Journal of Cancer Therapy
Vol. 4 No. 6 (2013) , Article ID: 35272 , 16 pages DOI:10.4236/jct.2013.46127
Development of Oncolytic Reovirus for Cancer Therapy
![]()
1Department of Microbiology and Immunology, University of Louisville, School of Medicine, Louisville, USA; 2Department of Surgery, University of Louisville, School of Medicine, Louisville, USA; 3James Graham Brown Cancer Center, University of Louisville, School of Medicine, Louisville, USA.
Email: *erginsahin@ymail.com, *hszhou01@louisville.edu
Copyright © 2013 Ergin Sahin et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 21st, 2013; revised June 22nd, 2013; accepted June 30th, 2013
Keywords: Reovirus; Oncolysis; Cancer; Cancer Gene Therapy; dsRNA
ABSTRACT
Reovirus, a double-stranded RNA virus, can infect many types of cancer cells and cause oncolysis. Mammalian reovirus has exhibited promising anticancer activity in clinical trials and holds great advantages and promise as an anticancer agent. Reovirus is not associated with any serious human diseases, naturally targets and destroys tumors, and lacks the DNA synthesis stage, thus avoiding potential DNA insertion mutations. This review discusses the properties of reovirus related to oncolysis and the mechanisms of oncolytic selection, and summarizes the preclinical and clinical studies that have led to the current Phase III trial. In addition, three major challenges in the development of reovirus-mediated oncolytic therapy are discussed. These are: the mechanisms of reovirus oncolysis remain to be fully characterized; the host immune responses should be manipulated to enhance viral anti-tumor effects; and the efficacy of reovirus oncolysis may be further improved by developing new vectors and studying other double-stranded RNA viruses.
1. Introduction
Reoviruses are non-enveloped icosahedral viruses with double-stranded RNA (dsRNA) genome. There are total ten dsRNA virus families (Table 1): reoviridae, birnaviridae, totiviridae, partitiviridae, hypoviridae, cystoviridae, chrysoviridae, endornaviridae, picobirnaviridae, and alternaviridae. Reoviruses belong to the reoviridae family. Only some viruses in the reoviridae family are known to infect mammals and thus have the potential to be developed for cancer oncolytic therapy. Although most humans have been infected with reoviruses, infections are usually sub-clinical. Reoviruses are not known to be associated with any serious human diseases, implying their safety in cancer virotherapy. Reovirus has been used in clinical trials and has exhibited promising anticancer activity in cancer patients.
TABLE I.
The reovirus name was coined from the word reo referring for Respiratory Enteric Orphan, reflecting the initial isolation from a child with recurrent pulmonary infections and malnutrition in 1951 [1,2]. The initial isolates were later grouped in the orthoreovirus genus in the reoviridae family, which includes 15 other recognized genera (Table 1). Although all virus members in the reoviridae family can be called reoviruses, the initial isolates classified in the orthreovirus genus are still commonly referred to as reoviruses in the literature. In the orthoreovirus genus, there are three distinct serotypes of mammalian reoviruses identified by neutralization and hemagglutination-inhibition tests. An isolate from a healthy child is the prototype for reovirus type 1 (type 1 Lang); an isolate from a child with diarrhea is the prototype for reovirus type 2 (type 2 Jones); and isolates from a child with diarrhea (type 3 Dearing) and a child with an upper respiratory illness (type 3 Abney) are prototypes for reovirus type 3 (Table 1). The strain of type 3 Dearing (T3D) is currently being evaluated in clinical trials, but all reovirus subtypes have oncolytic properties [3]. In this article, the term reovirus is used to refer to strains of mammalian orthoreovirus unless otherwise specified.
The capsid of reoviruses is composed of an outer and inner shell and the virion has a diameter of approximately 80 nm (Figure 1). The genome of reovirus consists of 10 separate segments that are divided among large (L), medium (M), and small (S) classes according to the dsRNA size. Reovirus genome segments have a total size of about 23.5 kb and encode for 12 viral proteins which contains 8 structural and 4 non-structural viral proteins [4]. The reovirus structural proteins consist of the outer capsid components, sigma 1, sigma 3, lambda 2, and
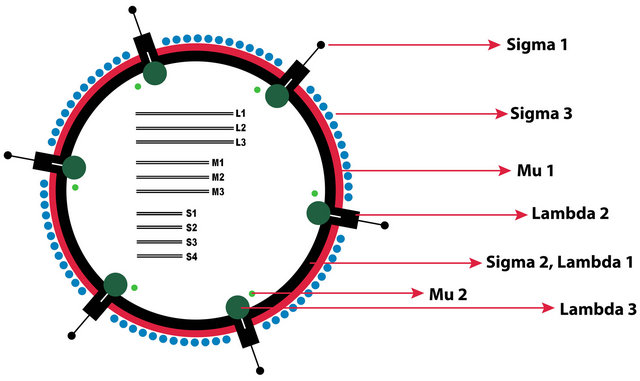
Figure 1. The schematic representation of the mammalian orthoreovirus virion. S: small genome segments, M: medium genome segments, L: large genome segments.

Table 1. dsRNA Virus Families and Serotypes of Mammalian Reoviruses.
Mu1, The inner capsid components sigma 2 and lambda 1, and the virus replication enzymes lambda 3 and Mu2. The reovirus non structural proteins include sigma1s which may have a role in reovirus virulence [5], and sigmaNS, MuNS, and MuNSC which are involved in viral inclusion formation [6,7] (Figure 1).
Reovirus holds many advantages and shows great promise as an anticancer agent. First, it results in only benign infections with few minor symptoms. Clinical trials with T3D for cancer oncolytic therapy have shown only low pathogenesis after systematic delivery. Secondwild type reoviruses naturally display tumor selective replication and cytotoxicity in cancers. In other words, reoviruses have tumor cell tropism which leads to efficient virus replication and consequent cancer specific cytotoxicity. Third, unlike other viruses, the reovirus life cycle takes place in the cytoplasm and does not include a stage of viral DNA synthesis. Therefore, it is highly unlikely that the reoviral genome fragments would integrate into human DNA and cause deleterious mutations.
2. Reovirus Infectious Life Cycle
The reovirus infectious life cycle starts with the binding of the virus particle to the target cell surface via viral protein sigma 1 (σ1) [8] (Figure 2). The binding is initiated with the attachment of the body domain of σ1 to the cell surface glycans which have been shown to be α- linked 5-N-acetyl neuraminic acid (Neu5Ac) for reovirus T3D strain and ganglioside GM2 glycan for reovirus T1L strain [9,10]. The cell surface glycans function as the coreceptors by low affinity binding to σ1, and by enabling the virus particle to diffuse laterally on the cell surface to facilitate the high affinity binding of the σ1 head domain to the junctional adhesion molecule-A (JAM-A) [11]. The σ1 carboxyl terminal region forms the compact head domain responsible for binding to the JAM-A which serves as the main receptor for reovirus infections in all three serotypes [12-14]. After cell attachment, reovirus particles are internalized into cell via cellular transmembrane receptor β1-integrin-mediated endocytosis [15]. β1-integrin-mediated endocytosis of reovirus particles occurs via a clathrin dependent manner [16-20]. The reovirus intermediate-subviral particles (ISVP) which could be formed with extracellular proteolysis can also directly enter into cells by penetration of the plasma membrane [20]. In addition, recent evidence indicates that the reovirus cell entry could occur via caveolin mediated endocytosis [21].
Upon cell entry, reovirus particles are transported through early, late, and recycling endosomes of which only the virus particles in the early and late endosomes become infectious [22]. The disassembly of the reovirus particles takes place in the late endosome compartments and requires a low pH environment and the cystein protease type of cathepsins that are cathepsin L, B, and S [19, 23-26]. In the presence of both a low pH environment and cathepsin proteases, reovirus particles go through a series of conformational changes in the late endosomes and give rise to the intermediate subviral particles (ISVP). ISVPs are formed with the proteolytic degradation of the main outer capsid protein sigma 3 (σ3) and with the proteolytic cleavage of the outer capsid protein mu1 (µ1) [27]. To penetrate the endosomal membranes, ISVPs require further proteolytic processing and major conforma-
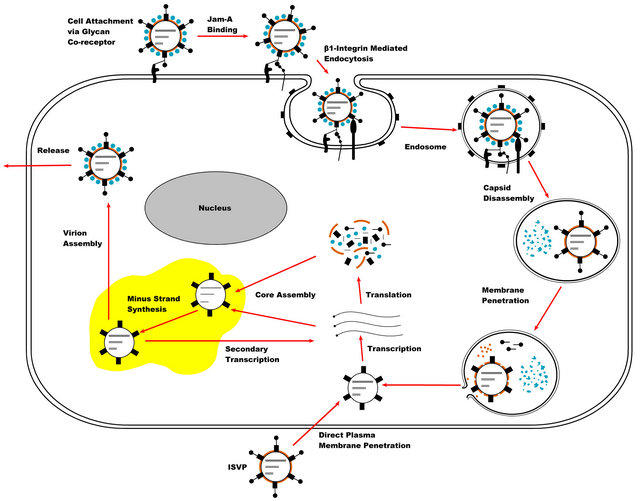
Figure 2. Diagram of the reovirus replication cycle: Reovirus replication is initiated with the attachment of the virus particles to the cell surface via the sigma1-receptor interaction, followed by cell entry via beta-1 integrin mediated endocytosis. Reovirus capsid disassembly takes place in the acidified late endosomes by the proteolytic activities of cathepsins. The formed reovirus intermediate subviral particles (ISVPs) penetrate the endosomal membranes by the aid of Mu1 membrane fusion protein. In the cytoplasm, transcriptionally active core particles initiate viral transcription. The synthesized viral mRNAs are released from the core particles and translated into the viral proteins. Reovirus core assembly and minus strand synthesis takes place in the viral inclusions (viral factories) which are composed of viral proteins and RNAs, complete and nascent core particles, and certain elements of the cell cytoskeleton. In the newly formed core particles a secondary transcription may take place which amplifies the synthesis of viral mRNAs and proteins. Reovirus virions are formed with the outer capsid assembly and released from the infected cell. Reovirus ISVPs can also be generated from virions by extracellular proteolysis and then enter cells through direct penetration of cell membrane.
tional rearrangements in the µ1 protein which result in exposure of the myristoylated N terminus and forms the distinct particle type called ISVP* [28-30].
The exposed N terminal fragment of µ1 goes through an auto cleavage and forms the membrane penetrating fragment µ1N. µ1N is then released from the particles along with shedding of the cell attachment protein σ1 [28, 31-33]. The released hydrophobic µ1N peptide interacts with the membrane lipids and mediates the release of viral core particles to the cytoplasm by forming size selective pores on the endosome membranes, thus ISVP* functions as transient cell entry intermediates [31,32,34].
Upon release to the cytoplasm, transcriptionally active reovirus core particles initiate virus replication. The transcription of the viral genome segments takes place within the core particles which contain the necessary virus replication components including the guanylyltransferase, methyltransferase, and RNA dependent RNA polymerase (RdRp) [35,36]. The reovirus transcripts which are capped from the 5’ end and lack a 3’ polyA trait [37,38] are released to the cytoplasm. The released positive sense reovirus transcripts are translated into the viral proteins in the ribosomes and upon encapsidation with newly synthesized core proteins serve as a template for the synthesis of minus strand RNAs to form the viral genome [35]. In the new viral core particles, a second viral transcription cycle may take place. With the condensation of the outer capsid proteins onto the nascent core particles, the progeny reovirus virions are formed and released from the infected cell by a process that is yet to be clarified [4]. Reovirus replication induces cell death and the released virions may further infect neighboring cells.
