested for both copies to be inactivated.
Abnormal expression of imprinted genes during development may result in severe paediatric disorders such as Prader-Willi syndrome (PWS), Angelman syndrome (AS), and Beckwith-Wiedemann syndrome (BWS) [35, 75], where epigenetic alterations have an important contributory or causative role.
Moreover, imprinted gene dysregulation can also occur in somatic cells, either by epigenetic or genetic mutations, causing cancer [76,77]; therefore, with specific reference to cancer development, the inheritance of an epigenetically imprinted gene can be equated, as previously mentioned, to Knudson’s first hit, although in this case no structural DNA alteration is involved.
Given all the above, finding that the Rb1 gene can be imprinted in retinoblastoma may add important clues to the probable epigenetic nature of the disease. At this regard it is important to mention that, according to the most recent evidence, with only a few exceptions, hypermethylation of CpG islands is acknowledged as a major epigenetic inactivation mechanism for tumour suppressor genes, representing a major contributor to neoplastic transformation [78]. Accordingly, recent data show that Rb1 gene is imprinted in retinoblastoma with a shift of expression in favour of the maternal allele [58,79], while previous reports had already significantly shown that hypermethylation with loss of function occurs in 18% of sporadic retinoblastoma [80].
Imprinting is, by definition, a process by which human genes are “functionally” inactivated and its detection in retinoblastoma represents another argument against the mutational model, which instead assumes that gene expression can be altered only by structural DNA modifications.
7. Retinoblastoma: Epigenetics Rather than Inheritance
As we have seen, with the only exception of familial retinoblastoma (8% - 10% of all cases), in which the disease is found in the “proband” and in some of his/her relatives, “hereditary” retinoblastoma is, according to the “two hit” model, a sporadic retinoblastoma (since no other affected family member can be identified) determined by a germline mutation.
As a matter of fact, transgenerational inheritance involves the transmission of biological traits to subsequent generations through the germline.
Epigenetic alterations of the genome, as it has been shown, can be inherited (transmitted from one generation to another), and since environmental factors can alter the epigenome, their ability to influence the disease risk might involve epigenetic transgenerational inheritance.
We can speak of transgenerational inheritance of environmental effects, when the effects themselves are maintained and detectable in at least F3 generation, where F0 is the gestating mother exposed, F1 is the embryo and F2 are the embryo’s germ cells.
It is clear that, when the gestating female (F0) is exposed to toxicants, both F1 (embryo) and F2 (embryo’s germ cells) are also directly exposed. Therefore, disease phenotypes in the F1 and F2 generations might still be due to the direct exposure of F0, F1 and F2 to environmental toxicants [33-36].
This line of reasoning alone would be more than sufficient to demonstrate that hereditary (bilateral) retinoblastoma, is not a true hereditary disease, but an epigenetic disorder most probably linked to the gestational exposure to environmental harmful agents. As a matter of fact, clinical reports on retinoblastoma are almost invariably limited to retinoblastoma patients (F1) and, very rarely, to their first generation descendants (F2), while a retinoblastoma occurring in the F3 generation, according to the mutational model proposed by Knudson, belongs to the “familial” group.
Notwithstanding all the above mentioned considerations, epigenetic alterations of gene expression have been reported in F3 generations [81-85], thus demonstrating that the environment may stably imprint its effects on the genome, thus mimicking a “genetic disease” even if no mutations are detectable, as reported in many cases of “hereditary” retinoblastoma [23].
As we have seen, a poor diet and infectious diseases are presently considered risk factors for the development of retinoblastoma in less affluent populations throughout the world, but even radiation may play an important role. Of extreme interest, at this regard, is the case of the American-Indian Navajo population who has represented the main working force in the uranium mines of southwest America, from World War II until 1971 [86], and still live in villages located near the mines. The incidence of retinoblastoma among these populations is more than twice when compared to other world populations [87].
More importantly, the incidence seem to arise to twenty times in the village of Seascale, situated in the vicinity of a nuclear reprocessing plant, and best known in epidemiological circles for its longstanding high incidence of malignant diseases in young people [88].
All the reported data represent a clear demonstration of the role of environmental factors in the genesis and development of retinoblastoma and, as a consequence, the role of Epigenetics rather than genetics in the determinism of this eye tumour.
