Advances in Bioscience and Biotechnology
Vol.5 No.7(2014), Article
ID:47419,10
pages
DOI:10.4236/abb.2014.57077
Improved Recovery of Erythropoietin and Darbepoetin from Equine Plasma by the Application of a Wheat Germ Agglutinin Mediated Pre-Extraction Prior to Immunoaffinity Chromatography
Shawn M. R. Stanley*, Danny Chua
The Singapore Turf Club Laboratory, 1 Turf Club Ave, Singapore, Singapore
Email: *Shawn_stanley@turfclub.com.sg
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

![]()

Received 29 April 2014; revised 31 May 2014; accepted 14 June 2014
ABSTRACT
We describe a two-step method that uses wheat germ agglutinin immobilized on Sepharose gel followed by immunoaffinity chromatography (IAC) to extract recombinant human erythropoietin and Darbepoetin from equine plasma. Lectin affinity chromatography was shown to be an effective approach for isolating these epoetins from plasma and in combination with IAC; this method gave superior recovery when compared to the use of the latter technique alone. Moreover, due to the ease with which it can be scaled up, it is particularly well suited for pre-concentrating larger volumes of samples prior to IAC and this provides a facile way of improving the overall sensitivity with which these foreign proteins can be detected in equine plasma.
Keywords:Erythropoietin, Darbepoetin Alfa, Immunoaffinity Extraction, Lectin Affinity Extraction, Wheat Germ Agglutinin, Horseracing