Reovirus replication and virion assembly takes place in viral inclusions (also called viral factories) which are formed in the cytoplasm of infected cells and consist of viral dsRNAs, viral proteins, complete and nascent virus particles, cell microtubules, and intermediate filaments [39,40-42]. Virus inclusion formation is mainly regulated by the reovirus nonstructural proteins µNS and σNS and the structural protein µ2 [6,7]. In contrast to the well identified early events of the reovirus infection, the precise roles of viral inclusions in the reovirus replication process including the formation of dsRNA replication machinery, gene segment assortment, genome packaging, and virion assembly have not been well understood and require further studies.
3. Selective Oncolysis of Reovirus
It has been known for a long time that wild-type reoviruses preferentially replicate in tumor cells and are naturally oncolytic. As early as 1977, the potential of selectively destroying cancers with reoviruses was demonstrated when transformed cell lines exhibited increased susceptibility to the human reoviruses [43,44]. Expression of epidermal growth factor receptors (EGFR) significantly enhanced reoviral infection and replication in otherwise nonpermissive cells [45]. Further studies showed that murine cells transformed with Ras oncogenes manifested enhanced susceptibility to reovirus infection and killing [46]. Further analysis suggested that the activated Ras/RalGEF/p38 pathway, a signaling pathway downstream of EGFR signaling, may be involved in cancer cell permissiveness to reovirus replication and oncolysis [47]. Ras is a GTP-binding protein that forms part of the Ras/RalGEF/p38 signal transduction pathway. When the EGFR is stimulated by signals on the cell surface, Ras is activated which further activates other proteins in the signal transduction pathway that ultimately induce expression of genes involved in cell growth and survival [48]. Over activation of the Ras/RalGEF/p38 pathway in cells by mutations of the Ras gene or other genes in the pathway can ultimately lead to cancer. Abnormal activation of the Ras pathway occurs commonly in human cancers, including pancreatic cancer (90%), colorectal cancer (50%), and lung cancer (40%) [49]. Reovirus oncolysis takes advantage of abnormal ras activation by targeting this pathway allowing efficient reoviral replication in ras-active human cancer cells.
Detailed studies of the reovirus life cycle have further shown that Ras transformation affects three important steps in viral replication: promoting reovirus proteolytic disassembly (uncoating) that is required for onset of infection; increasing the infectivity of progeny viruses, as shown virions purified from Ras-transformed cells are more infectious than those purified from non-transformed cells; and increasing reovirus release through caspaseinduced apoptosis [50,51]. Ras enhanced reovirus spread in subsequent rounds of infection by suppressing viral RNA-induced IFN-beta production through negative regulation of RIG-I signaling [52].
The reovirus infectious life cycle may be inhibited in normal cells by dsRNA-activated protein kinase (PKR), which functions as a defense mechanism against viral infection [46,53,54]. Reovirus replication produces dsRNA that activates PKR by binding to the N-terminal domain of PKR [53,54]. Once activated, PKR is able to inhibit viral mRNA translation, thereby preventing reoviral protein synthesis in normal cells. It has been hypothesized that PKR phosphorylation activation is blocked by an element(s) in the Ras pathway within viral-infected cancer cells, allowing viral protein synthesis and cancer selection [46]. Additionally the reovirus dsRNA binding protein sigma 3 (σ3) has been shown to inhibit PKR activation by sequestering the reovirus genome [55].
A recent report has argued that reovirus selective replication is not due to the activated Ras pathway in cancer cells [56]. In this report, the authors examined the reovirus-induced cytopathic effect (CPE) in eight human tumor cell lines and two untransformed cell lines, NIH/3T3 and LLC-MK2. They observed CPE in eight tumor cell lines and LLC-MK2 cells, but not in NIH/3T3 cells, even though NIH/3T3 expressed the same levels of Ras as other tested cancer cell lines. The expression of total PKR or phosphorylated PKR correlated with neither CPE nor Ras expression. The authors believe that the lack of reovirus receptor(s) on the cell surface (of NIH/3T3) is the critical reason for cell resistance to reovirus infection [56]. More reports have indicated that the over expression of the reovirus co-receptor glycan molecule in cancers could be a factor that affects the cancer selection of reoviruses by means of increasing the efficiency of reovirus infectivity [57-59]. The role of the Ras pathway in virus-mediated cytotoxicity was also studied recently in 15 head and neck cancer cell lines [60]. This study showed that oncolysis and reoviral replication were unaffected by inhibition or stimulation of EGFR signaling, and that inhibition of signaling downstream of Ras did not abrogate reoviral oncolysis. In addition, modulation of PKR by using 2-aminopurine did not alter the sensitivity of resistant cell lines to reovirus infection. Thus, this study also questioned the role of Ras signaling in reoviral selective oncolysis [60].
There is a recent report studying the importance of cell cycle on tumor sensitivity to reovirus oncolysis [61]. By treating with hydroxyurea and thymidine, which alter the normal cell cycle distribution, cells in S-phase exhibited enhanced sensitivity to reovirus, likely associated with increased viral replication. Cells that were devoid of an S-phase by mitotic shake-off were less susceptible to reovirus-induced cell killing. Studying adenovirus replication, we have shown that expression of several cell cycle-regulating genes, including cyclin E, is markedly induced in cells after adenovirus infection [62]. Cyclin E is an important regulator of entry into the S-phase of the cell cycle. Cyclin E overexpression and S-phase entry are associated with adenovirus selective replication in cancer cells [63]. As replication of all viruses is somehow dependent on the synthetic machinery of host cells, aggressively growing cancer cells are likely to provide a more suitable cellular environment for efficient virus replication and thus are more selectively destroyed.
The molecular basis for reovirus oncolytic selection, specifically the role of the Ras pathway, PKR activation, and cell cycle in reovirus replication needs further investtigation.
4. Mechanisms of Reovirus-Induced Cell Death
Oncolytic reovirus T3D is considered a strong candidate in cancer therapy. It is important to elucidate the mechanisms of reovirus-induced cell death. Many studies have indicated that reovirus replication induces apoptosis in cultured cells and in vivo. Reovirus-induced death of melanoma cell lines and Ras-transformed NIH-3T3 cells has been shown to be caspase-dependent, indicating that reovirus can cause apoptosis [51,64]. Reovirus release is mediated by caspase-induced apoptosis in Ras-transformed cells [51]. Caspase inhibition has been shown to block reovirus-induced melanoma cell death [64]. Reovirus-induced apoptosis depends on both the extrinsic pathway regulated by dead receptors and the intrinsic pathway which involves the mitochondrial release of apoptosis activator smac/DIABLO [65,66].
The transcription factor nuclear factor kappa B (NFKappaB) may play a significant role in reovirus-induced apoptosis in susceptible host cells [67]. In vitro studies have shown that during apoptosis in infected cells, reovirus activates NF-KappaB mainly via the classical pathway that is marked by the formation of p50 and RelA dimers [68]. Furthermore, the reovirus-induced NF-KappaB activation has been shown to involve the select components of both classical and alternative pathways in which the IKKα rather than IKKβ is required in the complex formation with IKKγ/Nemo [68].
The finding that the reovirus-induced apoptosis of infected cells requires virion disassembly upon cell entry indicates that the viral component(s) may be involved in this process [69]. One reovirus component that has been shown to play a role in the induction of apoptosis in infected cells is the outer capsid protein mu1, more specifically the C terminal φ domain of the protein [70]. The reovirus mu1 has been shown to induce apoptosis in reovirus-infected or mu1 expressing cells via both the caspase 8 activation regulated by dead receptors and the caspase 9 activation modulated by mitochondrial membrane permeability [71]. Additionally Mu1 induced apoptosis of infected cells or cells ectopically expressing mu1 has been shown to be independent of proapoptotic Bcl-2 family members Bak and Bax [71].
With regards to the reovirus activated intrinsic apoptotic pathway, it has been shown in vivo that the proapoptotic protein Bax has a vital role in the reovirusinduced apoptosis of infected neurons and thus in the CNS pathogenesis of infected mice [72]. The important roles of the proapoptotic protein Bid in the reovirus induced apoptosis and neurovirulence has been demonstrated in vitro and in vivo [73]. The activation of Bid upon reovirus infection has been shown to require NF-KappaB signaling and stimulation of TRAIL receptor [73]. Stabilization of p53 can enhance reovirus-induced apoptosis through p53-dependent NF-kappaB activation [74].
Additionally the role of interferon regulatory factor-3 (IRF-3) in the efficient induction of apoptosis in reovirus infected cells has been demonstrated in vitro [75]. It has been shown that reovirus activates IRF-3 in infected cells via a mechanism involving the retinoic acid inducible gene-1 (RIG-1) which is a cellular sensor for dsRNA, and the adaptor molecule interferon-β promoter stimulator-1 (IPS-1) [75]. A recent study has demonstrated that the reovirus-induced apoptosis of infected cells is IFN-β independent and involves the activation of IRF3 and NFKappaB which ultimately induces Bcl-2 family member pro apoptotic protein NOXA expression [76]. It has also been shown recently that reovirus induces apoptosis via stimulating endoplasmic reticular stress and up regulating the expression of Bcl-2 family member pro apoptotic protein NOXA in vitro in infected multiple myeloma cell lines and primary tumor specimens [77].
With regards to the reovirus activated extrinsic apoptotic pathway, a report has shown that reovirus-induced apoptosis is mediated by TRAIL (tumor necrosis factorrelated apoptosis inducing ligand) and associated with its release from infected cells [78]. The same group has further demonstrated that reovirus causes down regulation of CFLIP, which is a caspase 8 inhibitory protein and thus increases the susceptibility of cancer cells to TRAILinduced apoptosis [79]. A recent study demonstrated that reovirus infection down-regulates Akt activation, leading to the apoptosis of TRAIL-resistant gastric cancer cells [80].
It has been demonstrated that the reovirus T3D induces the apoptosis of neurons by up regulating the expression of Fas dead receptor which depends on the c-Jun N-terminal kinase (JNK) signaling in the brain tissue of infected mice [81]. Upon reovirus infection, the adaptor molecule dead associated protein 6 (Daxx) is upregulated in neuron cells via a type I interferon mediated mechanism in vitro [82]. Daxx has been shown to be involved in the reovirus-induced apoptosis of neurons by linking the Fas and JNK signaling and by up regulating the caspase 3 expression in infected mice [82].
One mechanism utilized by reovirus in the induction of apoptosis may involve the inactivation of microRNA-let-7d in infected cells, possibly via the RNA interfering mechanisms of the reovirus genome [83]. The increased expression of caspase 3 in reovirus infected cells has been attributed to the decreased levels of microRNA-let-7d which is known to target caspase 3 expression [83,84].
