Applied Mathematics
Vol.4 No.4(2013), Article ID:30436,12 pages DOI:10.4236/am.2013.44091
The Effects of a Backward Bifurcation on a Continuous Time Markov Chain Model for the Transmission Dynamics of Single Strain Dengue Virus
Centre for Advanced Studies in Mathematics, Lahore University of Management Sciences, Lahore, Pakistan
Email: *mudassar.imran@lums.edu.pk
Copyright © 2013 Adnan Khan et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received February 19, 2013; revised March 4, 2013; accepted March 11, 2013
Keywords: Epidemiology; Dengue Fever; Backward Bifurcation; Stochastic Model
ABSTRACT
Global incidence of dengue, a vector-borne tropical disease, has seen a dramatic increase with several major outbreaks in the past few decades. We formulate and analyze a stochastic epidemic model for the transmission dynamics of a single strain of dengue virus. The stochastic model is constructed using a continuous time Markov chain (CTMC) and is based on an existing deterministic model that suggests the existence of a backward bifurcation for some values of the model parameters. The dynamics of the stochastic model are explored through numerical simulations in this region of bistability. The mean of each random variable is numerically estimated and these are compared to the dynamics of the deterministic model. It is observed that the stochastic model also predicts the co-existence of a locally asymptotically stable disease-free equilibrium along with a locally stable endemic equilibrium. This co-existence of equilibria is important from a public health perspective because it implies that dengue can persist in populations even if the value of the basic reproduction number is less than unity.
1. Introduction
Dengue, a vector transmitted disease, has seen a dramatic increase in global incidence over the past decades [1,2]. Originally restricted to a handful of countries, dengue is now endemic in more than a hundred tropical and subtropical countries worldwide [1-3]. With an estimated 50 - 100 million cases and nearly 10,000 - 20,000 deaths annually, dengue ranks second to Malaria amongst deadly mosquito-born diseases [1,2,4-6]. The disease is caused by one of four virus serotypes (strains) of the genus Flavivirus [2,3,7]. Most infected individuals suffer from dengue fever, a severe flu-like illness characterized by high fever, which poses only a limited threat to mortality [2,8]. The symptoms usually last for one to two weeks, after an initial incubation period of about 4 - 7 days [9]. A minority of infected individuals however, develops dengue hemorrhagic fever (DHF) resulting in bleeding, low levels of blood platelets and blood plasma leakage, or dengue shock syndrome (DSS) resulting in extremely low blood pressures. The risk associated with DHF and DSS is considerably higher, with mortality ranging from 5% - 15% [3,5,9,10].
Dengue is transmitted to humans through mosquito bites. Female mosquitos of the Aedas genus, primarily Aedas aegypti, acquire the dengue virus through a blood meal from infected humans [2,11]. The dengue virus has an incubation period of about 7 - 10 days in the vector, and is then spread to susceptible humans who are bitten by the infected mosquito [9]. The virus also has an incubation period of 4 - 7 days in the host [9]. While vectors never recover from infection with the dengue virus, the infection in hosts lasts only about one to two weeks [2]. Recovery from infection with one serotype of the dengue virus gives life-long immunity to that serotype but only temporary and partial immunity to other serotypes [2,4, 12-14]. Secondary infection with a different serotype may result in Antibody Dependent Enhancement (ADE), which is speculated to increase the chances of DHF and DSS [4,12,13,15]. In this study however, we will consider infection involving only a single serotype of the dengue virus.
Historical records indicate the occurrence of dengue epidemics in North America, Asia and Africa in the late 18th century [3]. Since then and up until the middle of the 20th century, incidences of dengue fever have been rare [3]. Since the 1970’s however, there has been a marked increase in the number of dengue cases, as well as dengue epidemics, with the WHO claiming a 30-fold increase in the incidence of dengue between 1960 and 2010 [2,3,8]. This dramatic increase is attributed to rapid urbanization, population growth and increase international travel [8]. Dengue disease is currently endemic in nearly 110 countries in Southeast Asia, the Americas, Africa and the Eastern Mediterranean [2]. It is estimated by the WHO, that nearly 2.5 billion people are at risk of contracting the disease. Furthermore, nearly 50 - 100 million cases and almost 20,000 deaths due to more severe forms of dengue fever are reported globally every year, making dengue one of the deadliest mosquitotransmitted diseases [1,2,4-6].
The dengue virus (DENV), which causes dengue fever in humans, is a single positive-stranded RNA virus of the family Flaviviridae and genus Flavivirus [2,3,7]. The virus has four distinct serotypes, DENV1, DENV2, DENV3 and DENV4, each of which can cause the full spectrum of the disease [2,3,7]. Owing to the difficulty of developing an immunization against all four serotypes, there is currently no vaccine against the disease [6,8], [11]. Infection with and recovery from a particular serotype of the dengue virus grants life-long immunity to that serotype but only gives temporary and/or partial immunity to the other serotypes [2]. This partial cross-immunity is the cause of antibody-dependent enhancement (ADE) in the setting of a secondary infection with a different serotype of DENV. ADE is hypothesized to be one factors leading to DHF and DSS, the more severe form of dengue disease [4,12,13,15].
The clinical symptoms and effects of dengue disease vary greatly. Nearly 80% of individuals suffering from a primary infection with DENV are asymptomatic or display only a mild, uncomplicated fever [2,8]. A minority of infected individuals suffer from DHF and DSS, the more severe forms of dengue disease [3,5,9]. As mentioned previously however, risk of DHF and DSS is associated primarily with secondary infection with a heterologous serotype of DENV [4,12,13,15]. In general, dengue disease is marked by three separate phases: febrile, critical and recovery. The characteristic symptoms of dengue in the febrile phase are the sudden onset of high fever, rash, headaches and muscle and joint pains, which lead to the alternative name “breakbone fever” for dengue disease [8]. This phase of the disease is rarely life threatening and the associated mortality is quite low. Most individuals then progress to the recovery phase. However, a minority of individuals first pass through the critical phase of the disease. This phase lasts for one or two days and is marked by low blood pressure, leakage of blood plasma from the capillaries and decreased blood supply to organs. Severe cases of these symptoms are associated with DHF and DSS and the mortality in this phase of the disease is estimated to be as high as 5% - 15% [3,5,8,9].
Over the past several years, a number of deterministic mathematical models have been proposed to analyze the transmission dynamics of dengue in urban communities [5,11-17]. L. Esteva and C. Vargas [14] have investigated the coexistence of two serotypes of dengue virus using a deterministic ODE model. Moreover, Ferguson et al. [15] have investigated the effects of ADE on the transmission of multiple serotypes of dengue virus. In addition, Garba et al. [11] have shown the existence of a backward bifurcation in a standard incidence ODE model for a single strain of dengue virus. Garba et al. [12] have also explored the effects of cross-immunity on the transmission dynamics of two strains of dengue virus. Similarly, H. Wearing and P. Rohani [13] have investigated the effects of both ADE and cross immunity on multiple serotypes of dengue virus. Finally, Chowel et al. [18] have estimated the basic reproduction number for dengue using spatial epidemic data.
In addition, over the past few decades, several stochastic epidemic models for the spread of infectious diseases have also been proposed and analyzed [19-27]. An important qualitative difference between deterministic and stochastic epidemic models in general is the asymptotic dynamics [28]. Furthermore, stochastic models also allow for the possibility of disease extinction in finite time and therefore the expected time to disease extinction can be calculated [19,28,29]. It is also observed that stochastic models better capture the uncertainty and variability that is inherent in real-life epidemics due to factors such as the unpredictability of person-to-person contact [27, 29]. L. J. S. Allen [28,29] has explored the utility of stochastic epidemic models by comparing them with deterministic models. Despite, the utility of stochastic models, however, very little stochastic modeling has been performed for the transmission dynamics of dengue virus (see [26] and the references therein).