8. Concluding Remarks
Epigenetics can be defined as the study of changes that influence the phenotype without causing alterations of the genotype. It involves changes in the properties of a cell that are inherited in the absence of structural changes of its DNA [89]. Although epigenetic regulation of gene expression is the mechanism through which the extraordinary variety of specialized cells of the body differentiate starting from a single undifferentiated ancestor, the relevance of epigenetic factors in disease in humans was first detected only in 1983 when Feinberg and Vogelstein [90] found that gene hypomethylation could distinguish some human cancers from their normal counterparts [91].
Today, deregulation of gene expression is widely considered a hallmark of cancer, and although genetic lesions have been the focus of cancer research for many years, as in the case of retinoblastoma, it has become increasingly recognized that aberrant epigenetic modifications play major roles in cancer development [92,93].
This represents a great revolution and advancement with respect to the understanding of the pathogenesis of cancer we have gained so far, by applying the concepts and principles of Mendelian (or classic) genetics. In fact, Mendelian genetics has been proven largely insufficient to explain the diversity of phenotypes within a population, nor it explains how, despite their identical DNA sequences, monozygotic twins or cloned animals can have different phenotypes and different disease susceptibilities [94].
On this line of reasoning, we have been able to show herein and elsewhere [16-19] that the mutational model is largely inadequate to explain the variegated phenotypic expression of retinoblastoma, and there is an increasing agreement among researchers worldwide that the mutational (“two hit”) model is outdated and another paradigm has to be adopted for a better understanding of the pathogenesis of retinoblastoma [95].
Epigenetics allows to explain the inconsistencies of the mutational (“two hit”) model as applied to the pathogenesis of retinoblastoma (see Table 1), but it also has other important advantages which promise to revolutionize the field of both ophthalmology and oncology [96]. We refer herein to the following:
1) The potential reversibility of epigenetic states offers exciting opportunities for novel cancer drugs that can restore epigenetically silenced cancer genes. DNA methyltransferases and histone deacetylases are the two major drug targets for epigenetic inhibition to date, although others are expected to be added in the near future [97-100].
2) Epigenetic changes in cancer cells not only provide novel targets for drug therapy but also offer unique prospects for cancer diagnostics [100], through the study of gene expression, the evaluation of histone modifications and chromatin protein composition, and also the analysis of the promoter DNA methylation status;
3) Finally and more importantly, by shifting the focus on the environment and the complex interactions between the environmental regulation of gene expression and the genome, rather than on the genes themselves, Epigenetics stresses the importance of cancer prevention [101] and the changes of our lifestyles, including diet and behaviour [53,63,66,102,103].
9. Acknowledgements
Tha Authors thank Dr. Theodora Hadjistilianou, chief of the Ocular Oncology Unit of the Department of Ophthalmology of the University of Siena for supplying clinical data concerning retinoblastoma.
REFERENCES
- D. Mastrangelo, A. Di Leonardo, L. Lentini, S. De Francesco and T. Hadjistilianou, “Missing Evidences in Cancer Genetics: The Retinoblastoma Paradigm,” Cellular Oncology, Vol. 30, No. 6, 2008. pp. 509-510.
- A. G. Knudson, “Mutation and Cancer: Statistical Study of Rb,” Proceedings of the National Academy of Sciences, Vol. 68, No. 4, 1971, pp. 820-823. doi:10.1073/pnas.68.4.820
- S. H. Friend, R. Bernards, S. Rogelj, R. A. Weinberg, J. M. Rapaport, D. M. Albert, et al., “A Human DNA Segment with Properties of the Gene that Predisposes to Retinoblastoma and Osteosarcoma,” Nature, Vol. 323, 1986, pp. 643-646. doi:10.1038/323643a0
- D. R. Lohmann and B. L. Gallie, “Retinoblastoma,” In: R. A. Pagon, T. C. Bird, C. R. Dolan and K. Stephens, Eds., Gene Reviews, University of Washington, Seattle, 2010.
- G. Gaidano, A. Serra, A. Guerrasio, G. Rege-Cambrin, U. Mazza and G. Saglio, “Genetic Analysis of p53 and RB1 Tumor-Suppressor Genes in Blast Crisis of Chronic Myeloid Leukaemia,” Annals of Hematology, Vol. 68, No. 1, 1994, pp. 3-7. doi:10.1007/BF01695912
- A. G. Knudson, “Heredity and Human Cancer,” American Journal of Pathology, Vol. 77, No. 1, 1974, pp. 77-84.