Growing evidence indicates that reovirus infection leads to autophagy development in some cancer cells in vitro. It has been shown cytologically that reovirus induces autophagy in infected multiple myeloma cells during oncolysis [85,86]. Autophagy is a conserved catabolic process which develops in response to various cellular stress conditions and involves the lysosomal degradation of intracellular proteins and organelles [87]. In general autophagy is a cell death process and is called type II programmed cell death. However in certain cellular conditions, for instance in established cancers, autophagy could lead to cell survival by protecting cancer cells against therapeutic and metabolic stresses [88]. In the viral infection setting, autophagy contributes to host innate immune responses and prevents the virus replication in infected cells [89]. In this context some viruses have adapted mechanisms to exploit the autophagic defense for their propagation in the host [89]. Reovirus induced autophagy in cancer cells may involve the induction of endoplasmic reticular stress [77,85] which has been shown to lead to autophagic cell death via the Akt-mTOR signaling [90,91]. Besides this, autophagy induced by avian reovirus in infected cells may involve the Akt-mTOR pathway and promotes virus production in vitro [92]. However, the significance of reovirus induced autophagy in the induction of cancer cell death should be further studied to gain better insight into reovirus-mediated oncolysis.
Reovirus has also been shown to cause tumor cell necrosis in head and neck squamous carcinoma cells in vivo [93]. Necrotic cell death is defined by swelling of the cell cytoplasm and organelle dysfunction that consequently leads to membrane destabilization and lysis of the cell [94]. Recent findings have revealed that necrosis is indeed a regulated process [95]. Programmed necrosis (also called necroptosis) is regulated by a series of cellular mediators including receptor interacting protein kinase 1 and 3 (RIPK1,3), and Poly ADP-ribose polymerases (PARPs). These key elements of programmed necrosis manage cell death by increasing mitochondrial membrane permeability [95]. With regards to reovirus oncolysis, whether the induction of necrosis in infected head and neck squamous carcinoma cells involves the recruitment of RIPK1 and RIPK3 remains to be determined.
The exact mechanism of reovirus-induced tumor cell death remains controversial. Reovirus may induce cancer cell death with multiple mechanisms. These include the apoptotic pathway which is considered the fundamental pathway in reovirus-induced cancer cell death, the autophagy pathway which is significantly induced in infected myeloma cells, and the necrotic cell death pathway demonstrated in head and neck cancer cells. One example of reovirus affecting multiple cell death mechanisms is the finding that reovirus induces different programmed cell death pathways in cell lines of squamous cell carcinoma of head and neck in vitro [60]. It has been shown that in some head and neck cancer cells, reovirus induced oncolysis is independent of Ras signaling and caspase activation [60]. This finding could be the result of reovirus induced necrosis, as in fact the programmed necrosis has been shown to involve the apoptosis inducing factor (AIF) which is activated via a caspase independent manner [96]. In this context, identifying the complex interactions between these different programmed cell death pathways and their modulations by the cancer cells will be useful for gaining insight into reovirus oncolysis.
5. Clinical Studies with Oncolytic Reovirus
Reovirus therapy efficacy has been demonstrated in animal models of many human tumor types with reovirus T3D delivered by intratumoral and intravenous injection. These cancer types include breast, colon, ovarian, lung, neurological, hematological, pancreatic, sarcoma, and bladder neoplasms [97-104]. Virus replication has also been documented in ex vivo human tumor surgical specimens [98,102]. These preclinical studies provided the rationale for using reoviruses in human studies and ultimately have been translated into clinical trials. Preclinical studies are essential for demonstrating the concept of reovirus-mediated oncolysis in different tumor models to justify prospective clinical trials and for understanding the cellular and molecular mechanisms of reovirus induced cancer cell death.
Oncolytics Biotech, a biotechnology company based in Calgary, Canada, focuses on the development of oncolytic viruses as potential cancer therapeutics and its clinical program includes 31 completed and ongoing clinical trials using reovirus spanning over a decade. These clinical trials include phase I studies which are conducted to determine toxicity aspects associated with dose escalation, and to obtain preliminary results for efficacy, phase II studies which are conducted with predetermined treatment regimes on patients with disease, primarily for effectiveness evaluation and secondly for determining toxicity aspects, and phase III studies conducted on expanded groups consisting of patients with certain disease conditions and controls, with the aim of assessing overall benefit-risk relationship for the treatment, the effectiveness of which has been previously demonstrated. Oncolytics Biotech is conducting clinical studies using Reolysin, which is the company’s proprietary formulation of the human reovirus T3D. There are several excellent reviews covering clinical studies of reoviruses [105-107]. We will attempt to focus on how these studies have led to the current Phase III trial in head and neck cancers.
Of the 31 clinical trials of Reolysin, there are 10 phase I, 4 phase I/II, 14 phase II, 2 translational and 1 phase III studies. Currently, 13 of them have been completed and 18 are still ongoing. All clinical trials completed or on going with reported preliminary results indicate that Reolysin has been well tolerated with low grade toxicity and has shown measureable antitumor efficacy.
Therapeutic reovirus was first studied in humans via intratumoral injection. The first three clinical trials were local monotherapy of Reolysin for patients with subcutaneous tumors (REO 001), T2 prostate cancer (REO 002) [108] or recurrent malignant gliomas (REO 003) [105, 109]. These studies demonstrated that direct intratumoral delivery of reovirus was safe and well tolerated, with no dose-limiting toxicities observed. Modest clinical responses were observed in these studies. The results of these studies supported the necessity of further investigation of reovirus therapy via systemic administration of Reolysin for patients with metastatic tumors.
Two early phase I studies of intravenous administration of wild type reovirus T3D for patients with various metastatic tumors have been completed, one in the United Kingdom [110] and one in the United States [111]. The primary objective of the studies was to determine the safety of Reolysin when administered intravenously, and the secondary objectives were to observe tumor and immune system responses to intravenous infusion of T3D to determine dosage levels in further clinical studies. In the United Kingdom study, 33 patients received escalating doses of reovirus up to 3 × 1010 TCID(50) for 5 consecutive days every 4 weeks. There was no dose-limiting toxicity observed [110]. Reolysin was shown to be safely delivered to various tumor types via intravenous injections and resulted in virus-mediated tumor responses. One of the important findings of this study was that replication-competent virus was recovered from tumor biopsy samples, indicating the ability of the virus to reach the tumor after systemic administration [110]. In the clinical trial conducted in United States, a total of 18 patients were treated in the escalating dosage trial to a maximum daily dose of 3 ´ 1010 TCID(50) in a one-hour infusion [111]. One patient with breast cancer experienced a partial response (PR) with tumor shrinking of 34% in volume and 7 patients experienced stable disease (SD). The overall clinical benefit (1 PR + 7 SD) rate was 44% in this study. These two studies, as well as other intravenous studies, indicated that systematic administration is safe and well tolerated, even in multiple doses. The observed anti-tumor activity of Reolysin as a single agent warranted further evaluation.
To increase the therapeutic efficacy, the reolysis approach has been combined with standard chemotherapy or radiation therapy. Multiple phase I and II studies using combinations of reovirus and a conventional therapy have been conducted on patients with various pretreated advanced cancers. The combination treatments of intratumoral reovirus with chemotherapy or radiotherapy were well tolerated and tumor responses were observed in patients with various advanced cancers [105-107]. These studies evaluated the safety aspects of the combination therapy and its therapeutic effects on various tumors and determined the optimal dose of Reolysin for combination therapy.
According to Oncolytics Biotech reports, two studies of intravenously administered reovirus in combination with both paclitaxel and carboplatin in patients with head and neck cancer have been completed in the UK (REO 011 trial) [112] and in the US (REO 015 trial) (http://www.oncolyticsbiotech.com/clinical-trials). The cancers treated were mostly squamous cell carcinoma refractory to prior platinum-based chemotherapy for recurrent and metastatic disease. The REO 011 trial was a phase I/II study with the primary objectives to determine the maximum tolerated dose (MTD), dose-limiting toxicity (DLT), optimal dose and dosing schedule and safety profile of Reolysin when administered in combination with both paclitaxel and carboplatin in patients with advanced head and neck cancers. The final results showed that the three-way combination of Reolysin with paclitaxel and carboplatin was well tolerated. Of 19 evaluable patients, 1 had complete response, 8 had partial responses (42%) and six had stable disease (32%) [112]. The independent Phase II REO 015 trial was a confirmatory trial of an earlier REO 011 study, using the same combination of Reolysin and carboplatin/paclitaxel for patients with head and neck cancers. This study has been completed recently in the U.S. The primary objectives of the Phase II trial were to measure tumor responses and duration of response, and to describe any evidence of antitumor activity. All patients had previously received standard chemotherapy and/or radiotherapy for their metastatic or recurrent disease. Of the 13 patients evaluable for response, 4 patients had partial responses (31%) and 6 patients had stable disease or better for 12 weeks or longer (46%). The total clinical benefit rate (partial response + stable disease) was 77% in the trial, similar to the results of the REO 011 trial. The results of the two clinical trials are promising because head and neck cancers generally have a very low rate (3% to 7%) of success in treatment of patients who are refractory to first-line treatments. Based on these promising results, a phase III study (REO 018) is currently being conducted to investigate the combination of reovirus with carboplatin and paclitaxel in this patient population.
In September 2012, Oncolytics Biotech provided an update of the phase III trial of intravenous administration of Reolysin in combination with carboplatin and paclitaxel for the treatment of head and neck cancers (http://www.oncolyticsbiotech.com/clinical-trials). The company had conducted an internal analysis of the blinded combined clinical data for all 80 patients enrolled in the first stage of the study. The median evolving progression free survival (PFS) of the 80 patients, including the control and test groups, was greater than expected. Patients in whom only metastatic disease was measured had a median evolving PFS of 120 days. The response was statistically significant, suggesting that the combination treatment may provide greater benefit to patients with metastatic disease. Based on these promising results and discussions with the US Food and Drug Administration, the company expanded enrollment in the phase III trial to add additional 160 patients. The company expects this will provide a sufficient number of patients to conduct a meaningful statistical analysis.
It has become clear that to reach maximal therapeutic efficacy, reovirus will be used in combination with other anti-cancer agents. Preclinical and clinical studies will focus on identifying the synergistic antitumor efficacy of reovirus therapy combined with chemotherapy, radiotherapy, and targeted therapies. Some of the combinational studies and such synergies have been reported [103,113-117]. On the other hand, synergistic antitumor effects of reovirus oncolysis combined with targeted therapies (inhibitory small-molecules and antibody-based therapies) are also plausible and particularly appealing from a toxicity standpoint.
6. Immune Response against Reovirus and Tumors
Animal and clinical studies have indicated that immune responses may have less effect on reovirus oncolytic treatment following intratumoral delivery [118,119]. However, an antiviral immune response can be a critical obstacle to viral tumor targeting following intravenous delivery. It has been demonstrated that suppression of the immune system improved the efficacy of reovirus therapy. Intrahepatic administration of reovirus to liver tumor models has shown that reovirus inhibits tumor growth more efficiently in mice treated with the immunosuppressive drug cyclosporin A [120]. The combination of reovirus and cisplatin significantly delayed tumor growth and prolonged survival compared to reovirus or cisplatin alone treatment in a mouse melanoma model [115]. Cisplatin, used in the treatment of a variety of cancers, has been found to down regulate inflammatory cytokine gene expression induced by reovirus infection.