The purpose of this study is to formulate and analyze a stochastic model for the transmission dynamics of a single strain of dengue virus using a continuous time Markov chain (CTMC). The stochastic model is based on an existing deterministic model proposed by Garba et al. [11] with one minor but nevertheless important difference: contrary to the original deterministic model [11] and in line with previous studies of dengue virus such as [12,13], we will assume that exposed hosts and exposed vectors do not transmit the disease. In addition, the deterministic model [11] postulates the existence of a backward bifurcation for a subset of the model parameters space. Therefore, a major aim of this study is to estimate the mean of each random variable in this region of bistability using numerical simulations. These will be compared to the dynamics of the deterministic model [11]. A previous study on the effect of a backward bifurcation on the dynamics of a stochastic epidemic model is given in [26].
This paper is organized as follows. The second section contains a description of the deterministic model formulated in [11] along with a discussion of the basic reproduction number 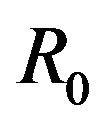 of the model as well as the conditions for the existence of the backward bifurcation. In Section 3 we formulate the stochastic model as a continuous time Markov chain (CTMC) and discuss some basic properties of the stochastic model. Finally, Section 4 contains the numerical simulations of the stochastic model. We conclude the paper by presenting a discussion of various directions in which to extend the current study.
of the model as well as the conditions for the existence of the backward bifurcation. In Section 3 we formulate the stochastic model as a continuous time Markov chain (CTMC) and discuss some basic properties of the stochastic model. Finally, Section 4 contains the numerical simulations of the stochastic model. We conclude the paper by presenting a discussion of various directions in which to extend the current study.
2. The Deterministic Model
2.1. Model Formulation
The deterministic model we have considered is a deterministic vector-host ODE model that assumes a homogenous mixing of the host (human) and vector (mosquito) populations. The total human population at time t, denoted by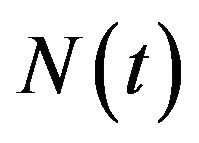 , is divided into four mutually exclusive classes comprising of susceptible humans
, is divided into four mutually exclusive classes comprising of susceptible humans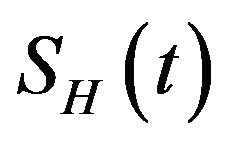 , exposed humans
, exposed humans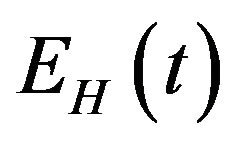 , infected humans
, infected humans 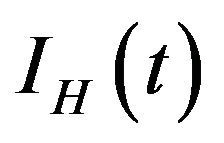 and recovered humans
and recovered humans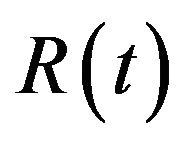 . It is assumed that individuals who recover from infection with a particular serotype of Dengue gain lifelong immunity to it [12]. Similarly, the total vector population at time t is denoted by
. It is assumed that individuals who recover from infection with a particular serotype of Dengue gain lifelong immunity to it [12]. Similarly, the total vector population at time t is denoted by 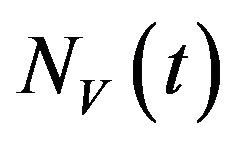 and is divided into three mutually exclusive classes comprising susceptible of susceptible vectors
and is divided into three mutually exclusive classes comprising susceptible of susceptible vectors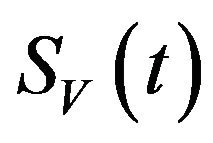 , exposed vectors
, exposed vectors 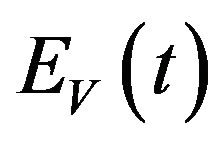 and infected vectors
and infected vectors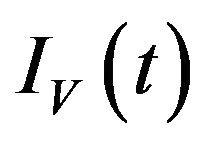 . It is assumed that vectors (mosquitoes) infected with a particular serotype of Dengue never recover [12]. As mentioned previously in the introduction, we will modify the original model of Garba et al. [11] by assuming that exposed humans and exposed vectors do not transmit the disease.
. It is assumed that vectors (mosquitoes) infected with a particular serotype of Dengue never recover [12]. As mentioned previously in the introduction, we will modify the original model of Garba et al. [11] by assuming that exposed humans and exposed vectors do not transmit the disease.
The model assumes that the susceptible human population  has a constant recruitment rate
has a constant recruitment rate  and natural death rate
and natural death rate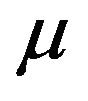 . Susceptible individuals are infected with Dengue virus (due to contact with infected vectors) at a rate
. Susceptible individuals are infected with Dengue virus (due to contact with infected vectors) at a rate 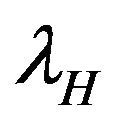 and thus enter the exposed class EH. The exposed population
and thus enter the exposed class EH. The exposed population 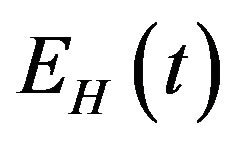 is depleted at the natural death rate
is depleted at the natural death rate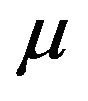 . Additionally, exposed individuals develop symptoms and move into the infected class IH at a rate
. Additionally, exposed individuals develop symptoms and move into the infected class IH at a rate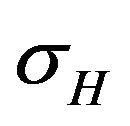 . The infected population
. The infected population 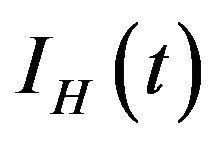 is depleted via the natural death rate mu, the disease-induced death rate
is depleted via the natural death rate mu, the disease-induced death rate 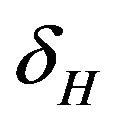 and the recovery rate of infected individuals
and the recovery rate of infected individuals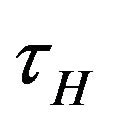 . Finally, the recovered population
. Finally, the recovered population 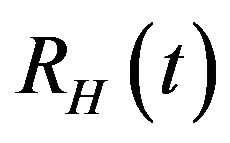 decreases due to the natural death rate mu.
decreases due to the natural death rate mu.
Similarly, the susceptible vector population 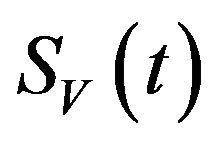 has a constant recruitment rate
has a constant recruitment rate 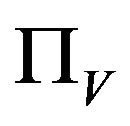 and a natural death rate
and a natural death rate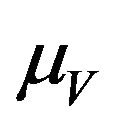 . Susceptible vectors are infected with Dengue virus (due to effective contact with infected humans) at a rate
. Susceptible vectors are infected with Dengue virus (due to effective contact with infected humans) at a rate 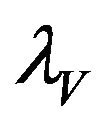 and thus move to the exposed vector class
and thus move to the exposed vector class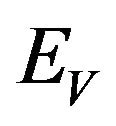 . The exposed vector class
. The exposed vector class  is depleted at the natural death rate
is depleted at the natural death rate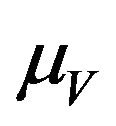 . In addition exposed vectors develop symptoms and move to the infected vector class
. In addition exposed vectors develop symptoms and move to the infected vector class 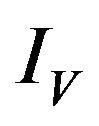 at a rate
at a rate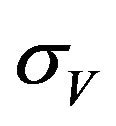 . Infected vectors, in addition to the natural death rate
. Infected vectors, in addition to the natural death rate 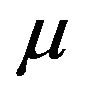 die at a disease induced death rate
die at a disease induced death rate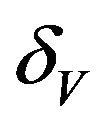 .
.