- A. G. Knudson, “A Personal Sixty-Year Tour of Genetics and Medicine,” Annual Review of Genomics and Human Genetics, Vol. 6, 2005, pp. 1-14. doi:10.1146/annurev.genom.6.080604.162320
- R. H. Li, A. Sonik, R. Stindl, D. Rasnick and P Duesberg, “Aneuploidy vs. Gene Mutation Hypothesis of Cancer: Recent Study Claims Mutation but is Found to Support Aneuploidy,” Proceedings of the National Academy of Sciences USA, Vol. 97, No. 7, 2000, pp. 3236-3241. doi:10.1073/pnas.040529797
- P. Duesberg, C. Rausch, D. Rasnick and R. Hehlmann, “Genetic Instability of Cancer Cells is Proportional to Their Degree of Aneuploidy,” Proceedings of the National Academy of Sciences USA, Vol. 95, No. 23, 1998, pp. 13692-13697. doi:10.1073/pnas.95.23.13692
- P. Duesberg, R. Li, D. Rasnik, C. Rausch, A. Willer, A. Kraemer, et al., “Aneuploidy Precedes and Segregates with Chemical Carcinogenesis,” Cancer Genetics and Cytogenetics, Vol. 119, No. 2, 2000, pp. 83-93. doi:10.1016/S0165-4608(99)00236-8
- P. Duesberg, R. Stindl, R. H. Li, R. Hehlmann and D. Rasnick, “Aneuploidy versus Gene Mutation as Cause of Cancer,” Current Science, Vol. 81, No. 5, 2001, pp. 490- 500.
- P. Duesberg, R. Li and D. Rasnick, “Aneuploidy Approaching a Perfect Score in Predicting and Preventing Cancer,” Cell Cycle, Vol. 3, No. 6, 2004, pp. 823-828. doi:10.4161/cc.3.6.938
- P. Duesberg, “Does Aneuploidy or Mutation Start Cancer?” Science, Vol. 307, No. 41, 2005, p. 5706.
- P. Duesberg, R. Li, A. Fabarius, and R. Hehlmann, “The Chromosomal Basis of Cancer,” Cellular Oncology, Vol. 27, No. 5-6, 2005, pp. 293-318.
- P. Duesberg, “Chromosomal Chaos and Cancer,” Scientific American, Vol. 296, 2007, pp. 52-59. doi:10.1038/scientificamerican0507-52
- D. Mastrangelo, S. De Francesco, A. Di Leonardo, L. Lentini and T. Hadjistilianou, “Does the Evidence Matter in Medicine? The Rb Paradigm,” International Journal of Cancer, Vol. 121, No. 11, 2007, pp. 2501-2505. doi:10.1002/ijc.22944
- D. Mastrangelo, S. De Francesco, A. Di Leonardo, L. Lentini and T. Hadjistilianou, “Rb Epidemiology: Does the Evidence Matter?” European Journal of Cancer, Vol. 43, No. 10, 2007, pp. 1596-603. doi:10.1016/j.ejca.2007.04.019
- D. Mastrangelo, T. Hadjistilianou, S. De Francesco and C. Loré, “Retinoblastoma and the Genetic Theory of Cancer: an Old Paradigm Trying to Survive to the Evidence,” Journal of Cancer Epidemiology, Vol. 2009, 2009. doi:10.1155/2009/301973
- D. Mastrangelo, S. De Francesco, A. Di Leonardo, L. Lentini and T. Hadjistilianou, “The Retinoblastoma Paradigm Revisited,” Medical Science Monitor, Vol. 14, No. 12, 2008, pp. 231-240.
- C. H. Waddington, “Organisers and Genes,” Cambridge University Press, Cambridge, 1940.
- R. Holliday and J. C. Pugh, “DNA Modification Mechanisms and Gene Activity during Development,” Science, Vol. 187, 1975, pp. 226-232.
- R. Holliday, “DNA Methylation and Epigenotypes,” Biochemistry, Vol. 70, No. 5, 2005, pp. 500-504.
- V. Blanquet, C. Turleau, M.S. Gross-Morand, C. Semanaud-Beaufort, F. Doz and C. Besmond, “Spectrum of Germline Mutations in the RB1 Gene: A Study of 232 Patients with Hereditary and Non Hereditary Rb,” Human Molecular Genetics, Vol. 4, No. 3, 1995, pp. 383-388. doi:10.1093/hmg/4.3.383
- Cancer Genetics Web: “Rb1, Retinoblastoma,” 2009. http://www.cancerindex.org/geneweb//RB1.htm
- F. Cetani, E. Pardi, P. Vacava, G. Di Pollina, G. Fanelli, A. Picone, et al., “A Reappraisal of the Rb1 Gene Abnormalities in the Diagnosis of Parathyroid Cancer,” Clinical Endocrinology, Vol. 60, No. 1, 2004, pp. 99-106.