In order to improve the systemic delivery and antitumor efficacy of reovirus, the effects of cyclophosphamide (CPA) on immune-modulation were investigated with an immunocompetent syngenic tumor animal model [121]. The study has shown that CPA selectively inhibits the T regulatory cell activity and antibody response to reovirus. CPA may modulate the adaptive immunity in favor of tumor specific T-cell response, increasing the therapeutic efficacy of the reovirus. In combination with IL2, CPA has been shown to enhance the NK cell activity that may also enhance reovirus therapy [121].
Although repression of immune systems increases reovirus tumor targeting, virus infection-activated immune responses may promote systematic anti-tumor activity. A recent study has shown that reovirus therapy overrides tumor immune evasion and establishes clinically meaningful antitumor immunity capable of protecting against subsequent tumor challenge [122]. A report has shown that reovirus-infected tumor cells released a variety of inflammatory cytokines and chemokines which can cause bystander toxicity against tumors and activates human myeloid dendritic cells (DCs) [64]. These activated DCs may then stimulate innate killing by natural killer cells and T cell mediated adaptive immunity, complementing the direct oncolytic effects of reovirus. Reovirus-mediated oncolysis may also promote the display of otherwise inaccessible tumor-specific immunogenic peptides on the surface of DCs and facilitate the recognition of tumor antigens. DCs produce cytokines (such as IL-1 and IL-6), undergo maturation, and migrate into the tumor microenvironment along with CD8 T cells. Reovirus virotherapy augments the efficacy of DCand T cell-based anticancer immunotherapies and increases survival in tumor-bearing mice.
It has been shown that the immune-mediated antitumor activity of reovirus is independent of direct viral oncolysis and replication [123]. In immunocompetent mice, but not in severely combined immunodeficient mice bearing lymph node melanoma, systemic delivery of non-specific T cells infected with reovirus is able to clear lymph node tumors which are resistant to reovirus replication. Additionally it has been demonstrated that UV-inactivated non-replicating reovirus, when added to dendritic cells already loaded with melanoma cells, can function as an adjuvant to prime specific antitumor cytotoxic lymphocytes [123].
A recently completed phase I trial of intravenous reovirus has conducted a detailed analysis of neutralizing anti-reoviral antibodies, peripheral blood mononuclear cells, and cytokines [119]. Significant increases in antireoviral antibodies were seen with a median fold increase of 250. A significant number of patients had increases in CD3+CD4+ and CD3+CD8+ T cells, and CD3−CD56+ natural killer cells following treatment. Additionally, combined Th1 and Th2 cytokine expression increased in 38% of patients. These data indicate the possibility that reovirus intravenous injection may stimulate the cellular immune response against tumors in patients [119]. Combination of reovirus with gemcitabine attenuated the neutralizing antibody responses to reovirus and this could impact efficacy of this treatment [124].
It appears that immune response to reovirus intravenous delivery is a double-edged sword. On the one hand, the response inhibits the efficacy of tumor cell infection by reovirus, but on the other hand the response may stimulate antitumor immune reactions. Therefore, it will be very important to understand how to selectively repress the immune response that decrease reovirus targeting tumor cells, but enhance the antitumor immune reactions. Specifically, chemotherapy agents that repress the development of the anti-reovirus antibody or attenuate the neutralizing antibody response, but enhance cellular immunity favoring reovirus-initiated antitumor response, may greatly improve reovirus tumor targeting and enhance therapeutic efficacy.
7. Oncolytic Reovirus Vector Development
Until now only the unmodified wild-type reovirus T3D has been extensively studied in clinical trials and shown significant potential as an oncolytic agent. Although the naturally occurring reovirus causes only mild symptoms in immunocompetent humans, the virus may infect healthy tissues and cause adverse side effects especially in immunosuppressed patients. A study has shown that immunocompromised mice exhibit “Black Foot” syndrome (discoloration and necrosis of extremities including feet, distal leg, tail and ears) after reovirus injection, likely due to reovirus infection-caused myocarditis and heart failure [125]. Therefore, it is important to further promote reovirus oncolytic properties while improve its safety. However, because the reoviral genome is formed by 10 separate dsRNA segments, it is difficult to modify its genes with DNA genetic engineering approaches. Two approaches, reverse genetic and classical genetic, may be applied to modify reovirus genes.
A reverse genetic system for modification of reovirus genes has been described [126]. The system, consisting of transcripts of the genetically modified cDNA form of a reovirus genome segment, was used to incorporate the CAT gene into the reovirus genome. With a similar approach, another report has shown that reovirus can be genetically modified to target cancer cells [127]. The study demonstrated that the C-terminus of the σ1 protein of reovirus is a suitable locale for the insertion of oligopeptide ligands for cancer targeting. Recently a plasmid based reverse genetic system for reovirus strains T1L and T3D has been established which does not require helper virus or antibody selection [128]. In this system each reovirus genome segment is cloned into separate plasmids in a region flanked by the bacteriophage T7 promoter and the hepatitis delta virus (HDV) ribozyme with the aim of obtaining viruses with native genome segment terminals. The progeny virus particles can be rescued from cells transfected with these plasmids containing all reovirus genome segments along with supplying the bacteriophage T7 RNA polymerase with recombinant vaccinia virus infection [128]. By using this system, it has been shown that up to 120 nucleotides can be added to the σ1 carboxyl terminal end coding region of the S1 genome segment [129]. The efficacy of the system has been improved with reducing the number of plasmids simply by cloning two or more reovirus genome segments in a single plasmid and with using a cell system that constitutively express the bacteriophage T7 RNA polymerase [130].
A classical genetic approach has been applied to generate and isolate attenuated reovirus from cells after persistent infection [131]. The mutant strain has a premature STOP codon in its σ1 gene, resulting in truncation of the cell attachment protein. The mutant reovirus did not kill healthy stem cells and showed reduced toxicity when administered to immunodeficient hosts, indicating improved safety in comparison to wild-type reovirus. Despite containing a truncated attachment protein, the reovirus still preferentially targets cancer cells and retains its oncolytic activity in vivo [131]. In a recent study, also via a classical genetic approach, two mutant reoviruses with enhanced infectivity and oncolytic activity have been isolated [132]. The two mutants exhibit a large plaqueforming phenotype associated with unique mutations in viral lambda2 vertex protein and σ1 cell attachment protein, respectively. An analysis of virus replication revealed that the mutants were more efficient than wildtype reovirus in initiating productive infection and thus produced significantly higher levels of early viral RNAs. In a syngenic mouse model of melanoma, both mutants increased mouse survival compared with the wild-type reovirus [132].
The strain T3D belonging to orthoreovirus genus is the only reovirus that has been extensively studied and is currently being evaluated in clinical trials. However, other reovirus subtypes in the orthoreovirus genus all showed oncolytic activity in cancer cells [3]. These subtypes should be further investigated and compared with T3D. In addition to orthoreovirus, there are four other genera, rotaviruses, orbiviruses, coltivirus and seadornavirus in the reoviridae family that also infect animals including humans (Table 1) [133]. Strains in these genera may also be capable of selectively destroying cancer cells, and thus should be explored for oncolytic virus therapy.
8. Summary and Perspectives
Reovirus is a dsRNA virus that naturally targets and replicates within cancer cells, causing oncolysis. Reovirus holds great advantages as an anticancer agent over other viruses because of its low pathogenesis and the lack of a DNA synthesis stage in its viral life cycle, avoiding integration mutation. Reolysin, the human reovirus serotype 3 Dearing, has been extensively studied in preclinical and clinical trials. In all reported clinical trials, Reolysin is well tolerated with low grade toxicities and shows recognizable antitumor efficacy. However, there are at least 3 major challenges in the development of reovirus-mediated oncolytic therapy.
First, the mechanisms underlying reoviral oncolysis remain to be fully characterized. Specifically, the role of the Ras pathway and PKR activation in reovirus selective replication and cancer cell death needs further investigation. It should be noted that this Ras-PKR hypothesis is yet to be substantiated with convincing evidence. A similar situation was faced by the scientific community when E1b-deleted adenoviruses were developed for oncolytic therapy. The antitumor selection of the virus was originally proposed to be dependent on p53 inactivation in cancer cells, but extensive studies have revealed that selective replication of the mutated adenoviruses is unlikely dependent on p53 activity or its pathway in cancer cells. Recent studies have shown that cyclin E overexpression may be associated with the selective replication of E1b-mutated adenovirus in cancer cells [63]. Aggressive proliferation of cancer cells may be related to cancer selection of oncolytic viruses. Although reovirus, like adenovirus, has been used in cancer therapy and has already exhibited encouraging results in clinical trials, a better understanding of the molecular basis of reovirusmediated oncolysis will allow us to develop better oncolytic viruses and therapeutic strategies.
Second, it is important to study how to promote the immune responses against cancers but repress the immune responses against reovirus tumor targeting. Antiviral immune responses, existing before or activated after virus injection, can decrease viral tumor targeting and spreading. Selective suppression of the immune responses that inactivate reovirus tumor infection and spread into tumors will improve the efficacy of reovirus therapy. However, suppression of antivirus activation may increase the possibility of reovirus infection of healthy tissues and cause adverse side effects in cancer patients. In addition, it is likely that without activating cancer patients’ immune systems to fight tumors, reovirus-mediated therapeutic effects may not last long. Virus infection-activated immune responses may benefit induction of systematic anti-tumor activity. Recent studies have indicated that reovirus oncolytic therapy may promote the display of tumor immune-antigens to override a tumor immune evasion and establish clinically meaningful antitumor immunity [122]. Chemotherapy agents that enhance immunity in favor of antitumor responses may immediately lead to greater efficacy and patient benefits. However, the exact role of innate and adaptive immune responses in virus-mediated tumor regression has not yet been clearly defined.
The third challenge, mainly for virologists, is to improve oncolytic reovirus and develop new vectors. Currently only the wild-type reovirus T3D strain has been extensively studied in preclinical and clinical trials. To improve the therapeutic efficacy and decrease potential toxicities of reovirus, wild-type reovirus may be genetically modified, so the vector may more selectively infect and destroy cancer cells. Genetic engineering methods have been efficiently used in the modification of adenoviruses and other DNA viruses, but have not been applied extensively in the development of oncolytic reovirus, due to the difficulties in manipulating the reovirus genome which consists of 10 separate dsRNA segments. Reverse genetic and classical genetic approaches must be further investigated for modification and selection of oncolytic reoviruses. In addition, other virus strains in the reoviridae family may also have the potential to infect and destroy cancer cells, and therefore should be systematically studied.
9. Acknowledgements
This work was partially supported by NIH Grant R01 CA129975 (HSZ). We thank Stephen L. Wechman for critically reading the manuscript and helpful suggestions.
REFERENCES
- M. Ramos-Alvarez and A. B. Sabin, “Characteristics of Poliomyelitis and Other Enteric Viruses Recovered in Tissue Culture from Healthy American Children,” Proceedings of the Society for Experimental Biology and Medicine, Vol. 87, No. 3, 1954, pp. 655-661. doi:10.3181/00379727-87-21474
- A. B. Sabin, “Reoviruses. A New Group of Respiratory and Enteric Viruses Formerly Classified as ECHO Type 10 Is Described,” Science, Vol. 130, No. 3386, 1959, pp. 1387-1389. doi:10.1126/science.130.3386.1387
- S. H. Alloussi, M. Alkassar, S. Urbschat, N. Graf and B. Gartner, “All Reovirus Subtypes Show Oncolytic Potential in Primary Cells of Human High-Grade Glioma,” Oncology Reports, Vol. 26, No. 3, 2011, pp. 645-649.