Mathematically, the modified determinstic model is as follows:
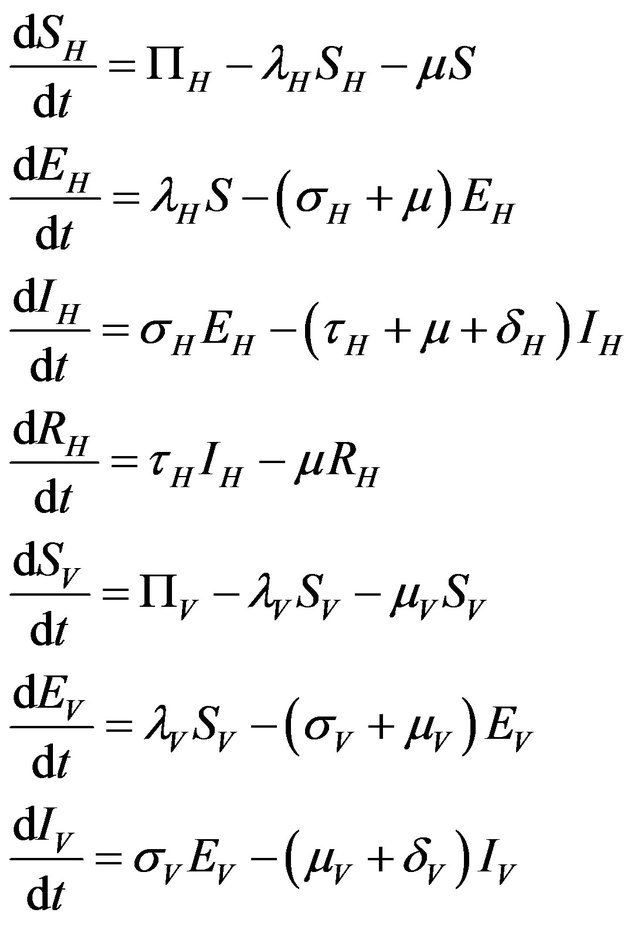 (2.1)
(2.1)
where,
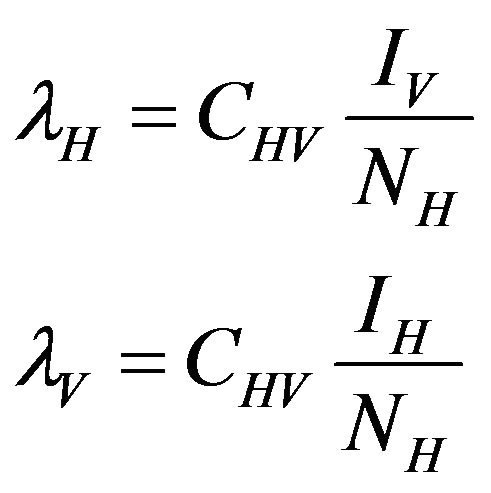
The basic reproduction number for the model (2.1) can be calculated using the next generation operator method given in [30]. Hence, for the model (2.1),  is given by
is given by
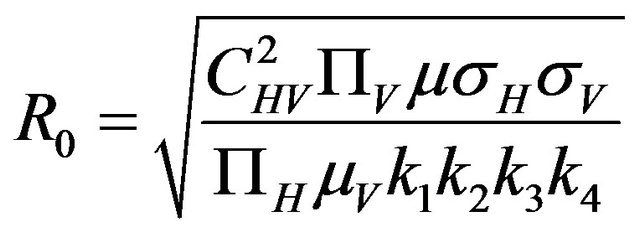 (2.2)
(2.2)
where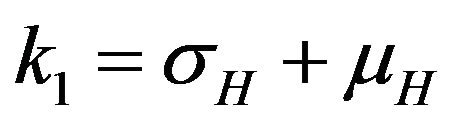 ,
, 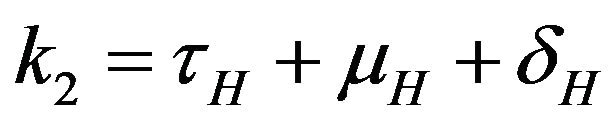 ,
, 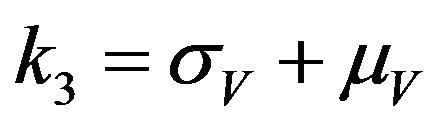 and
and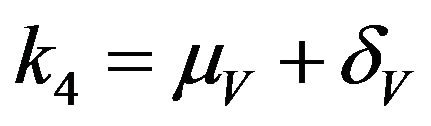 .
.
It follows from [30] that the following lemma holds.
Lemma 1. The system (2.1) has a locally asymptotically stable disease-free equilibrium (DFE) given by
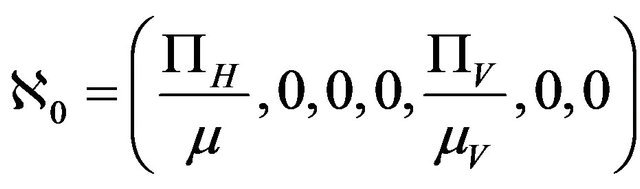 , whenever
, whenever . The DFE given by
. The DFE given by 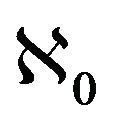 is locally unstable whenever
is locally unstable whenever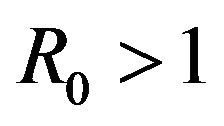 .
.
It should be pointed out however, that we have not proven that the DFE is globally asymptotically stable for values of the basic reproduction number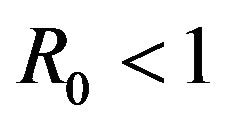 . Indeed, as shown in the next section, this is not true.
. Indeed, as shown in the next section, this is not true.
2.2. Backward Bifurcation
As mentioned in the introduction, Garba et al. proved that the deterministic model presented in [11] undergoes a backward bifurcation for certain values of the model parameters. Since the model (2.1) considered in this study is a special case of the model considered by Garba et al. in [11], it follows easily that model (2.1) also undergoes a backward bifurcation. This is detailed in Theorem 1.
Theorem 1. Define the constants 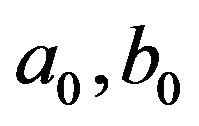 and
and 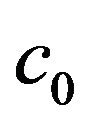 as follows:
as follows:
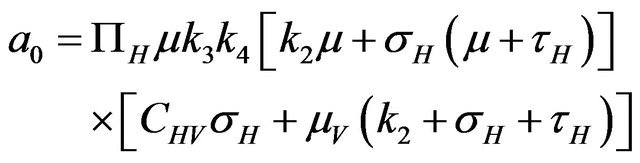
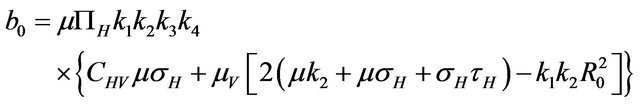
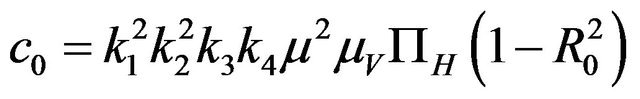
Then, the system (2.1) has:
• A unique endemic equilibrium if .
.
• A unique endemic equilibrium if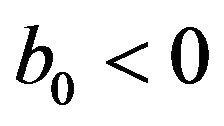 , and
, and 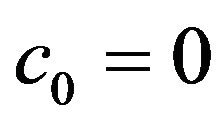 or
or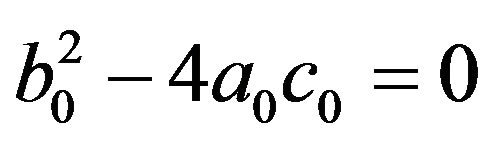 .
.
• Two endemic equilibria if  and
and 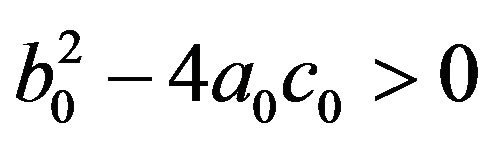 .
.
• No endemic equilibrium otherwise.
Furthermore, if case 3) from above holds and we define 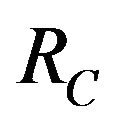 as follows
as follows
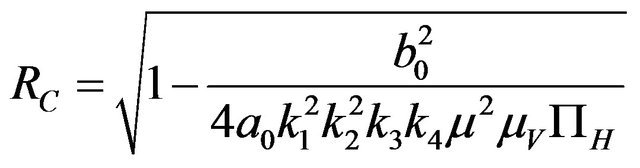 (2.3)
(2.3)
then a backward bifurcation of the system (2.1) occurs for values of the basic reproduction 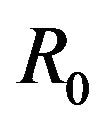 such that
such that
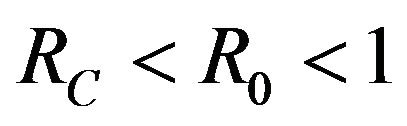 .