- J. I. Herschkowitz, X He, C. Fan, C. M. Perou, “The Functional Loss of the Retinoblastoma Tumour Suppressor is a Common Event in Basal-Like and Luminal B Breast Carcinomas,” Breast Cancer Research, Vol. 10, No. 5, 2008, pp. 1-13. doi:10.1186/bcr2142
- P. S. Lai, P. Y. Cheah, P. Kadam, C. Li-Ming Chua, D. Khun Hong Lie, H. H. Li, et al., “Overexpression of RB1 Transcript is Significantly Correlated with 13q14 Allelic Imbalance in Colorectal Carcinomas,” International Journal of Cancer, Nol. 119, No. 5, 2006, pp. 1061-1066.
- D. M. Parkin and C. A. Stiller, “Childhood Cancer in Developing Countries: Environmental Factors,” International Journal of Pediatric Hematology/Oncology, Vol. 2, 1995, pp. 411-417.
- D. M. Parkin, P. Pisani and J. Ferlay, “Global Cancer Statistics,” CA Cancer Journal for Clinicians, Vol. 49, No. 1, 1999, pp. 33-64. doi:10.3322/canjclin.49.1.33
- C. A. Stiller and D. M. Parkin, “Geographic and Ethnic Variations in the Incidence of Childhood Cancer,” British Medical Bulletin, Vol. 52, No. 4, 1996, pp. 682-703.
- I. Ajioka, R. A. Martins, I. T. Bayazitov, S. Donovan, D. A. Johnson, S. Frase, et al., “Differentiated Horizontal Interneurons Clonally Expand to Form Metastatic Retinoblastoma in Mice,” Cell, Vol. 131, No. 2, 2007, pp. 378-390. doi:10.1016/j.cell.2007.09.036
- J. Madhavan, A. Ganesh, J. Roy, J. Biswas and G. Kumaramanickavel, “The Relationship between Tumor Cell Differentiation and Age at Diagnosis in Retinoblastoma,” Journal of Pediatric Ophthalmology & Strabismus, Vol. 45, No. 1, 2008, pp. 22-25. doi:10.3928/01913913-20080101-16
- D. C. Dolinoy, “Epigenetic Gene Regulation: Early Environmental Exposures,” Pharmacogenomics, Vol. 8, No. 1, 2007, pp. 5-10. doi:10.2217/14622416.8.1.5
- R. L. Jirtle and M. K. Skinner, “Environmental Epigenomics and Disease Susceptibility,” Nature Reviews Genetics, Vol. 8, 2007, pp. 253-262. doi:10.1038/nrg2045
- D. C. Dolinoy, J. R. Weidman and R. L. Jirtle, “Epigenetic Gene Regulation: Linking Early Developmental Environment to Adult Disease,” Reproductive Toxicology, Vol. 23, No. 3, 2007, pp. 297-307. doi:10.1016/j.reprotox.2006.08.012
- D. C. Dolinoy, R. Das, J. R. Weidman and R. L. Jirtle, “Metastable Epialleles, Imprinting, and the Fetal Origins of Adult Diseases,” Pediatric Research, Vol. 61, No. 5, 2007, pp. 31-37.
- Genetics Home Reference. http://ghr.nlm.nih.gov/glossary=housekeepinggene
- F. Iovino, L. Lentini, A. Amato and A. Di Leonardo, “RB Acute Loss Induces Centrosomes Amplification and aneuploidy in Murine Primary Fi Broblasts,” Molecular Cancer, Vol. 5, 2006, p. 38. doi:10.1186/1476-4598-5-38
- E. Hernando, Z. Nahle, G. Juan, E. Diaz-Rodriguez, M. Alaminos, M. Hemann, et al., “Rb Inactivation Promotes Genomic Instability by Uncoupling Cell Cycle Progression from Mitotic Control,” Nature, Vol. 430, No. 7001, 2004, pp. 797-802. doi:10.1038/nature02820
- N. Ohtani-Fujita, T. Fujita, A. Aoike, N. E. Osifchin, P. D. Robbins and T. Sakai, “CpG Methylation Inactivates the Promoter Activity of the Human Retinoblastoma Tumor-Suppressor Gene,” Oncogene, Vol. 8, No. 4, 1993, pp. 1063-1067.