- L. A. Schiff, M. L. Nibert and K. L. Tyler, “Orthoreoviruses and Their Replication,” In: Fields Virology, 5th Edition, Lippincott Williams and Wilkins, 2007, pp. 1853-1915.
- K. W. Boehme, K. M. Guglielmi and T. S. Dermody, “Reovirus Nonstructural Protein Signa1 Is Required for Establishment of Viremia and Systemic Dissemination,” Proceedings of the National Academy of Science USA, Vol. 106, No. 47, 2009, pp. 19986-19991.
- M. M. Becker, M. I. Goral, P. R. Hazelton, G. S. Baer, S. E. Rodgers, E. G. Brown, et al., “Reovirus sigmaNS Protein Is Required for Nucleation of Viral Assembly Complexes and Formation of Viral Inclusions,” Journal of Virology, Vol. 75, No. 3, 2001, pp. 1459-1475. doi:10.1128/JVI.75.3.1459-1475.2001
- T. Kobayashi, J. D. Chappell, P. Danthi and T. S. Dermody, “Gene-Specific Inhibition of Reovirus Replication by RNA Interference,” Journal of Virology, Vol. 80, No. 18, 2006, pp. 9053-9063.
- P. W. Lee, E. C. Hayes and W. K. Joklik, “Protein Sigma 1 Is the Reovirus Cell Attachment Protein,” Virology, Vol. 108, No. 1, 1981, pp. 156-163. doi:10.1016/0042-6822(81)90535-3
- D. M. Reiter, J. M. Frierson, E. E. Halvorson, T. Kobayashi, T. S. Dermody and T. Stehle, “Crystal Structure of Reovirus Attachment Protein Sigma1 in Complex with Sialylated Oligosaccharides,” PLoS Pathogens, Vol. 7, No. 8, 2011. doi:10.1371/journal.ppat.1002166
- K. Reiss, J. E. Stencel, Y. Liu, B. S. Blaum, D. M. Reiter, T. Feizi, et al., “The GM2 Glycan Serves as a Functional Coreceptor for Serotype 1 Reovirus,” PLoS Pathogens, Vol. 8, No. 12, 2012. doi:10.1371/journal.ppat.1003078
- E. S. Barton, J. L. Connolly, J. C. Forrest, J. D. Chappell and T. S. Dermody, “Utilization of Sialic Acid as a Coreceptor Enhances Reovirus Attachment by Multistep Adhesion Strengthening,” Journal of Biological Chemistry, Vol. 276, No. 3, 2001, pp. 2200-2211. doi:10.1074/jbc.M004680200
- J. D. Chappell, A. E. Prota, T. S. Dermody and T. Stehle, “Crystal Structure of Reovirus Attachment Protein Sigma1 Reveals Evolutionary Relationship to Adenovirus Fiber,” EMBO Journal, Vol. 21, No. 1-2, 2002, pp. 1-11. doi:10.1093/emboj/21.1.1
- E. S. Barton, J. C. Forrest, J. L. Connolly, J. D. Chappell, Y. Liu, F. J. Schnell, et al., “Junction Adhesion Molecule Is a Receptor for Reovirus,” Cell, Vol. 104, No. 3, 2001, pp. 441-451. doi:10.1016/S0092-8674(01)00231-8
- J. A. Campbell, P. Schelling, J. D. Wetzel, E. M. Johnson, J. C. Forrest, G. A. Wilson, et al., “Junctional Adhesion Molecule a Serves as a Receptor for Prototype and FieldIsolate Strains of Mammalian Reovirus,” Journal of Virology, Vol. 79, No. 13, 2005, pp. 7967-7978. doi:10.1128/JVI.79.13.7967-7978.2005
- M. S. Maginnis, J. C. Forrest, S. A. Kopecky-Bromberg, S. K. Dickeson, S. A. Santoro, M. M. Zutter, et al., “Beta1 Integrin Mediates Internalization of Mammalian Reovirus,” Journal of Virology, Vol. 80, No. 6, 2006, pp. 2760-2770. doi:10.1128/JVI.80.6.2760-2770.2006
- M. S. Maginnis, B. A. Mainou, A. Derdowski, E. M. Johnson, R. Zent and T. S. Dermody, “NPXY Motifs in the Beta1 Integrin Cytoplasmic Tail Are Required for Functional Reovirus Entry,” Journal of Virology, Vol. 82, No. 7, 2008, pp. 3181-3191. doi:10.1128/JVI.01612-07
- M. Ehrlich, W. Boll, A. Van Oijen, R. Hariharan, K. Chandran, M.L. Nibert, et al., “Endocytosis by Random Initiation and Stabilization of Clathrin-Coated Pits,” Cell, Vol. 118, No. 5, 2004, pp. 591-605. doi:10.1016/j.cell.2004.08.017
- D. H. Rubin, D. B. Weiner, C. Dworkin, M. I. Greene, G. G. Maul and W. V. Williams, “Receptor Utilization by Reovirus Type 3: Distinct Binding Sites on Thymoma and Fibroblast Cell Lines Result in Differential Compartmentalization of Virions,” Microbial Pathogens, Vol. 12, No. 5, 1992, pp. 351-365. doi:10.1016/0882-4010(92)90098-9
- L. J. Sturzenbecker, M. Nibert, D. Furlong and B. N. Fields, “Intracellular Digestion of Reovirus Particles Requires a Low pH and Is an Essential Step in the Viral Infectious Cycle,” Journal of Virology, Vol. 61, No. 8, 1987, pp. 2351-2361.
- J. Borsa, B. D. Morash, M. D. Sargent, T. P. Copps, P. A. Lievaart and J. G. Szekely, “Two Modes of Entry of Reovirus Particles into L Cells,” Journal of General Virology, Vol. 45, No. 1, 1979, pp. 161-170. doi:10.1099/0022-1317-45-1-161
- W. L. Schulz, A. K. Haj and L. A. Schiff, “Reovirus Uses Multiple Endocytic Pathways for Cell Entry,” Journal of Virology, Vol. 86, No. 23, 2012, pp. 12665-12675. doi:10.1128/JVI.01861-12
- B. A. Mainou and T. S. Dermody, “Transport to Late Endosomes Is Required for Efficient Reovirus Infection,” Journal of Virology, Vol. 86, No. 16, 2012, pp. 8346- 8358. doi:10.1128/JVI.00100-12
- E. Maratos-Flier, M. J. Goodman, A. H. Murray and C. R. Kahn, “Ammonium Inhibits Processing and Cytotoxicity of Reovirus, a Nonenveloped Virus,” Journal of Clinical Investigation, Vol. 78, No. 4, 1986, pp. 1003-1007. doi:10.1172/JCI112653
- D. H. Ebert, J. Deussing, C. Peters and T. S. Dermody, “Cathepsin L and Cathepsin B Mediate Reovirus Disassembly in Murine Fibroblast Cells,” Journal of Biological Chemistry, Vol. 277, No. 27, 2002, pp. 24609-24617. doi:10.1074/jbc.M201107200
- J. W. Golden, J. A. Bahe, W. T. Lucas, M. L. Nibert and L. A. Schiff, “Cathepsin S Supports Acid-Independent Infection by Some Reoviruses,” Journal of Biological Chemistry, Vol. 279, No. 10, 2004, pp. 8547-8557. doi:10.1074/jbc.M309758200
- G. S. Baer, D. H. Ebert, C. J. Chung, A. H. Erickson and T. S. Dermody, “Mutant Cells Selected during Persistent Reovirus Infection Do Not Express Mature Cathepsin L and Do Not Support Reovirus Disassembly,” Journal of Virology, Vol. 73, No. 11, 1999, pp. 9532-9543.
- J. A. Madren, P. Sarkar and P. Danthi, “Cell Entry-Associated Conformational Changes in Reovirus Particles Are Controlled by Host Protease Activity,” Journal of Virology, Vol. 86, No. 7, 2012, pp. 3466-3473. doi:10.1128/JVI.06659-11
- K. Chandran, D. L. Farsetta and M. L. Nibert, “Strategy for Nonenveloped Virus Entry: A Hydrophobic Conformer of the Reovirus Membrane Penetration Protein Micro 1 Mediates Membrane Disruption,” Journal of Virology, Vol. 76, No. 19, 2002, pp. 9920-9933. doi:10.1128/JVI.76.19.9920-9933.2002
- S. Liemann, K. Chandran, T. S. Baker, M. L. Nibert and S. C. Harrison, “Structure of the Reovirus MembranePenetration Protein, Mu1, in a Complex with Is Protector Protein, Sigma3,” Cell, Vol. 108, No. 2, 2002, pp. 283- 295. doi:10.1016/S0092-8674(02)00612-8
- L. Zhang, K. Chandran, M. L. Nibert and S. C. Harrison, “Reovirus Mu1 Structural Rearrangements That Mediate Membrane Penetration,” Journal of Virology, Vol. 80, No. 24, 2006, pp.12367-12376. doi:10.1128/JVI.01343-06
- T. Ivanovic, M. A. Agosto, L. Zhang, K. Chandran, S. C. Harrison and M. L. Nibert, “Peptides Released from Reovirus Outer Capsid Form Membrane Pores That Recruit virus Particles,” EMBO Journal, Vol. 27, No. 8, 2008, pp. 1289-1298. doi:10.1038/emboj.2008.60
- M. A. Agosto, T. Ivanovic and M. L. Nibert, “Mammalian Reovirus, a Nonfusogenic Nonenveloped Virus, Forms Size-Selective Pores in a Model Membrane,” Proceedings of the National Academy of Science USA, Vol. 103, No. 44, 2006, pp. 16496-16501. doi:10.1073/pnas.0605835103
- M. L. Nibert, A. L. Odegard, M. A. Agosto, K. Chandran and L. A. Schiff, “Putative Autocleavage of Reovirus Mu1 Protein in Concert with Outer-Capsid Disassembly and Activation for Membrane Permeabilization,” Journal of Molecular Biology, Vol. 345, No. 3, 2005, pp. 461-474. doi:10.1016/j.jmb.2004.10.026
- L. Zhang, M. A. Agosto, T. Ivanovic, D. S. King, M. L. Nibert and S. C. Harrison, “Requirements for the Formation of Membrane Pores by the Reovirus Myristoylated Micro1N Peptide,” Journal of Virology, Vol. 83, No. 14, 2009, pp. 7004-7014. doi:10.1128/JVI.00377-09
- M. Schonberg, S. C. Silverstein, D. H. Levin and G. Acs, “Asynchronous Synthesis of the Complementary Strands of the Reovirus Genome,” Proceedings of the National Academy of Science USA, Vol. 68, No. 2, 1971, pp. 505- 508. doi:10.1073/pnas.68.2.505
- M. Yamakawa, Y. Furuichi and A. J. Shatkin, “Reovirus Transcriptase and Capping Enzymes Are Active in Intact Virions,” Virology, Vol. 118, No. 1, 1982, pp. 157-168. doi:10.1016/0042-6822(82)90329-4
- Y. Furuichi, M. Morgan, S. Muthukrishnan and A. J. Shatkin, “Reovirus Messenger RNA Contains a Methylated, Blocked 5'-Terminal Structure: m-7G(5')ppp (5') GMpCp,” Proceedings of the National Academy of Science United States of America, Vol. 72, No. 1, 1975, pp. 362-366. doi:10.1073/pnas.72.1.362
- M. A. McCrae, “Terminal Structure of Reovirus RNAs,” Journal of General Virololgy, Vol. 55, No. 2, 1981, pp. 393-403. doi:10.1099/0022-1317-55-2-393
- B. N. Fields, C. S. Raine and S. G. Baum, “TemperatureSensitive Mutants of Reovirus Type 3: Defects in Viral Maturation as Studied by Immunofluorescence and Electron Microscopy,” Virology, Vol. 43, No. 3, 1971, pp. 569-578. doi:10.1016/0042-6822(71)90282-0
- S. C. Silverstein and P. H. Schur, “Immunofluorescent Localization of Double-Stranded RNA in Reovirus-Infected Cells,” Virology, Vol. 41, No. 3, 1970, pp. 564- 566. doi:10.1016/0042-6822(70)90178-9
- A. H. Sharpe, L. B. Chen and B. N. Fields, “The Interaction of Mammalian Reoviruses with the Cytoskeleton of Monkey Kidney CV-1 Cells,” Virology, Vol. 120, No. 2, 1982, pp. 399-411. doi:10.1016/0042-6822(82)90040-X
- J. S. Parker, T. J. Broering, J. Kim, D. E. Higgins and M. L. Nibert, “Reovirus Core Protein μ2 Determines the Filamentous Morphology of Viral Inclusion Bodies by Interacting with and Stabilizing Microtubules,” Journal of Virology, Vol. 76, No. 9, 2002, pp. 4483-4496. doi:10.1128/JVI.76.9.4483-4496.2002
- G. Hashiro, P. C. Loh and J. T. Yau, “The Preferential Cytotoxicity of Reovirus for Certain Transformed Cell Lines,” Archives of Virology, Vol. 54, No. 4, 1977, pp. 307-315. doi:10.1007/BF01314776
- M. R. Duncan, S. M. Stanish and D. C. Cox, “Differential Sensitivity of Normal and Transformed Human Cells to Reovirus Infection,” Journal of Virology, Vol. 28, No. 2, 1978, pp. 444-449.