.
Proof. By setting , the result follows readily from the proof of Theorem 1 in [11].
, the result follows readily from the proof of Theorem 1 in [11].
As a consequence of Theorem 1, the model (2.1) does not possess a globally asymptotically stable DFE for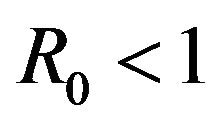 . Indeed, it is clear that for certain values of
. Indeed, it is clear that for certain values of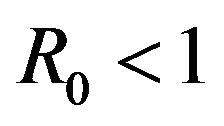 , there is a simultaneous co-existence of a locally asymptotically stable DFE alongside a locally asymptotically stable endemic equilibrium. Thus, lowering the value of the basic reproduction number
, there is a simultaneous co-existence of a locally asymptotically stable DFE alongside a locally asymptotically stable endemic equilibrium. Thus, lowering the value of the basic reproduction number 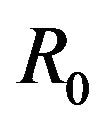 to less than unity is no longer a sufficient condition to ensure disease elimination. From an epidemiological perspective, this possibility is extremely important because it implies that the dengue virus can persist in a population even when
to less than unity is no longer a sufficient condition to ensure disease elimination. From an epidemiological perspective, this possibility is extremely important because it implies that the dengue virus can persist in a population even when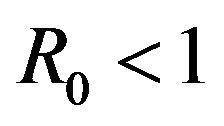 .
.
3. The Stochastic Model
We now formulate an analogous stochastic model using a continuous time Markov chain. Our main purpose in this study will be to explore the dynamics of the stochastic model using numerical simulations in the region of bistability predicted by Theorem 1 and compare them to the corresponding results from the deterministic model (2.1). It has been observed [25] that for epidemic models exhibiting backward bifurcation, the deterministic and stochastic versions of the model do not always have similar dynamics. This difference is all the more important since, it is also observed that stochastic models better capture the uncertainty and variability that is inherent in real-life epidemics due to factors such as the unpredictability of person-to-person contact [27,28].
Model Formulation
Following [29], we assume the total population size to be bounded. We denote the bound by K. Let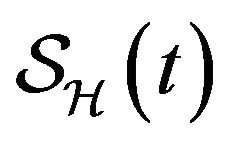 ,
, 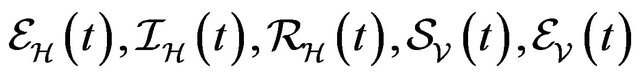 and
and 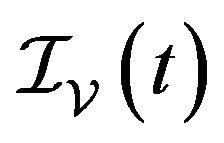 denote discrete random variables for the number of susceptible hosts, exposed hosts, infected hosts, recovered hosts, susceptible vectors, exposed vectors and infected vectors respectively. Furthermore, let
denote discrete random variables for the number of susceptible hosts, exposed hosts, infected hosts, recovered hosts, susceptible vectors, exposed vectors and infected vectors respectively. Furthermore, let 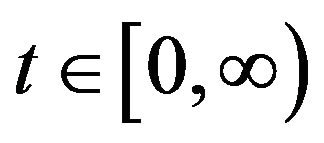 and
and
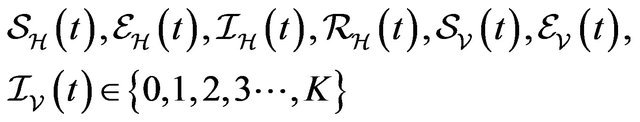
with
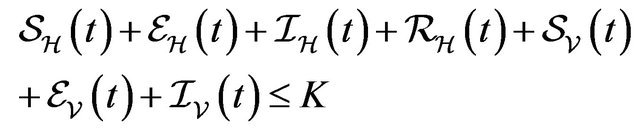
The continous-time stochastic dynamical system
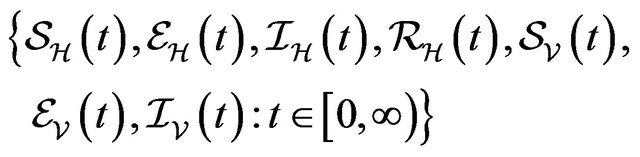
is a multivariate process with a joint probability function with
![]() (3.1)
(3.1)
where
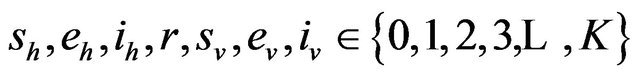 and
and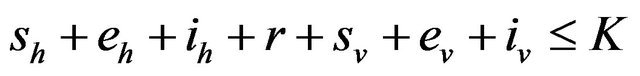 .
.
We further assume that the stochastic process is time homogeneous. Thus, using the notation of [28],
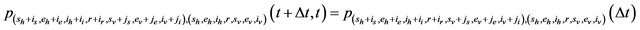 and therefore, the probability of transition from the state
and therefore, the probability of transition from the state 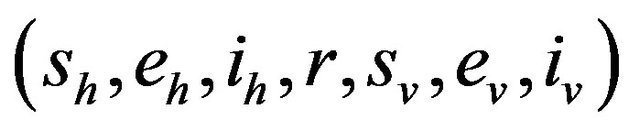 to the state
to the state
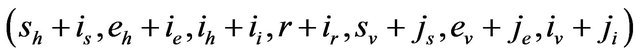
is defined by
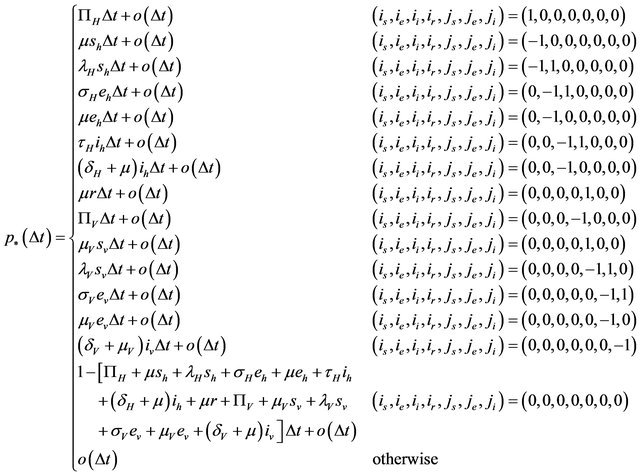
where
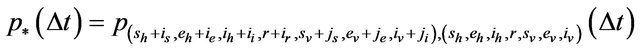
In order for the probabilities to be meaningful (non-negative and bounded by 1), the following is assumed:
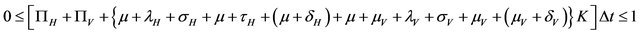 .
.
Since all the parameters are positive, a small value of 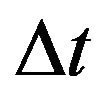 ensures that the above condition is satisfied.
ensures that the above condition is satisfied.
Although, we now have the infinitesimal transition probabilities in hand, it is difficult to express the transition matrix and the generator matrix in easily expressible forms. One possible solution to this problem is given in [19] and we will make use of this later. Our stochastic model has seven independent discrete random variables and it is therefore even more cumbersome to express the associated transition and generator matrices. We therefore desist from this exercise for now.
We now assume that the stochastic process satisfies the Markov property [28]:
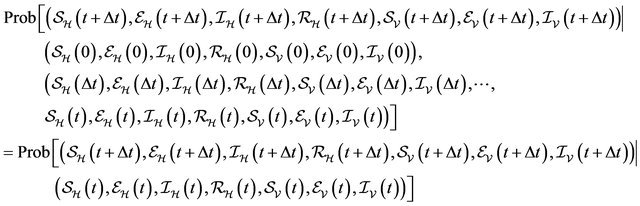
Using the Markov property and the infinitesimal transition probabilities, we can express the state probabilities at time 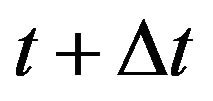 in terms of the state probabilities at time t. For the sake of simplicity, we assume that the total population at time t is less than the upper bound K and furthermore that none of the random variables is zero. The purpose of these two assumptions is to ignore numerous tedious sub-cases.
in terms of the state probabilities at time t. For the sake of simplicity, we assume that the total population at time t is less than the upper bound K and furthermore that none of the random variables is zero. The purpose of these two assumptions is to ignore numerous tedious sub-cases.