- N. Ohtani-Fujita, T. P. Dryja, J. M. Rapaport, T. Fujita, S. Matsumura, K. Ozasa, et al., “Hypermethylation in the Retinoblastoma Gene Is Associated with Unilateral, Sporadic Retinoblastoma,” Cancer Genetics and Cytogenetics, Vol. 98, No. 1, 1997, pp. 43-49. doi:10.1016/S0165-4608(96)00395-0
- M. J. Bello, P. Gonzalez-Gomez, M. Eva Alonso, N. P. Anselmo, D. Arjona, C. Amiñoso, et al., “Methylation Analysis of Cell Cycle Control Genes RB1, p14ARF and p16INK4a in Human Gliomas,” Cancer Therapy, Vol. 2, 2004, pp. 187-194.
- P. R. Moreira, M. M. Guimarães, C. C. Gomes, M. G. Diniz, J. A. Brito, W. H. de Castro, et al., “Methylation Frequencies of Cell-Cycle Associated Genes in Epithelial Odontogenic Tumours,” Archives of Oral Biology, Vol. 54, No. 10, 2009, pp. 893-897. doi:10.1016/j.archoralbio.2009.07.006
- K. Malekzadeh, R. C. Sobti, M. Nikbakht, M. Shekari, S. A.Hosseini, D. K. Tamandani, et al., “Methylation Patterns of Rb1 and Casp-8 Promoters and Their Impact on Their Expression in Bladder Cancer,” Cancer Investigation, Vol. 27, No. 1, 2009, pp. 70-80. doi:10.1080/07357900802172085
- K. Bastide, M. N. Guilly, J. F. Bernaudin, C. Joubert, B. Lectard, C. Levalois, et al., “Molecular Analysis of the Ink4a/Rb1-Arf/Tp53 Pathways in Radon-Induced Rat Lung Tumors,” Lung Cancer, Vol. 63, No. 3, 2009, pp. 348-353. doi:10.1016/j.lungcan.2008.06.007
- C. S. Chim, K. Y. Wong, F. Loong, W. W. Lam and G. Srivastava, “Frequent Epigenetic Inactivation of Rb1 in Addition to p15 and p16 in Mantle Cell and Follicular Lymphoma,” Human Pathology, Vol. 38, No. 12, 2007, pp. 1849-1857. doi:10.1016/j.humpath.2007.05.009
- K. Amara, M. Trimeche, S. Ziadi, A. Laatiri, M. Hachana, B. Sriha, et al., “Presence of Simian Virus 40 DNA Sequences in Diffuse Large B-cell Lymphomas in Tunisia Correlates with Aberrant Promoter Hypermethylation of Multiple Tumor Suppressor Genes,” International Journal of Cancer, Vol. 121, No. 12, 2007, pp. 2693-2702. doi:10.1002/ijc.23038
- Y. F. Zhao, Y. G. Zhang, X. X. Tian, J. Du and J. Zheng, “Aberrant Methylation of Multiple Genes in Gastric Carcinomas,” International Journal of Surgical Pathology, Vol. 15, No. 3, 2007, pp. 242-251. doi:10.1177/1066896907302117
- M. B. Michalowski, F. de Fraipont, D. Plantaz, S. Michelland, V. Combaret and M. C. Favrot, “Methylation of Tumor-Suppressor Genes in Neuroblastoma: The RASSF1A Gene is almost Always Methylated in Primary Tumors,” Pediatric Blood Cancer, Vol. 50, No. 1, 2008, 29-32. doi:10.1002/pbc.21279
- A. Yoshino, Y. Katayama, A. Ogino, T. Watanabe, K. Yachi, T. Ohta, et al., “Promoter Hypermethylation Profile of Cell Cycle Regulator Genes in Pituitary Adenomas,” Journal of Neurooncology, Vol. 83, No. 2, 2007, pp. 153-162. doi:10.1007/s11060-006-9316-9
- E. N. Tolmacheva, A. A. Kashevarova, N. N. Sukhanova, E. A. Sazhenova and I. N. Lebedev, “Epigenetic Inactivation of the RB1 Gene as a Factor of Genomic Instability: A Possible Contribution to Etiology of Chromosomal Mosaicism during Human Embryo Development,” Russian Journal of Genetics, Vol. 44, No. 11, 2008, pp. 1266-1271. doi:10.1134/S1022795408110033
- H. Shigematsu, M. Suzuki, T. Takahashi, K. Miyajima, S. Toyooka, N. Shivapurkar, et al., “Aberrant Methylation of HIN-1 (High in Normal-1) is a Frequent Event in Many Human Malignancies,” International Journal of Cancer, Vol. 113, No. 4, 2005, pp. 600-604. doi:10.1002/ijc.20622
- A. Rathi, A. K. Virmani, K. Harada, C. F.Timmons, K. Miyajima, R. J. Hay, et al., “Aberrant Methylation of the HIC1 Promoter Is a Frequent Event in Specific Pediatric Neoplasms,” Clinical Cancer Research, Vol. 9, No. 10, 2003, pp. 3674-3678.