- J. E. Strong, D. Tang and P. W. Lee, “Evidence That the Epidermal Growth Factor Receptor on Host Cells Confers Reovirus Infection Efficiency,” Virology, Vol. 197, No. 1, 1993, pp. 405-411. doi:10.1006/viro.1993.1602
- J. E. Strong, M. C. Coffey, D. Tang, P. Sabinin and P. W. Lee, “The Molecular Basis of Viral Oncolysis: Usurpation of the Ras Signaling Pathway by Reovirus,” EMBO Journal, Vol. 17, No. 12, 1998, pp. 3351-3362. doi:10.1093/emboj/17.12.3351
- K. L. Norman, K. Hirasawa, A. D. Yang, M. A. Shields and P. W. Lee, “Reovirus Oncolysis: the Ras/RalGEF/ p38 Pathway Dictates Host Cell Permissiveness to Reovirus Infection,” Proceedings of the National Academy of Science of the United States of America, Vol. 101, No. 30, 2004, pp. 11099-11104. doi:10.1073/pnas.0404310101
- J. A. McCubrey, L. S. Steelman, W. H. Chappell, S. L. Abrams, E. W. Wong, F. Chang, et al., “Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance,” Biochimica et Biophysica Acta, Vol. 1773, No. 8, 2007, pp. 1263-1284. doi:10.1016/j.bbamcr.2006.10.001
- J. L. Bos, “Ras Oncogenes in Human Cancer: A Review,” Cancer Research, Vol. 49, No. 17, 1989, pp. 4682-4689.
- T. Alain, T. S. Kim, X. Lun, A. Liacini, L. A. Schiff, D. L. Senger, et al., “Proteolytic Disassembly Is a Critical Determinant for Reovirus Oncolysis,” Molecular Therapy, Vol. 15, No. 8, 2007, pp. 1512-1521. doi:10.1038/sj.mt.6300207
- P. Marcato, M. Shmulevitz, D. Pan, D. Stoltz and P. W. Lee, “Ras Transformation Mediates Reovirus Oncolysis by Enhancing Virus Uncoating, Particle Infectivity and Apoptosis-Dependent Release,” Molecular Therapy, Vol. 15, No. 8, 2007, pp. 1522-1530. doi:10.1038/sj.mt.6300179
- M. Shmulevitz, L. Z. Pan, K. Garant, D. Pan and P. W. Lee, “Oncogenic Ras Promotes Reovirus Spread by Suppressing IFN-Beta Production Through Negative Regulation of RIG-I Signaling,” Cancer Research, Vol. 70, No. 12, 2010, pp. 4912-4921. doi:10.1158/0008-5472.CAN-09-4676
- G. S. Feng, K. Chong, A. Kumar and B. R. Williams, “Identification of Double-Stranded RNA-Binding Domains in the Interferon-Induced Double-Stranded RNAActivated p68 Kinase,” Proceedings of the National Academy of Science of the United States of America, Vol. 89, No. 12, 1992, pp. 5447-5451. doi:10.1073/pnas.89.12.5447
- S. A. Vorburger, A. Pataer, S. G. Swisher and K. K. Hunt, “Genetically Targeted Cancer Therapy: Tumor Destruction by PKR Activation,” American Journal of Pharmacogenomics, Vol. 4, No. 3, 2004, pp. 189-198. doi:10.2165/00129785-200404030-00006
- F. Imani and B. L. Jacobs, “Inhibitory Activity for the Interferon-Induced Protein Kinase Is Associated with the Reovirus Serotype 1 Sigma 3 Protein,” Proceedings of the National Academy of Science of the United States of America, Vol. 85, No. 21, 1988, pp. 7887-7891. doi:10.1073/pnas.85.21.7887
- L. Song, T. Ohnuma, I. H. Gelman and J. F. Holland, “Reovirus Infection of Cancer Cells Is Not Due to Activated Ras Pathway,” Cancer Gene Therapy, Vol. 16, No. 4, 2009, p. 382. doi:10.1038/cgt.2008.84
- H. Cui, Y. Lin, L. Yue, X. Zhao and J. Liu, “Differential Expression of the Alpha2,3-Sialic Acid Residues in Breast Cancer is Associated with Metastatic Potential,” Oncology Reports, Vol. 25, No. 5, 2011, pp. 1365-1371.
- D. Lopez-Morales, J. Reyes-Leyva, G. Santos-Lopez, E. Zenteno and V. Vallejo-Ruiz, “Increased Expression of Sialic Acid in Cervical Biopsies with Squamous Intraepithelial Lesions,” Diagnostic Pathology, Vol. 5, 2010, pp. 74. doi:10.1186/1746-1596-5-74
- G. Raval, S. Biswas, P. Rayman, K. Biswas, G. Sa, S. Ghosh, et al., “TNF-Alpha Induction of GM2 Expression on Renal Cell Carcinomas Promotes T Cell Dysfunction,” Journal of Immunology, Vol. 178, No. 10, 2007, pp. 6642- 6652.
- [61] K. Twigger, V. Roulstone, J. Kyula, E. M. Karapanagiotou, K. N. Syrigos, R. Morgan, et al., “Reovirus Exerts Potent Oncolytic Effects in Head and Neck Cancer Cell Lines that Are Independent of Signalling in the EGFR Pathway,” BMC Cancer, Vol. 12, No. 1, 2012, p. 368. doi:10.1186/1471-2407-12-368
- [62] L. Heinemann, G. R. Simpson, N. E. Annels, R. Vile, A. Melcher, R. Prestwich, et al., “The Effect of Cell Cycle Synchronization on Tumor Sensitivity to Reovirus Oncolysis,” Molecular Therapy, Vol. 18, No. 12, 2010, pp. 2085-2093. doi:10.1038/mt.2010.189
- [63] X. M. Rao, X. Zheng, S. Waigel, W. Zacharias, K. M. McMasters and H. S. Zhou, “Gene Expression Profiles of Normal Human Lung Cells Affected by Adenoviral E1B,” Virology, Vol. 350, No. 2, 2006, pp. 418-428. doi:10.1016/j.virol.2006.02.009
- [64] X. Zheng, X. M. Rao, J. G. Gomez-Gutierrez, H. Hao, K. M. McMasters and H. S. Zhou, “Adenovirus E1B55K Region is Required to Enhance Cyclin E Expression for Efficient Viral DNA Replication,” Journal of Virology, Vol. 82, No. 7, 2008, pp. 3415-3427. doi:10.1128/JVI.01708-07
- [65] F. Errington, C. L. White, K. R. Twigger, A. Rose, K. Scott, L. Steele, et al., “Inflammatory Tumour Cell Killing by Oncolytic Reovirus for the Treatment of Melanoma,” Gene Therapy, Vol. 15, No. 18, 2008, pp. 1257- 1270. doi:10.1038/gt.2008.58
- [66] D. J. Kominsky, R. J. Bickel and K. L. Tyler, “Reovirus-Induced Apoptosis Requires Both Death Receptorand Mitochondrial-Mediated Caspase-Dependent Pathways of Cell Death,” Cell Death and Differentiation, Vol. 9, No. 9, 2002, pp. 926-933.