Proposition 1. Assume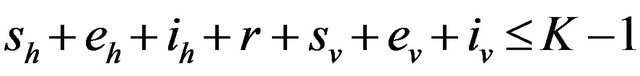 . Further assume that none of the variables sh, eh, ih, r, sv, ev, iv is zero. Then, the state probabilities
. Further assume that none of the variables sh, eh, ih, r, sv, ev, iv is zero. Then, the state probabilities 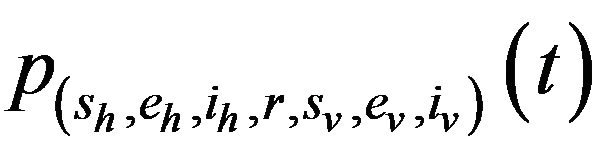 satisfy the following difference equations
satisfy the following difference equations
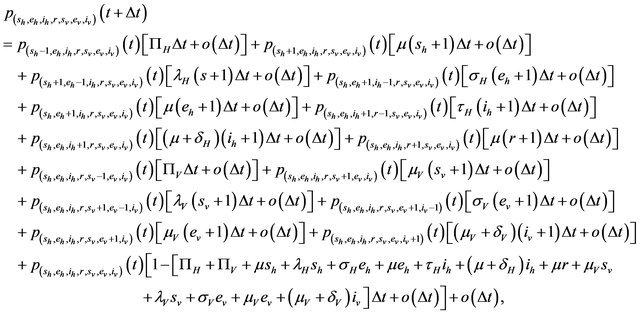
where 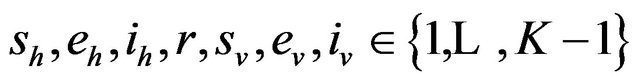
The remaining cases, which result from removing the previous assumptions, are straightforward to calculate.
Some properties of the Markov chain can now be discussed. The Markov chain is reducible, with two important communication classes [28], given by
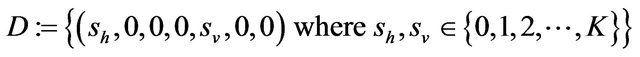
and
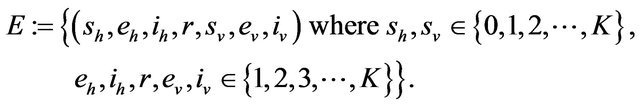
Furthermore, E is not closed since, for example,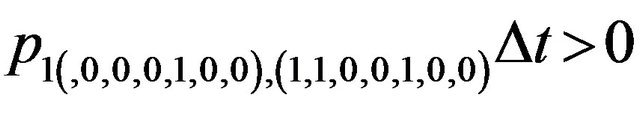 . On the other hand, D is closed since
. On the other hand, D is closed since
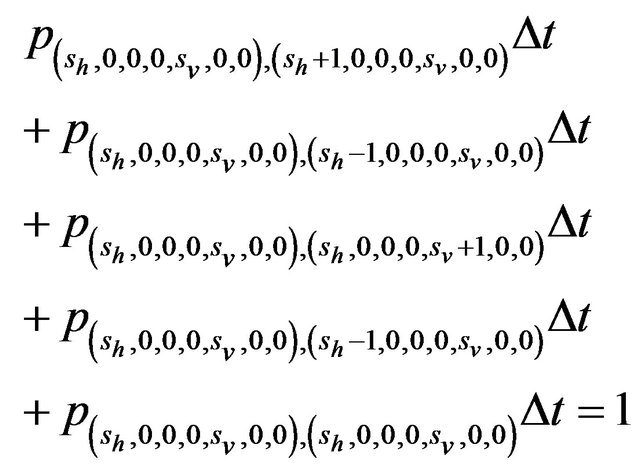
Since E is an open class, all states in E are transient. Hence and in view of D being the only closed communication class, all sample paths will eventually be absorbed into the communication class D. However, while D is indeed a closed class, it does not contain an absorbing state since 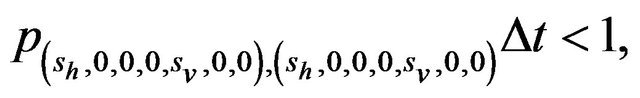
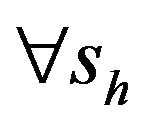 ,
, 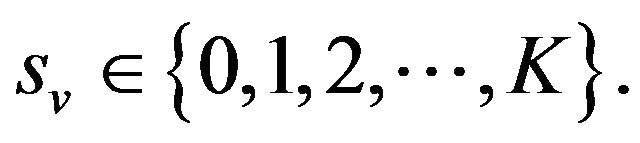
In view of the above discussion, the following theorem is established.
Theorem 2. The CTMC is a reducible chain with no absorbing states. However, all sample paths are eventually absorbed in the closed class
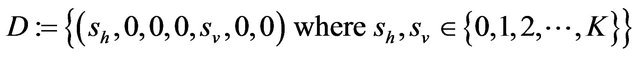 .
.
The two communication classes D and E correspond to the disease-free and endemic stages of the disease respectively. Therefore, the above result indicates that every sample path is eventually absorbed into the disease-free class and therefore, irrespective of parameter values, the stochastic model will always converge to a disease-free state. This is in direct contrast to the deterministic case in which a unique endemic steady state solution always exists for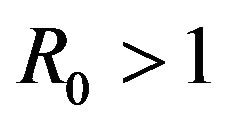 . Thus, the deterministic case allows for the possibility of the disease having an endemic equilibrium, depending on the value of a threshold parameter, while the stochastic model always predicts an eventual disease-free equilibrium. Furthermore, disease extinction takes place in finite time in the stochastic case while in the deterministic case a disease-free equilibrium is only approached asymptotically. Depending on the value of
. Thus, the deterministic case allows for the possibility of the disease having an endemic equilibrium, depending on the value of a threshold parameter, while the stochastic model always predicts an eventual disease-free equilibrium. Furthermore, disease extinction takes place in finite time in the stochastic case while in the deterministic case a disease-free equilibrium is only approached asymptotically. Depending on the value of 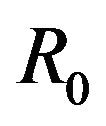 however, the sample paths for the stochastic model might remain in the endemic class E for a very long time. As a consequence, for most practical purposes, the stochastic model follows closely the behavior of the corresponding deterministic model for the majority of time. This can be demonstrated using numerical simulations.
however, the sample paths for the stochastic model might remain in the endemic class E for a very long time. As a consequence, for most practical purposes, the stochastic model follows closely the behavior of the corresponding deterministic model for the majority of time. This can be demonstrated using numerical simulations.
4. Numerical Simulations
Our purpose in this section is to explore the dynamical behavior of the stochastic model for values of the model parameter, which result in a backward bifurcation and produce a region of bistability. The behavior of the stochastic model in this region is important due to the public health implications of the backward bifurcation. As mentioned previously, the existence of a backward bifurcation in model (2.1) results in the possibility of a locally asymptotically stable endemic equilibrium even for values of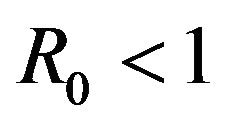 . Therefore, lowering the value of the basic reproduction number below unity is no longer a sufficient condition for disease elimination. Moreover, as mentioned in [25], the dynamics of a stochastic epidemic model does not necessarily agree with its deterministic counterpart in this region of bistability.
. Therefore, lowering the value of the basic reproduction number below unity is no longer a sufficient condition for disease elimination. Moreover, as mentioned in [25], the dynamics of a stochastic epidemic model does not necessarily agree with its deterministic counterpart in this region of bistability.