- K. Harada, S. Toyooka, N. Shivapurkar, A. Maitra, J. L. Reddy, H. Matta, et al., “Deregulation of Caspase 8 and 10 Expression in Pediatric Tumors and Cell Lines,” Cancer Research, Vol. 62, No. 20, 2002, pp. 5897-5901.
- K. Harada, S. Toyooka, A. Maitra, R. Maruyama, K. O. Toyooka, C. F. Timmons, et al., “Aberrant Promoter Methylation and Silencing of the RASSF1A Gene in Pediatric Tumors and Cell Lines,” Oncogene, Vol. 21, No. 27, 2002, pp. 4345-4349. doi:10.1038/sj.onc.1205446
- D. C. Dolinoy and R. L. Jirtle, “Environmental Epigenomics in Human Health and Disease,” Environmental Molecular Mutagenesis, Vol. 49, No. 1, 2008, pp. 4-8. doi:10.1002/em.20366
- K. E. Nichols, S. Walther, E. Chao, C. Shields and A. Ganguly, “Recent Advances in Retinoblastoma Genetic Research,” Current Opinion in Ophthalmology, Vol. 20, No. 5, 2009, pp. 351-355. doi:10.1097/ICU.0b013e32832f7f25
- D. Knaber, T. Berulava, O. Ammerpohl, D. Mitter, J Richter, R Siebert, et al., “The Human Retinoblastoma Gene Is Imprinted,” PLoS Genetics, Vol. 5, No. 12, 2009.
- G. E. Sanbor, J. J. Augsburger and J. A. Shields, “Spontaneous Regression of Bilateral Retinoblastoma,” British Journal of Ophthalmology, Vol. 66, No. 11, 1982, pp. 685-690. doi:10.1136/bjo.66.11.685
- R. C. Eagle, J. A. Shields, L. Donoso and R. S. Milner, “Malignant Transformation of Spontaneously Regressed Retinoblastoma, Retinoma/Retinocytoma Variant,” Ophthalmology, Vol. 96, No. 9, 1989, pp. 1389-1395.
- J. K. Steward, J. L. S. Smith and E. L. Arnold, “Spontaneous Regression of Retinoblastoma,” British Journal of Ophthalmology, Vol. 40, 1956, pp. 449-461. doi:10.1136/bjo.40.8.449
- A. Raj, S. A. Rifkin, E. Andersen and A. van Oudenaarden, “Variability in Gene Expression Underlies Incomplete Penetrance,” Nature, Vol. 463, 2010, pp. 913-918. doi:10.1038/nature08781
- N. Zamudio and D. Bourc’his, “Transposable Elements in the Mammalian Germline: A Comfortable Niche or a Deadly Trap?” Heredity, Vol. 105, No. 1, 2010, pp. 92- 104. doi:10.1038/hdy.2010.53
- T. H. Bestor, “Cytosine Methylation Mediates Sexual Conflict,” Trends in Genetics, Vol. 19, No. 4, 2003, pp. 185-190. doi:10.1016/S0168-9525(03)00049-0
- J. A. Yoder, C. P. Walsh and T. H. Bestor, “Cytosine Methylation and the Ecology of Intragenomic Parasite,” Trends in Genetics, Vol. 13, No. 8, 1997, pp. 335-340. doi:10.1016/S0168-9525(97)01181-5
- D. C. Dolinoy, J. Wiedman, R. Waterland and R. L. Jirtle, “Maternal Genistein Alters Coat Color And Protects Avy Mouse Offspring from Obesity by Modifying the Fetal Epigenome,” Environmental Health Perspectives, Vol. 114, 2006, pp. 567-572. doi:10.1289/ehp.8700
- H. Abouzeid, D. F. Schorderet, A. Balmer and F. L. Munier, “Germline Mutations in Retinoma Patients: Relevance to Low Penetrance and Low-Expressivity Molecular Basis,” Molecular Vision, Vol. 15, 2009, pp. 771-777.