- [67] D. J. Kominsky, R. J. Bickel and K. L. Tyler, “Reovirus-Induced Apoptosis Requires Mitochondrial Release of Smac/DIABLO and Involves Reduction of Cellular Inhibitor of Apoptosis Protein Levels,” Journal of Virology, Vol. 76, No. 22, 2002, pp. 11414-11424. doi:10.1128/JVI.76.22.11414-11424.2002
- [68] J. L. Connolly, S. E. Rodgers, P. Clarke, D. W. Ballard, L. D. Kerr, K. L. Tyler, et al., “Reovirus-Induced Apoptosis Requires Activation of Transcription Factor NF-KappaB,” Journal of Virology, Vol. 74, No. 7, 2000, pp. 2981-2989. doi:10.1128/JVI.74.7.2981-2989.2000
- [69] M. W. Hansberger, J. A. Campbell, P. Danthi, P. Arrate, K. N. Pennington, K. B. Marcu, et al., “IkappaB Kinase Subunits Alpha and Gamma Are Required for Activation of NF-kappaB and Induction of Apoptosis by Mammalian Reovirus,” Journal of Virology, Vol. 81, No. 3, 2007, pp. 1360-1371. doi:10.1128/JVI.01860-06
- [70] J. L. Connolly and T. S. Dermody, “Virion Disassembly Is Required for Apoptosis Induced by Reovirus,” Journal of Virology, Vol. 76, No. 4, 2002, pp. 1632-1641. doi:10.1128/JVI.76.4.1632-1641.2002
- [71] C. M. Coffey, A. Sheh, I. S. Kim, K. Chandran, M. L. Nibert and J. S. Parker, “Reovirus Outer Capsid Protein Micro1 Induces Apoptosis and Associates with Lipid Droplets, Endoplasmic Reticulum and Mitochondria,” Journal of Virology, Vol. 80, No. 17, 2006, pp. 8422- 8438. doi:10.1128/JVI.02601-05
- [72] M. L. Wisniewski, B. G. Werner, L. G. Hom, L. J. Anguish, C. M. Coffey and J. S. Parker, “Reovirus Infection or Ectopic Expression of Outer Capsid Protein Micro1 Induces Apoptosis Independently of the Cellular Proapoptotic Proteins Bax and Bak,” Journal of Virology, Vol. 85, No. 1, 2011, pp. 296-304. doi:10.1128/JVI.01982-10
- [73] H. M. Berens and K. L. Tyler, “The Proapoptotic Bcl-2 Protein Bax Plays an Important Role in the Pathogenesis of Reovirus Encephalitis,” Journal of Virology, Vol. 85, No. 8, 2011, pp. 3858-3871. doi:10.1128/JVI.01958-10
- [74] P. Danthi, A. J. Pruijssers, A. K. Berger, G. H. Holm, S. S. Zinkel and T. S. Dermody, “Bid Regulates the Pathogenesis of Neurotropic Reovirus,” PLoS Pathogens, Vol. 6, No. 7, 2010, Article ID: e1000980. doi:10.1371/journal.ppat.1000980
- [75] D. Pan, L.-Z. Pan, R. Hill, P. Marcato, M. Shmulevitz, L. T. Vassilev and P. W. K. Lee, “Stabilisation of p53 Enhances Reovirus-Induced Apoptosis and Virus Spread through p53-Dependent NF-kappaB Activation,” British Journal of Cancer, Vol. 105, No. 7, 2011, pp. 1012-1022. doi:10.1038/bjc.2011.325
- [76] G. H. Holm, J. Zurney, V. Tumilasci, S. Leveille, P. Danthi, J. Hiscott, et al., “Retinoic Acid-Inducible Gene-I and Interferon-Beta Promoter Stimulator-1 Augment Proapoptotic Responses Following Mammalian Reovirus Infection via Interferon Regulatory Factor-3,” Journal of Biological Chemistry, Vol. 282, No. 30, 2007, pp. 21953- 21961. doi:10.1074/jbc.M702112200
- [77] J. J. Knowlton, T. S. Dermody and G. H. Holm, “Apoptosis Induced by Mammalian Reovirus Is Beta Interferon (IFN) Independent and Enhanced by IFN Regulatory Factor 3- and NF-kappaB-Dependent Expression of Noxa,” Journal of Virology, Vol. 86, No. 3, 2012, pp. 1650-1660. doi:10.1128/JVI.05924-11
- [78] K. R. Kelly, C. M. Espitia, D. Mahalingam, B. O. Oyajobi, M. Coffey, F. J. Giles, et al., “Reovirus Therapy Stimulates Endoplasmic Reticular Stress, NOXA Induction, and Augments Bortezomib-Mediated Apoptosis in Multiple Myeloma,” Oncogene, Vol. 31, No. 25, 2012, pp. 3023-3038. doi:10.1038/onc.2011.478
- [79] P. Clarke, S. M. Meintzer, A. C. Spalding, G. L. Johnson and K. L. Tyler, “Caspase 8-Dependent Sensitization of Cancer Cells to TRAIL-Induced Apoptosis Following Reovirus-Infection,” Oncogene, Vol. 20, No. 47, 2001, pp. 6910-6919. doi:10.1038/sj.onc.1204842
- [80] P. Clarke and K. L. Tyler, “Down-Regulation of cFLIP Following Reovirus Infection Sensitizes Human Ovarian Cancer Cells to TRAIL-Induced Apoptosis,” Apoptosis, Vol. 12, No. 1, 2007, pp. 211-223. doi:10.1007/s10495-006-0528-4
- [81] I. R. Cho, S. S. Koh, H. J. Min, E. H. Park, R. Srisuttee, B. H. Jhun, et al., “Reovirus Infection Induces Apoptosis of TRAIL-Resistant Gastric Cancer Cells by Down-Regulation of Akt Activation,” International Journal of Oncology, Vol. 36, No. 4, 2010, pp. 1023-1030.
- [82] P. Clarke, J. D. Beckham, J. S. Leser, C. C. Hoyt and K. L. Tyler, “Fas-Mediated Apoptotic Signaling in the Mouse Brain Following Reovirus Infection,” Journal of Virology, Vol. 83, No. 12, 2009, pp. 6161-6170. doi:10.1128/JVI.02488-08
- [83] K. R. Dionne, Y. Zhuang, J. S. Leser, K. L. Tyler and P. Clarke, “Daxx Upregulation within the Cytoplasm of Reovirus-Infected Cells Is Mediated by Interferon and Contributes to Apoptosis,” Journal of Virology, Vol. 87, No. 6, 2013, pp. 3447-3460. doi:10.1128/JVI.02324-12
- [84] G. J. Nuovo, M. Garofalo, N. Valeri, V. Roulstone, S. Volinia, D. E. Cohn, et al., “Reovirus-Associated Reduction of microRNA-let-7d is Related to the Increased Apoptotic Death of Cancer Cells in Clinical Samples,” Modern Pathology, Vol. 25, No. 10, 2012, pp. 1333-1344. doi:10.1038/modpathol.2012.95
- [85] W. P. Tsang and T. T. Kwok, “Let-7a microRNA Suppresses Therapeutics-Induced Cancer Cell Death by Targeting Caspase-3,” Apoptosis, Vol. 13, No. 10, 2008, pp. 1215-1222. doi:10.1007/s10495-008-0256-z
- [86] C. M. Thirukkumaran, Z. Q. Shi, J. Luider, K. Kopciuk, H. Gao, N. Bahlis, et al., “Reovirus as a Viable Therapeutic Option for the Treatment of Multiple Myeloma,” Clinical Cancer Research, Vol. 18, No. 18, 2012, pp. 4962-4972. doi:10.1158/1078-0432.CCR-11-3085
- [87] C. M. Thirukkumaran, Z. Q. Shi, J. Luider, K. Kopciuk, H. Gao, N. Bahlis, et al., “Reovirus Modulates Autophagy during Oncolysis of Multiple Myeloma,” Autophagy, Vol. 9, No. 3, 2013, pp. 413-414. doi:10.4161/auto.22867
- [88] B. Levine and D. J. Klionsky, “Development by SelfDigestion: Molecular Mechanisms and Biological Functions of Autophagy,” Developmental Cell, Vol. 6, No. 4, 2004, pp. 463-477. doi:10.1016/S1534-5807(04)00099-1
- [89] J. S. Carew, K. R. Kelly and S. T. Nawrocki, “Autophagy as a Target for Cancer Therapy: New Developments,” Cancer Management and Research, Vol. 4, 2012, pp. 357-365.
- [90] C. Richetta and M. Faure, “Autophagy in Antiviral Innate Immunity,” Celullar Microbiology, Vol. 15, No. 3, 2013, pp. 368-376. doi:10.1111/cmi.12043
- [91] L. Qin, Z. Wang, L. Tao and Y. Wang, “ER Stress Negatively Regulates AKT/TSC/mTOR Pathway to Enhance Autophagy,” Autophagy, Vol. 6, No. 2, 2010, pp. 239-247. doi:10.4161/auto.6.2.11062
- [92] C. Appenzeller-Herzog and M. N. Hall, “Bidirectional Crosstalk between Endoplasmic Reticulum Stress and mTOR Signaling,” Trends in Cell Biology, Vol. 22, No. 5, 2012, pp. 274-282. doi:10.1016/j.tcb.2012.02.006
- [93] [92] S. Meng, K. Jiang, X. Zhang, M. Zhang, Z. Zhou, M. Hu, et al., “Avian Reovirus Triggers Autophagy in Primary Chicken Fibroblast Cells and Vero Cells to Promote Virus Production,” Archives of Virology, Vol. 157, No. 4, 2012, pp. 661-668.
- [94] Y. Ikeda, G. Nishimura, S. Yanoma, A. Kubota, M. Furukawa and M. Tsukuda, “Reovirus Oncolysis in Human Head and Neck Squamous Carcinoma Cells,” Auris Nasus Larynx, Vol. 31, No. 4, 2004, pp. 407-412.
- [95] J. Yuan and G. Kroemer, “Alternative Cell Death Mechanisms in Development and Beyond,” Genes and Development, Vol. 24, No. 23, 2010, pp. 2592-2602. doi:10.1101/gad.1984410
- [96] W. Wu, P. Liu and J. Li, “Necroptosis: An Emerging form of Programmed Cell Death,” Critical Reviews in Oncology/Hematology, Vol. 82, No. 3, 2012, pp. 249-258. doi:10.1016/j.critrevonc.2011.08.004
- [97] J. M. Penninger and G. Kroemer, “Mitochondria, AIF and Caspases—Rivaling for Cell Death Execution,” Nature Cell Biology, Vol. 5, No. 2, 2003, pp. 97-99. doi:10.1038/ncb0203-97
- [98] W. Q. Yang, X. Lun, C. A. Palmer, M. E. Wilcox, H. Muzik, Z. Q. Shi, et al., “Efficacy and Safety Evaluation of Human Reovirus Type 3 in Immunocompetent Animals: Racine and Nonhuman Primates,” Clinical Cancer Research, Vol. 10, No. 24, 2004, pp. 8561-8576. doi:10.1158/1078-0432.CCR-04-0940
- [99] K. L. Norman, M. C. Coffey, K. Hirasawa, D. J. Demetrick, S. G. Nishikawa, L. M. DiFrancesco, et al., “Reovirus Oncolysis of Human Breast Cancer,” Human Gene Therapy, Vol. 13, No. 5, 2002, pp. 641-652. doi:10.1089/10430340252837233
- [100] T. Alain, J. F. Wong, R. Endersby, S. J. Urbanski, P. W. Lee, D. A. Muruve, et al., “Reovirus Decreases Azoxymethane-Induced Aberrant Crypt Foci and Colon Cancer in a Rodent Model,” Cancer Gene Therapy, Vol. 14, No. 10, 2007, pp. 867-872. doi:10.1038/sj.cgt.7701068
- [101] S. Hirano, T. Etoh, R. Okunaga, K. Shibata, M. Ohta, A. Nishizono, et al., “Reovirus Inhibits the Peritoneal Dissemination of Pancreatic Cancer Cells in an Immunocompetent Animal Model,” Oncology Reports, Vol. 21, No. 6, 2009, pp. 1381-1384. doi:10.3892/or_00000364
- [102] K. Hirasawa, S. G. Nishikawa, K. L. Norman, M. C. Coffey, B. G. Thompson, C. S. Yoon, et al., “Systemic Reovirus Therapy of Metastatic Cancer in Immune-Competent Mice,” Cancer Research, Vol. 63, No. 2, 2003, pp. 348-353.