For the purpose of numerical simulations, we will formulate our stochastic model as a discrete time Markov chain (DTMC) and employ a constant time step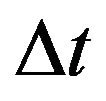 . This simplifies many of the numerical simulations and allows us to calculate the numerical mean, which is not possible when using a CTMC with random, exponentially distributed interevent times. As mentioned in [19], if the time step
. This simplifies many of the numerical simulations and allows us to calculate the numerical mean, which is not possible when using a CTMC with random, exponentially distributed interevent times. As mentioned in [19], if the time step 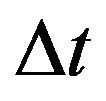 is small enough, the DTMC provides an excellent approximation to the original formulation of the stochastic model as CTMC.
is small enough, the DTMC provides an excellent approximation to the original formulation of the stochastic model as CTMC.
For the numerical simulations in this section, the following parameter values were chosen [11]:
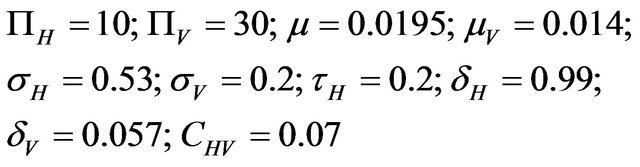
This results in a value of R0 = 0.855 and Rc = 0.571 and thus we have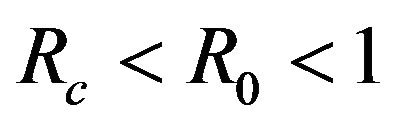 . Therefore, in view of Lemma 1 and Theorem 1, the deterministic model (2.1) now has a locally asymptotically stable disease-free equilibrium (DFE) as well as a locally asymptotically stable endemic equilibrium. We point out however, that these parameter values are not necessarily realistic from a biological and epidemiological point of view.
. Therefore, in view of Lemma 1 and Theorem 1, the deterministic model (2.1) now has a locally asymptotically stable disease-free equilibrium (DFE) as well as a locally asymptotically stable endemic equilibrium. We point out however, that these parameter values are not necessarily realistic from a biological and epidemiological point of view.
We numerically simulate 500 sample paths to estimate the mean of each discrete random variable ,
,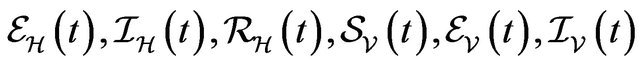 . The following initial conditions were chosen:
. The following initial conditions were chosen:
Initial Population:
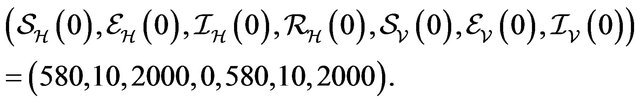
For these initial conditions, the behavior of the stochastic model closely follows that of the deterministic model (2.1) as shown in Figure 1. Despite the value of R0 being less than unity, we see that the stochastic model predicts that the disease will remain endemic in the host population. As a consequence, the behavior of the stochastic model suggests that lowering the value of R0 to less than unity is no longer a sufficient condition for disease elimination.
On the other hand, the following initial conditions result in the stochastic model as well as the deterministic model (2.1) tending towards the DFE:
Initial Population:
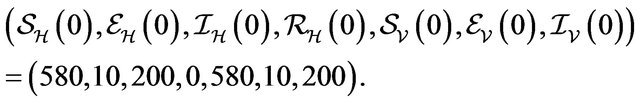
In this case, both the deterministic model (2.1) and the stochastic model predict that the disease will be eliminated from the host population in time, as shown in Figure 2.
The behavior of both the stochastic and the deterministic model is therefore highly dependent on the initial conditions. We therefore conclude that due to the existence of the backward bifurcation in model (2.1), disease elimination or persistence in the region of bistability is dependent on the initial conditions of the system. If a sufficient number of infectives are present in the population, then both the stochastic as well as the deterministic model predict that there is a possibility that the disease will persist, despite R0 being less than unity. Figures 3 and 4 display the forces of infection for the two different initial conditions.
5. Uncertainty Analysis of R0 and Rc
The values of R0 and Rc depend on the variables![]() ,
, 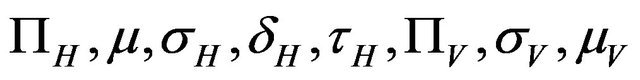 and
and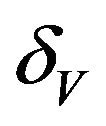 . While, deterministic models implicitly assume that the model parameters are not stochastic in nature, an element of uncertainty is always associated with estimates of these parameters due to factors such as natural variation, errors in measurements and lack of measuring techniques. In general, uncertainty analysis quantifies the degree of confidence in the parameter estimates by producing 95% confidence intervals (CI) which can be interpreted as intervals containing 95% of future estimates when the same assumptions are made and the only source of noise is observation error. This enables us to estimate the probability of
. While, deterministic models implicitly assume that the model parameters are not stochastic in nature, an element of uncertainty is always associated with estimates of these parameters due to factors such as natural variation, errors in measurements and lack of measuring techniques. In general, uncertainty analysis quantifies the degree of confidence in the parameter estimates by producing 95% confidence intervals (CI) which can be interpreted as intervals containing 95% of future estimates when the same assumptions are made and the only source of noise is observation error. This enables us to estimate the probability of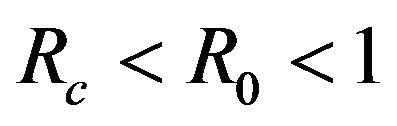 , while taking into account the uncertainty associated with estimates of the model parameters. Our purpose in this section is to estimate the likelihood of a backward bifurcation in model (2.1) for our chosen parameter values.
, while taking into account the uncertainty associated with estimates of the model parameters. Our purpose in this section is to estimate the likelihood of a backward bifurcation in model (2.1) for our chosen parameter values.
We use the Latin Hypercube Sampling (LHS) to quantify the uncertainty in R0 and Rc as a function of the 7 model parameters 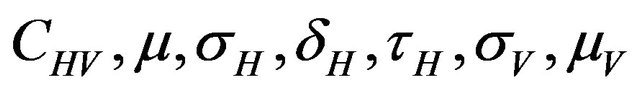 and
and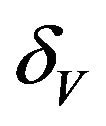 .
.
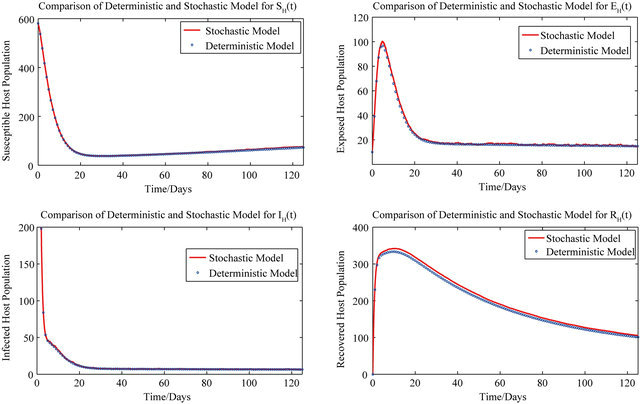
Figure 1. Comparison of the dynamics of the deterministic model (2.1) and the stochastic model.
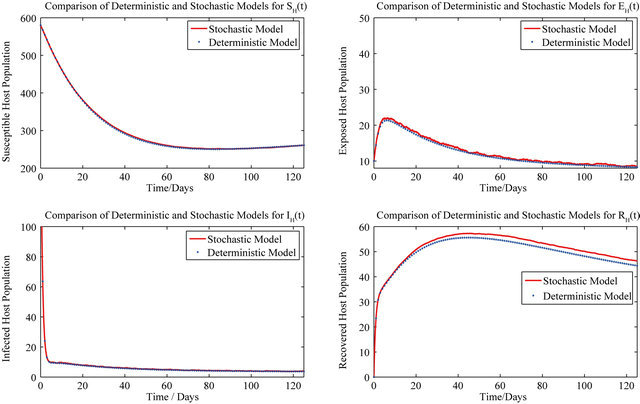
Figure 2. Comparison of the dynamics of the deterministic model (2.1) and the stochastic model.