- G. Morgan, “Diffuse Infiltrating Retinoblastoma,” British Journal of Ophthalmology, Vol. 55, No. 9, 1971, pp. 600-606. doi:10.1136/bjo.55.9.600
- C. L.Shields, F. Ghassemi, S. Tuncer, A. Thangappan and J. A. Shields, “Clinical Spectrum of Diffuse Infiltrating Retinoblastoma in 34 Consecutive Eyes,” Ophthalmology, Vol. 115, No. 12, 2008, pp. 2253-2258. doi:10.1016/j.ophtha.2008.07.003
- E. Y. Cho, Y. L. Suh and H. J. Shin, “Trilateral Retinoblastoma: A Case Report,” Journal of Korean Medical Science, Vol. 17, 2002, pp. 137-140.
- J. M. Provenzale, S. Gururangan and G. Klintworth, “Trilateral Retinoblastoma: Clinical and Radiologic Progression,” American Journal of Roentgenology, Vol. 183, No. 2, 2004, pp. 505-511.
- K. C. Sippel, R. E. Fraioli, G. D. Smith, M. E. Schalkoff, J. Sutherland, B. L. Gallie, et al., “Frequency of Somatic and Germ-Line Mosaicism in Retinoblastoma: Implications for Genetic Counseling,” American Journal of Human Genetics, Vol. 62, No. 3, 1998, pp. 610-619. doi:10.1086/301766
- D. Rushlow, B. Piovesan, K. Zhang, N. L. Prigoda-Lee, M. N. Marchong, R. D. Clark, et al., “Detection of Mosaic RB1 Mutations in Families with Retinoblastoma,” Vol. 30, No. 5, 2009, pp. 842-851.
- R. H. Barbosa, F. R. Vargas, F. C. Aguiar, S. Ferman, E. Lucena, C. R. Bonvicino, et al., “Hereditary Retinoblastoma Transmitted by Maternal Germline Mosaicism,” Pediatric Blood Cancer, Vol. 51, No. 5, 2008, pp. 598-602. doi:10.1002/pbc.21687
- S. K. Murphy, R. L. Jirtle, “Imprinting Evolution and the Price of Silence,” Bioessays, Vol. 25, No. 6, 2003, pp. 577-588. doi:10.1002/bies.10277
- A. P. Feinberg, B. Tycko, “The History of Cancer Epigenetics,” Nature Review Cancer, Vol. 4, 2004, pp. 143-153. doi:10.1038/nrc1279
- A. P. Feinberg, “The Epigenetics of Cancer Etiology,” Seminars in Cancer Biology, Vol. 14, No. 6, 2004, pp. 427-432. doi:10.1016/j.semcancer.2004.06.005
- S. S. Palii, K. D. Robertson, “Epigenetic Control of Tumour Suppression,” Critical Reviews in Eukaryotic Gene Expression, Vol. 17, No. 4, 2007, pp. 295-316.
- K. Buiting, D. Kanber, D. Lohmann, “Imprinting of RB1 (the New Kid on the Block),” Briefings in Functional Genomics, Vol. 9, No. 4, 2010, 347-353. doi:10.1093/bfgp/elq014
- V. Greger, E. Passarge, W. Höpping, M. Messmer and B. Horsthemke, “Epigenetic Changes may Contribute to the Formation and Spontaneous Regression of Retinoblastoma,” Human Genetics, Vol. 83, No. 2, 1989, pp. 155 -158. doi:10.1007/BF00286709
- M. D. Anway, A. S. Cupp, M. Uzumcu and M. K. Skinner, “Epigenetic Transgenerational Actions of Endocrine Disruptors and Male Fertility,” Science, Vol. 308, No. 5727, 2005, pp. 1466-1469. doi:10.1126/science.1108190
- V. S. Turusov, T. V. Nikonova and Y. Parfenov, “Increased Multiplicity of Lung Adenomas in Five Generations of Mice Treated with Benz(a)Pyrene When Pregnant,” Cancer Letters, Vol. 55, No. 3, 1990, pp. 227-231. doi:10.1016/0304-3835(90)90123-F
- M. D. Anway, C. Leathers and M. K. Skinner, “Endocrine Disruptor Vinclozolin Induced Epigenetic Transgenerational Adult-Onset Disease,” Endocrinology, Vol. 147, No. 12, 2006, pp. 5515-5523. doi:10.1210/en.2006-0640
- H. S. Chang, M. D. Anway, S. S. Rekow and M .K. Skinner, “Transgenerational Epigenetic Imprinting of the Male Germline by Endocrine Disruptor Exposure during Gonadal Sex Determination,” Endocrinology, Vol. 147, No. 12, 2006, pp. 5524-5541. doi:10.1210/en.2006-0987
- G. Durcova-Hills, P. Hajkova, S. Sullivan, S. Barton, M. Azim Surani and A. McLaren, “Influence of Sex Chromosome Constitution on the Genomic Imprinting of Germ Cells,” Proceedings of the National Academy Sciences USA, Vol. 103, No. 30, 2006, pp. 1184-1188. doi:10.1073/pnas.0602621103
- D. Brugge and R. Goble, “The History of Uranium Mining and the Navajo People,” American Journal of Public Health, Vol. 92, No. 9, 2002, pp. 1410-1419.