- [103] T. Alain, K. Hirasawa, K. J. Pon, S. G. Nishikawa, S. J. Urbanski, Y. Auer, et al., “Reovirus Therapy of Lymphoid Malignancies,” Blood, Vol. 100, No. 12, 2002, pp. 4146-4153. doi:10.1182/blood-2002-02-0503
- [104] P. Hingorani, W. Zhang, J. Lin, L. Liu, C. Guha and E. A. Kolb, “Systemic Administration of Reovirus (Reolysin) Inhibits Growth of Human Sarcoma Xenografts,” Cancer, Vol. 117, No. 8, 2011, pp. 1764-1774. doi:10.1002/cncr.25741
- [105] E. G. Hanel, Z. Xiao, K. K. Wong, P. W. Lee, R. A. Britten and R. B. Moore, “A Novel Intravesical Therapy for Superficial Bladder Cancer in an Orthotopic Model: Oncolytic Reovirus Therapy,” Journal of Urology, Vol. 172, No. 5, 2004, pp. 2018-2022. doi:10.1097/01.ju.0000142657.62689.f6
- [106] T. A. Yap, A. Brunetto, H. Pandha, K. Harrington and J. S. Debono, “Reovirus Therapy in Cancer: Has the Orphan Virus Found a Home?” Expert Opinion on Investigational Drugs, Vol. 17, No. 12, 2008, pp. 1925-1935. doi:10.1517/13543780802533401
- [107] K. Kelly, S. Nawrocki, A. Mita, M. Coffey, F. J. Giles and M. Mita, “Reovirus-Based Therapy for Cancer,” Expert Opinion on Biological Therapy, Vol. 9, No. 7, 2009, pp. 817-830. doi:10.1517/14712590903002039
- [108] K. J. Harrington, R. G. Vile, A. Melcher, J. Chester and H. S. Pandha, “Clinical Trials with Oncolytic Reovirus: Moving beyond Phase I into Combinations with Standard Therapeutics,” Cytokine and Growth Factor Reviews, Vol. 21, No. 2-3, 2010, pp. 91-98. doi:10.1016/j.cytogfr.2010.02.006
- [109] C. M. Thirukkumaran, M. J. Nodwell, K. Hirasawa, Z. Q. Shi, R. Diaz, J. Luider, et al., “Oncolytic Viral Therapy for Prostate Cancer: Efficacy of Reovirus as a Biological Therapeutic,” Cancer Research, Vol. 70, No. 6, 2010, pp. 2435-2444. doi:10.1158/0008-5472.CAN-09-2408
- [110] P. Forsyth, G. Roldan, D. George, C. Wallace, C. A. Palmer, D. Morris, G. et al., “A Phase I Trial of Intratumoral Administration of Reovirus in Patients with Histologically Confirmed Recurrent Malignant Gliomas,” Molecular Therapy, Vol. 16, No. 3, 2008, pp. 627-632. doi:10.1038/sj.mt.6300403
- [111] L. Vidal, H. S. Pandha, T. A. Yap, C. L. White, K. Twigger, R. G. Vile, et al., “A Phase I Study of Intravenous Oncolytic Reovirus Type 3 Dearing in Patients with Advanced Cancer,” Clinical Cancer Research, Vol. 14, No. 21, 2008, pp. 7127-7137. doi:10.1158/1078-0432.CCR-08-0524
- [112] R. Gollamudi, M. H. Ghalib, K. K. Desai, I. Chaudhary, B. Wong, M. Einstein, et al., “Intravenous Administration of Reolysin, a Live Replication Competent RNA Virus Is Safe in Patients with Advanced Solid Tumors,” Investigational New Drugs, Vol. 28, No. 5, 2009, pp. 641-649. doi:10.1007/s10637-009-9279-8
- [113] E. M. Karapanagiotou, V. Roulstone, K. Twigger, M. Ball, M. Tanay, C. Nutting, et al., “Phase I/II Trial of Carboplatin and Paclitaxel Chemotherapy in Combination with Intravenous Oncolytic Reovirus in Patients with Advanced Malignancies,” Clinical Cancer Research, Vol. 18, No. 7, 2012, pp. 2080-2089. doi:10.1158/1078-0432.CCR-11-2181
- [114] K. Twigger, L. Vidal, C. L. White, J. S. De Bono, S. Bhide, M. Coffey, et al., “Enhanced in Vitro and in Vivo Cytotoxicity of Combined Reovirus and Radiotherapy,” Clinical Cancer Research, Vol. 14, No. 3, 2008, pp. 912- 923. doi:10.1158/1078-0432.CCR-07-1400
- [115] J. Qiao, H. Wang, T. Kottke, C. White, K. Twigger, R. M. Diaz, et al., “Cyclophosphamide Facilitates Antitumor Efficacy against Subcutaneous Tumors Following Intravenous Delivery of Reovirus,” Clinical Cancer Research, Vol. 14, No. 1, 2008, pp. 259-269. doi:10.1158/1078-0432.CCR-07-1510
- [116] H. S. Pandha, L. Heinemann, G. R. Simpson, A. Melcher, R. Prestwich, F. Errington, et al., “Synergistic Effects of Oncolytic Reovirus and Cisplatin Chemotherapy in Murine Malignant Melanoma,” Clinical Cancer Research, Vol. 15, No. 19, 2009, pp. 6158-6166. doi:10.1158/1078-0432.CCR-09-0796
- [117] S. Sei, J. K. Mussio, Q. E. Yang, K. Nagashima, R. E. Parchment, M. C. Coffey, et al., “Synergistic Antitumor Activity of Oncolytic Reovirus and Chemotherapeutic Agents in Non-Small Cell Lung Cancer Cells,” Molecular Cancer, Vol. 8, 2009, p. 47. doi:10.1186/1476-4598-8-47
- [118] V. Roulstone, K. Twigger, S. Zaidi, T. Pencavel, J. N. Kyula, C. White, et al., “Synergistic Cytotoxicity of Oncolytic Reovirus in Combination with Cisplatin-Paclitaxel Doublet Chemotherapy,” Gene Therapy, Vol. 20, No. 5, 2012, pp. 521-528. doi:10.1038/gt.2012.68
- [119] M. C. Coffey, J. E. Strong, P. A. Forsyth and P. W. Lee, “Reovirus Therapy of Tumors with Activated Ras Pathway,” Science, Vol. 282, No. 5392, 1998, pp. 1332-1334. doi:10.1126/science.282.5392.1332
- [120] C. L. White, K. R. Twigger, L. Vidal, J. S. De Bono, M. Coffey, L. Heinemann, et al., “Characterization of the Adaptive and Innate Immune Response to Intravenous Oncolytic Reovirus (Dearing Type 3) during a Phase I Clinical Trial,” Gene Therapy, Vol. 15, No. 12, 2008, pp. 911-920. doi:10.1038/gt.2008.21
- [121] N. Smakman, J. D. van der Bilt, D. J. van den Wollenberg, R. C. Hoeben, I. H. Borel Rinkes and O. Kranenburg, “Immunosuppression Promotes Reovirus Therapy of Colorectal Liver Metastases,” Cancer Gene Therapy, Vol. 13, No. 8, 2006, pp. 815-818. doi:10.1038/sj.cgt.7700949
- [122] T. Kottke, J. Thompson, R. M. Diaz, J. Pulido, C. Willmon, M. Coffey, et al., “Improved Systemic Delivery of Oncolytic Reovirus to Established Tumors Using Preconditioning with Cyclophosphamide-Mediated Treg Modulation and Interleukin-2,” Clinical Cancer Research, Vol. 15, No. 2, 2009, pp. 561-569. doi:10.1158/1078-0432.CCR-08-1688
- [123] S. A. Gujar, P. Marcato, D. Pan and P. W. Lee, “Reovirus Virotherapy Overrides Tumor Antigen Presentation Evasion and Promotes Protective Antitumor Immunity,” Molecular Cancer Therapeutics, Vol. 9, No. 11, 2010, pp. 2924-2933. doi:10.1158/1535-7163.MCT-10-0590
- [124] R. J. Prestwich, E. J. Ilett, F. Errington, R. M. Diaz, L. P. Steele, T. Kottke, et al., “Immune-Mediated Antitumor Activity of Reovirus is Required for Therapy and Is Independent of Direct Viral Oncolysis and Replication,” Clinical Cancer Research, Vol. 15, No. 13, 2009, pp. 4374-4381. doi:10.1158/1078-0432.CCR-09-0334
- [125] M. P. Lolkema, H. T. Arkenau, K. Harrington, P. Roxburgh, R. Morrison, V. Roulstone, et al., “A Phase I Study of the Combination of Intravenous Reovirus Type 3 Dearing and Gemcitabine in Patients with Advanced Cancer,” Clinical Cancer Research, Vol. 17, No. 3, 2011, pp. 581-588. doi:10.1158/1078-0432.CCR-10-2159
- [126] S. D. Loken, K. Norman, K. Hirasawa, M. Nodwell, W. M. Lester and D. J. Demetrick, “Morbidity in Immunosuppressed (SCID/NOD) Mice Treated with Reovirus (Dearing 3) as an Anti-Cancer Biotherapeutic,” Cancer Biology and Therapy, Vol. 3, No. 8, 2004, pp. 734-738. doi:10.4161/cbt.3.8.963
- [127] M. R. Roner and W. K. Joklik, “Reovirus Reverse Genetics: Incorporation of the CAT Gene into the ReoVirus Genome,” Proceedings of the National Academy of Science United States of America, Vol. 98, No. 14, 2001, pp. 8036-8041. doi:10.1073/pnas.131203198
- [128] D. J. van den Wollenberg, S. K. van den Hengel, I. J. Dautzenberg, S. J. Cramer, O. Kranenburg and R. C. Hoeben, “A Strategy for Genetic Modification of the Spike-Encoding Segment of Human Reovirus T3D for Reovirus Targeting,” Gene Therapy, Vol. 15, No. 24, 2008, pp. 1567-1578. doi:10.1038/gt.2008.118
- [129] T. Kobayashi, A. A. Antar, K. W. Boehme, P. Danthi, E. A. Eby, K. M. Guglielmi, et al., “A Plasmid-Based Reverse Genetics System for Animal Double-Stranded RNA Viruses,” Cell Host and Microbe, Vol. 1, No. 2, 2007, pp. 147-157. doi:10.1016/j.chom.2007.03.003
- [130] V. Brochu-Lafontaine and G. Lemay, “Addition of Exogenous Polypeptides on the Mammalian Reovirus Outer Capsid Using Reverse Genetics,” Journal of Virological Methods, Vol. 179, No. 2, 2012, pp. 342-350. doi:10.1016/j.jviromet.2011.11.021
- [131] K. W. Boehme, M. Ikizler, T. Kobayashi and T. S. Dermody, “Reverse Genetics for Mammalian Reovirus,” Methods, Vol. 55, No. 2, 2011, pp. 109-113. doi:10.1016/j.ymeth.2011.07.002
- [132] M. Kim, K. A. Garant, N. I. zur Nieden, T. Alain, S. D. Loken, S. J. Urbanski, et al., “Attenuated Reovirus Displays Oncolysis with Reduced Host Toxicity,” British Journal of Cancer, Vol. 104, No. 2, 2010, pp. 290-299. doi:10.1038/sj.bjc.6606053
- [133] M. Shmulevitz, S. A. Gujar, D. G. Ahn, A. Mohamed and P. W. Lee, “Reovirus Variants with Mutations in Genome Segments S1 and L2 Exhibit Enhanced Virion Infectivity and Superior Oncolysis,” Journal of Virology, Vol. 86, No. 13, 2012, pp. 7403-7413. doi:10.1128/JVI.00304-12
- [134] M. L. Nibert and L. A. Schiff, “Reoviruses and Their Replication,” In: D. M. Knipe, Ed., Fields Virology, 4th Edition, Lippincott Williams and Wilkins, Philadephia, 2001, pp. 1679-1728.
NOTES
*Corresponding authors.