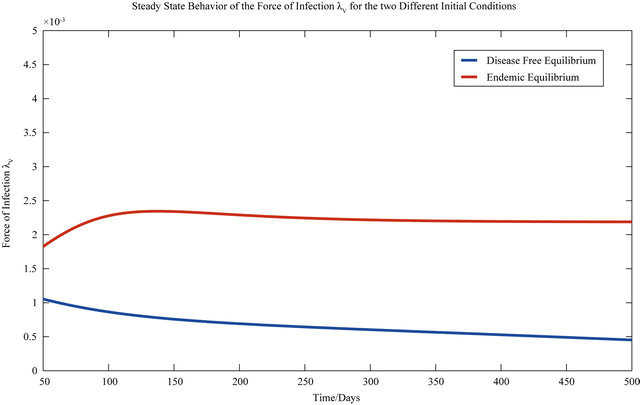
Figure 3. The steady state behavior of the force of infection λV for the two different initial conditions.
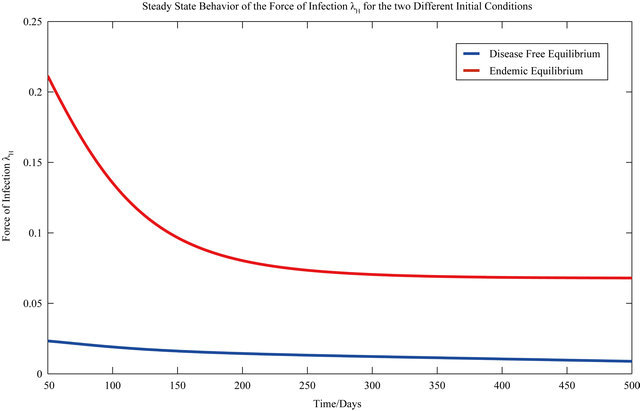
Figure 4. The steady state behavior of the force of infection λH for the two different initial conditions.
It is assumed that the recruitment rates 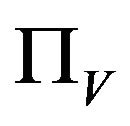 and
and 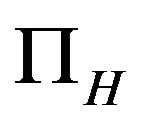 are constants. The assumed distributions of the model parameters used in the two analyses are mentioned in Table 1. The symbols
are constants. The assumed distributions of the model parameters used in the two analyses are mentioned in Table 1. The symbols 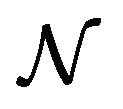 and
and ![]() stand for the normal and gamma distributions respectively. Figure 5 displays the assumed distributions of each of the seven model parameters along with the resultant distribution of
stand for the normal and gamma distributions respectively. Figure 5 displays the assumed distributions of each of the seven model parameters along with the resultant distribution of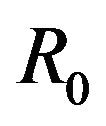 . The probability that
. The probability that 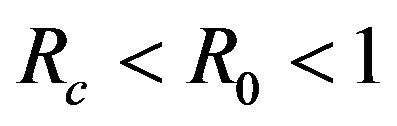 for the given parameter values and distributions is 15%. Thus, for the assumed parameter values, there is a non-trivial possibility of a backward bifurcation in the model (2.1).
for the given parameter values and distributions is 15%. Thus, for the assumed parameter values, there is a non-trivial possibility of a backward bifurcation in the model (2.1).
6. Discussion
We formulate a stochastic model, based on an existing deterministic ODE model, for the transmission dynamics of dengue virus using a continuous time Markov chain. The deterministic model is known to undergo a backward bifurcation for certain values of the model parameters and consequently presents the possibility of the co-existence of both a locally asymptotically stable DFE along with a locally asymptotically stable endemic equilibrium for certain values of the basic reproduction number less than unity. It is important to note that the deterministic model (2.1) undergoes a backward bifurcation as a consequence of both the use of standard incidence, as opposed to mass action, as well as inclusion of the vector dynamics. Thus, removing the vector dynamics from model (2.1) and using a direct transmission model as suggested in [13], or using a mass action model [11] will eliminate the phenomenon of the backward bifurcation.
The numerical mean of each discrete random variable is calculated using Monte Carlo simulations. It is found that the numerically calculated means are in excellent agreement with the corresponding results from the deterministic case for parameter values, which result in the backward bifurcation. Therefore, both the stochastic model as well as the deterministic model in [11] predict the co-existence of a locally asymptotically stable disease-free equilibrium along with an endemic equilibrium for certain values of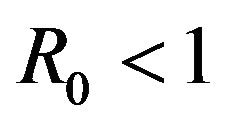 . As a consequence, the stochastic model indicates that the disease may persist in the population even if
. As a consequence, the stochastic model indicates that the disease may persist in the population even if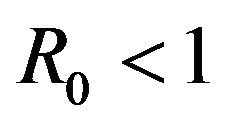 .
.
Table 1. Model parameters along with assumed values and distributions.
| Parameter | Mean, variance (dist) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
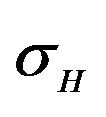
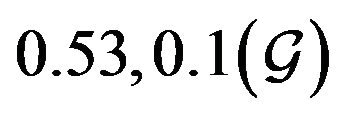
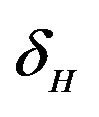
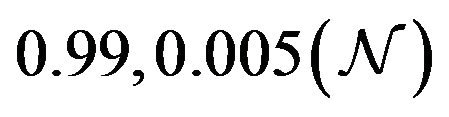
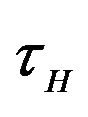
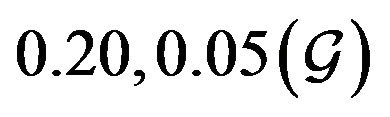
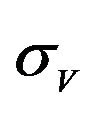
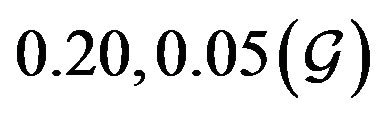
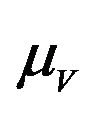
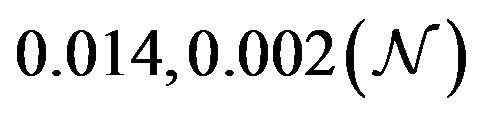
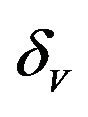
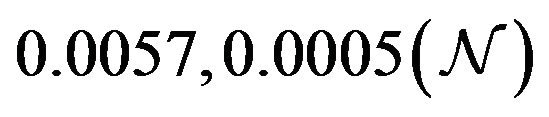

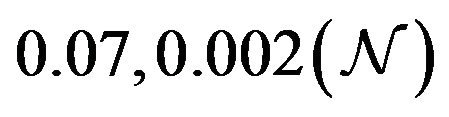
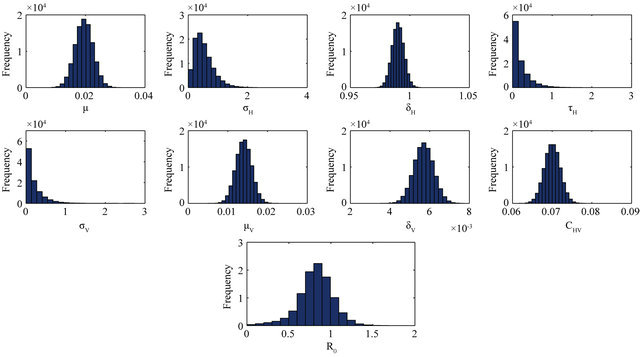
Figure 5. Assumed distributions of the model parameters generated using 100,000 iterations.
The current study focuses on a model for the spread of single strain dengue virus. However, as demonstrated by Garba et al. in [12], a model for the transmission dynamics of two strains of dengue with temporary cross immunity and ADE, also allows for the possibility of backward bifurcations. One possible extension of the current study therefore, is to formulate an analogous stochastic model corresponding to the deterministic model presented in [12] and explore the dynamics of the stochastic model for values of the model parameters which produce the bifurcation.