- R. L. Berkow and J. K. Fleshman, “Retinoblastoma in Navajo Indian Children,” American Journal of Diseases in Children, Vol. 137, No. 2, 1983, 137-138.
- C. A. Stiller, “Retinoblastoma and Low Level of Radiation,” British Medical Journal, Vol. 307, 1993, pp. 461-462. doi:10.1136/bmj.307.6902.461
- B. Lewin, “Genes VIII,” Pearson Prentice Hall, Upper Saddle River, 2004.
- A. P. Feinberg and B. Vogelstein, “Hypomethylation Distinguishes Genes of Some Human Cancers from Their Normal Counterparts,” Nature, Vol. 301, 1983, pp. 89-92. doi:10.1038/301089a0
- J. Peedicayil, “Epigenetic Therapy: A New Development in Pharmacology,” Indian Journal of Medical Research, Vol. 123, No. 1, 2006, pp. 17-24.
- E. N. Gal-Yam, Y. Saito, G. Egger and P. A. Jones, “Cancer Epigenetics: Modifications, Screening and Therapy,” Annual Review of Medicine, Vol. 59, 2008, pp. 267-280. doi:10.1146/annurev.med.59.061606.095816
- M. Nakao, “Epigenetics: Interaction of DNA Methylation and Chromatin,” Gene, Vol. 278, No. 1-2, 2001, pp. 25- 31. doi:10.1016/S0378-1119(01)00721-1
- M. Esteller, “Epigenetics in Cancer,” New England Journal of Medicine, Vol. 358, 2008, pp. 1148-1159. doi:10.1056/NEJMra072067
- A. C. Schefler, D. H. Abramson, “Retinoblastoma: What is New in 2007-2008,” Current Opinions in Ophthalmology, Vol. 19, No. 6, 2008, pp. 526-534. doi:10.1097/ICU.0b013e328312975b
- P. W. Laird, “Cencer Epigenetics,” Human Molecular Genetics, Vol. 14, No. 1, 2005, pp. R65-R76. doi:10.1093/hmg/ddi113
- R. W. Johnstone, “Histone-Deacetylase Inhibitors: Novel Drugs for the Treatment of Cancer,” Nature Review Drug Discovery, Vol. 1, 2002, pp. 287-299. doi:10.1038/nrd772
- G. Egger, G. Liang, A. Aparicio and P. A. Jones, “Epigenetics in Human Disease and Prospects for Epigenetic Therapy,” Nature, Vol. 429, 2004, pp. 457-463. doi:10.1038/nature02625
- M. Esteller, “DNA Methylation and Cancer Therapy: New Developments and Expectations,” Current Opinion Oncology, Vol. 17, No. 1, 2005, pp. 55-60. doi:10.1097/01.cco.0000147383.04709.10
- A. Zelent, S. Waxman, M. Carducci, J. Wright, J. Zweibel and S. D. Gore, “State of the Translational Science: Summary of Baltimore Workshop on Gene Re-expression as a Therapeutic Target in Cancer,” Clinical Cancer Research, Vol. 10, 2004, pp. 4622-4629. doi:10.1158/1078-0432.CCR-1219-03
- P. W. Laird, “The Power and the Promise of DNA Methylation Markers,” Nature, Vol. 3, No. 4, 2003, pp. 253-266.
- J. P. Issa, “Cancer Prevention: Epigenetics Steps up to the Plate,” Cancer Prevention Research, Vol. 1, No. 4, 2008, pp. 219-222. doi:10.1158/1940-6207.CAPR-08-0029
- D. Crews, “Epigenetics and Its Implications for Behavioural Neuroendocrinology,” Neuroendocrinology, Vol. 29, No. 3, 2008, pp. 344-357. doi:10.1016/j.yfrne.2008.01.003