REFERENCES
- S. Ranjit and N. Kissoon, “Dengue Hemorrhagic Fever and Shock Syndromes,” Pediatric Critical Care Medicine, Vol. 12, No. 1, 2011, pp. 90-100. doi:10.1097/PCC.0b013e3181e911a7
- World Health Organization, “Dengue and Dengue Hemorrhagic Fever,” 2012. http://www.who.int/mediacentre/factsheets/fs117/en/
- D. J. Gubler, “Dengue and Dengue Hemorrhagic Fever,” Clinical Microbiology Reviews, Vol. 11, No. 3, 1998, pp. 480-496.
- S. B. Halstead, S. Nimmannitya and S. N. Cohen, “Observations Related to Pathogenesis of Dengue Hemorrhagic Fever IV. Relation of Disease Severity to Antibody Response and Virus Recovered,” The Yale Journal of Biology and Medicine, Vol. 42, No. 5, 1970, pp. 311-322.
- I. Kautner, M. J. Robinson and U. Kuhnle, “Dengue Virus Infection: Epidemiology, Pathogenesis, Clinical Presentation, Diagnosis, and Prevention,” The Journal of Pediatrics, Vol. 131, No. 4, 1997, pp. 516-524.
- C. Shekhar, “Deadly Dengue: New Vaccines Promise to Tackle This Escalating Global Menace,” Chemistry and Biology, Vol. 14, No. 8, 2007, pp. 871-872. doi:10.1016/j.chembiol.2007.08.004
- E. C. Holmes and S. S. Twiddy, “The Origin, Emergence and Evolutionary Genetics of Dengue Virus,” Infection, Genetics and Evolution, Vol. 3, No. 1, 2003, pp. 19-28. doi:10.1016/S1567-1348(03)00004-2
- J. Whitehorn and J. Farrar, “Dengue,” British Medical Bulletin, Vol. 95, No. 1, 2010, pp. 161-173.
- D. J. Gubler and G. Kuno, “Dengue and Dengue Hemorrhagic Fever,” CAB International, London, 1997.
- I. Kawaguchi, A. Sasaki and M. Boots, “Why Are Dengue Virus Serotypes so Distantly Related? Enhancement and Limiting Serotype Similarity between Dengue Virus Srains,” Proceedings of the Royal Society of London, Series B: Biological Sciences, Vol. 270, No. 1530, 2003, pp. 2241-2247. doi:10.1098/rspb.2003.2440
- S. M. Garba, A. B. Gumel and M. R. Abu Bakar, “Backward Bifurcations in Dengue Transmission Dynamics,” Mathematical Biosciences, Vol. 215, No. 1, 2008, pp. 11- 25. doi:10.1016/j.mbs.2008.05.002
- S. M. Garba, A. B. Gumel and M. R. Abu Bakar, “Effect of Cross-Immunity on the Transmission Dynamics of Two Strains of Dengue,” International Journal of Computer Mathematics, Vol. 87, No. 10, 2010, pp. 2361- 2384. doi:10.1080/00207160802660608
- H. J. Wearing and P. Rohani, “Ecological and Immunological Determinants of Dengue Epidemics,” Proceedings of the National Academy of Sciences, Vol. 103, No. 31, 2006, pp. 11802-11807. doi:10.1073/pnas.0602960103
- L. Esteva and C. Vargas, “Coexistence of Different Serotypes of Dengue Virus,” Journal of Mathematical Biology, Vol. 46, No. 1, 2003, pp. 31-47. doi:10.1007/s00285-002-0168-4
- N. Ferguson, R. Anderson and S. Gupta, “The Effect of Antibody-Dependent Enhancement on the Transmission Dynamics and Persistence of Multiple-Strain Pathogens,” Proceedings of the National Academy of Sciences, Vol. 96, No. 2, 1999, pp. 790-794. doi:10.1073/pnas.96.2.790
- L. Esteva and C. Vargas, “A Model for Dengue Disease with Variable Human Population,” Journal of Mathematical Biology, Vol. 38, No. 3, 1999, pp. 220-240. doi:10.1007/s002850050147
- L. Esteva and C. Vargas, “Analysis of a Dengue Disease Transmission Model,” Mathematical Biosciences, Vol. 150, No. 2, 1998, pp. 131-151. doi:10.1016/S0025-5564(98)10003-2
- G. Chowell, P. Diaz-Duenas, J. C. Miller, A. AlcazarVelazco, J. M. Hyman, P. W. Fenimore and C. Castillo Chavez, “Estimation of the Reproduction Number of Dengue Fever from Spatial Epidemic Data,” Mathematical Biosciences, Vol. 208, No. 2, 2007, pp. 571-589. doi:10.1016/j.mbs.2006.11.011
- L. J. S. Allen, “An Introduction to Stochastic Epidemic Models,” In: F. Brauer, P. Van den Driessche and J. Wu, Eds., Mathematical Epidemiology, Springer-Verlag, Berlin, 2008, pp. 77-128. doi:10.1007/978-3-540-78911-6_3
- A. J. Keeling and J. V. Ross, “On Methods for Studying Stochastic Disease Dynamics,” Journal of The Royal Society Interface, Vol. 5, No. 19, 2008, pp. 171-181. doi:10.1098/rsif.2007.1106
- N. Bailey, “A Simple Stochastic Epidemic,” Biometrika, Vol. 37, No. 3-4, 1950, pp. 193-202. doi:10.2307/2332371
- L. J. S. Allen, D. A. Flores, R. K. Ratnayake and J. R. Herbold, “Discrete-Time Deterministic and Stochastic Models for the Spread of Rabies,” Applied Mathematics and Computation, Vol. 132, No. 2, 2002, pp. 271-292. doi:10.1016/S0096-3003(01)00192-8
- G. H. Weiss and M. Dishon, “On the Asymptotic Behavior of the Stochastic and Deterministic Models of an Epidemic,” Mathematical Biosciences, Vol. 11, No. 3, 1971, pp. 261-265. doi:10.1016/0025-5564(71)90087-3
- A. R. Tuite, J. Tien, M. Eisenberg, D. J. Earn, J. Ma and D. N. Fisman, “Cholera Epidemic in Haiti, 2010: Using a Transmission Model to Explain Spatial Spread of Disease and Identify Optimal Control Interventions,” Annals of Internal Medicine, Vol. 154, No. 9, 2011, pp. 593-601. doi:10.7326/0003-4819-154-9-201105030-00334
- L. J. S. Allen and P. Driessche, “Stochastic Epidemic Models with a Backward Bifurcation,” Mathematical Biosciences and Engineering, Vol. 3, No. 3, 2006, pp. 445- 458. doi:10.3934/mbe.2006.3.445
- D. R. de Souza, T. Tomé, S. T. Pinho, F. R. Barreto and M. J. de Oliveira, “Stochastic Dynamics of Dengue Epidemics,” Physical Review, Vol. 87, No. 1, 2013, Article ID: 012709.
- S. Spencer, “Stochastic Epidemic Models for Emerging Diseases,” Ph.D. Dissertation, University of Nottingham, Nottingham, 2008.
- L. J. S. Allen, “An Introduction to Stochastic Processes with Applications to Biology,” Chapman and Hall/CRC, Boca Ratton, 2010.
- L. J. S. Allen and A. M. Burgin, “Comparison of Deterministic and Stochastic SIS and SIR Models in Discrete Time,” Mathematical Biosciences, Vol. 163, No. 1, 2000, pp. 1-34. doi:10.1016/S0025-5564(99)00047-4
- P. van den Driessche and J. Watmough, “Reproduction Numbers and Sub-Threshold Endemic Equilibria for Compartmental Models of Disease Transmission,” Mathematical Biosciences, Vol. 180, No. 1, 2002, pp. 29-48. doi:10.1016/S0025-5564(02)00108-6
NOTES
*Corresponding author.