1. Introduction
Anecdotal evidence is that erythropoiesis stimulating agents (ESAs) have been widely abused to dope horses in the belief that increasing the production of red blood cells will have a beneficial effect on stamina and, hence, improve their chance of winning the race. Furthermore, these compounds are rapidly excreted, yet the effects last for weeks after administration, which maximizes the beneficial outcome while, at the same time, minimizes the risk of getting caught abusing these drugs.
The erythropoietin receptor can be stimulated by the various forms of recombinant human erythropoietin (EPO) and also by Darbepoetin (DPO), where substitution of five amino acids in the backbone has produced a substance with significantly more sugar attachment and an average mass that has been increased by approximately 6 kDa [1] . CERA (Micera™) represents a novel direction [2] in the development of these drugs, as the glycosylation of the EPO protein has been replaced by the chemical attachment of polyethylene glycol and this produces a longer period of in-vivo action. However, despite attempts to produce substances with prolonged excretion from the body, all of aforementioned ESAs are almost completely eliminated from blood within several days of administration.
In addition to modifications to the original human EPO protein structure, there are also the EPO mimetics. For example, Hematide® contains a dimeric peptide coupled to polyethylene glycol core [3] and this substance is able to elicit the desired response from the erythropoietin receptor.
Consequently doping control laboratories have to tackle two issues in regards to ESAs. Firstly, there is growing diversity in the chemical structure of the agents that can be used to stimulate the receptor to trigger erythropoiesis and that means that a single screening method is unlikely to be able to detect all of the available variants within a single analysis. A second and considerably more important factor is that drugs that are rapidly eliminated have a much lower probability of being encountered during the screening of the routinely collected samples. While this effect can be offset to some extent by collecting a larger number of out-of-competition testing (OOCT) samples in the weeks before the horse is due to run in a race, this needs to be implemented with a very high sample collection frequency to ensure that there is a reasonable probability that sampling will coincide with the window of detection following the administration of the ESA. However, since doping can take place at anytime within a time frame of several weeks, this translates into a large number of samples that must be collected and the reality is that this would be a prohibitively expensive exercise. In addition, for obvious reasons, visits by the sample collection staff must be unannounced and this unexpected event may cause some disruption to the normal running of the facility where the horses are stabled. Whilst the trainers in most jurisdictions are obligated by their Rules of Racing to accede to each and every directive to produce a horse for the collection of samples for testing, there are practical reasons why this cannot be done too frequently. In combination, these factors produce an environment where the sample collection frequency is almost always suboptimal and so it is probable that only a very low percentage of the samples received at the laboratory will have been obtained from a horse that has been treated with an ESA just (~<48 hours) before the sample was collected. Consequently, since this will be an infrequent event, it is important to maximize the chance of confirming each and every suspicious sample that has been detected on initial screening. This is particularly important as these epoetins do not have a legitimate place in the treatment of racehorses and cases of abuse should be detected and the offenders punished accordingly to act as a deterrent and thereby protect the integrity of the sport. This means it is essential to have a strategy to cope effectively with low levels of these substances and all aspects of the sample preparation and analytical methodology must be fully optimized to achieve this goal.
Various ways [1] [4] -[10] of using mass spectrometric detection to provide the confirmatory data required to prove the presence of recombinant human EPO have been published and these represent an improvement on methods [11] -[18] that provide somewhat ambiguous evidence relating to the presence of the drug in the sample. In particular the uses of techniques, such as immunoassay or isoelectric focusing followed by double blotting, fail to meet the minimum standard for proof of detection and are not considered suitable for confirmatory testing of thoroughbred racing samples. In this regard the laboratory should comply with the Association of Official Racing Chemists’ guide [19] for identification of a Prohibited Substance.
There have been a large number of publications [4] -[7] [9] [10] covering the isolation and analysis of EPO, DPO and CERA and these proteins all share a high degree of homology in their core structure. While it has been reported that the limit at which these substances can be confirmed is around 0.1 to 1 ng/ml using IAC, it is worth mentioning that in regards to the Guan et al. publication [4] , it was noted by other authors [10] that they were not able to achieve a similar limit of detection when applying the methodology in the manner described. In many cases the techniques described utilize paramagnetic beads with an anti-EPO antibody attached to the surface. Another option is for the anti-EPO antibody to be held on a monolithic polymer support inside a disposable holder [20] [21] or on a chromatographic gel bed [22] .
Despite the heavy focus on IAC, this is not the only variant of affinity chromatography that has been shown to be effective in the isolation of the EPO from biological matrices, as more than thirty years ago it was established that lectin-agarose derivatives could be used in the isolation and recovery of this family of compounds [23] -[25] . These authors demonstrated that wheat germ agglutinin (WGA) was the most effective of all of the lectins that they tested for this purpose and this is due to the strong affinity that it has for the sialic acid and N-acetylglucosamine sugars that are a significant feature [8] [26] [27] of the EPO & DPO glycoproteins.
The lectin purification approach can also be used as an additional means of purification and the use of WGA extraction has been reported as an effective process for reducing the chemical background when EPO is isolated from human urine by ultrafiltration [28] . It is also a simple and effective way of increasing the volume of sample that is extracted and this is a particularly useful attribute, as methods based on magnetic bead and/or monolithic support mediated extractions are difficult to scale up to cater for larger (>10 ml) volumes of plasma.
We present an optimized method for extracting EPO & DPO from equine plasma using WGA immobilized on Sepharose 4B-CL gel and a monolithic support based IAC extraction.
2. Materials and Methods
2.1. Reagents and Chemicals
Eprex™ recombinant human erythropoietin (Epoetinum α, 10,000 IU which is equivalent to 83 µg/ml) was purchased from Janssen-Cilag PTE Ltd. (Singapore). Polyclonal anti-rhEPO antibodies (purified rabbit IgG) were obtained from R&D systems (MN, USA). The EPO purification & quantification kits were supplied by Maiia Diagnostics (Uppsala, Sweden). Dynabeads M-280 tosylactivated magnetic beads and the particle separator were purchased from Invitrogen (Oslo, Norway). Molecular biology grade Igepal CA 630, N-acetylneuraminic acid (NANA), wheat germ agglutinin (WGA) from Triticum vulgaris immobilized on agarose CL-4B, sodium borate, bovine serum albumin (BSA), ethylenediaminetetracetic acid (EDTA), sodium azide and polyethylene glycol 6000 were purchased from Sigma-Aldrich (Singapore). Tris-HCl, sodium chloride, di-sodium hydrogen phosphate and sodium dihydrogen phosphate were purchased from Merck (Darmstadt, Germany). N-acetyl-D-glucosamine (GlucNAc) was purchased from Alfa Aesar (Lancashire, UK). Thermo Scientific (MA, USA) supplied the disposable gravity-flow columns (Prod No: 29922), Pierce protein research 99+% formic acid and Triton-X. Sequencing grade modified trypsin (100 μg) was procured from Promega (Singapore). PicoFrit™ columns were from Alpha Analytical (Singapore). Optima™ LCMS grade acetonitrile and water were from Fisher Scientific. The synthetic peptide T17 (VYSNFLR) was obtained from Auspep (VIC, Australia).
2.2. Preparation of Blank Plasma and the Spiking of Equine Plasma with Recombinant Human EPO Equine plasma that had previously been screened by the Maiia EPO quantification kit and had shown a low response for the analyte was pooled for use as the plasma blank. The pooled plasma (50 ml) was also used for preparing the spiked samples, where Eprex™ was added to bring the level of recombinant human EPO to required concentration. The buffer A (20 mM Tris, 0.15 M NaCl, 0.1% Trition-X) blank was used for all of the extraction methods listed below.
2.3. Measurement of EPO Using the Maiia Quantification Kit
The kit was used, in the manner described in the Maiia Diagnostics handbook, to measure the EPO level in 25 μl aliquots of the extracted material.
2.4. Extraction Methods Used to Isolate rhEPO from the Equine Plasma
2.4.1. Lectin Affinity Extraction of EPO from Equine Plasma Using WGA
Sample was mixed with buffer A before being filtered using 0.22 µm cellulose acetate syringe filter (Millipore, Singapore). The WGA slurry was washed three times with buffer A and after centrifugation at 1000 rcf the wash the supernatant was discarded. The sample and the gel were combined in a 10 ml polyethylene capped tube and this mixture was rotated slowly for between 4 & 16 h at ambient temperature before being transferred to a column and the liquid was allowed to elute under gravity. The column was washed once with buffer A followed by buffer B (20 mM Tris, 0.15 M NaCl). The column was centrifuged at 1000 rcf for 1 min and the liquid was discarded. The EPO was eluted with either (0.2 or 0.5 M) N-acetyl-D-glucosamine (2 × 1 ml) or 0.2 M NANA in buffer B and the eluate was recovered by centrifuged at 1000 rcf for 1 min.
2.4.2. Immunoaffinity Extraction of EPO from Equine Plasma
Samples were extracted following the procedures described in the EPO purification kit user manual supplied by Maiia Diagnostics, as well as those described by Guan et al. [4] and also Yu et al. [10] . In the experiments where these extraction methods were preceded by the WGA extraction of the plasma, the only deviation from the methods published by these authors was to substitute the eluate (0.5 M N-acetyl-D-glucosamine (GlucNAc)) for the plasma described in their procedure.
2.5. Trypsin Digestion
The trypsin was reconstituted in 100 µl of ammonium bicarbonate buffer (100 mM) and 5 µl added to the 55 µl eluate and adjustment buffer from the Maiia EPO purification kit. The reaction was allowed to proceed for 3 hours at 37 ˚C before 4 µl of 10% formic acid in acetonitrile was added to stop the process.
2.6. Nano LC ESI/MS of the Tryptic Peptides from the WGA + Maiia Extracted Plasma Samples
Twenty-two microlitre of the digest was injected using an Eksigent™ (CA, USA) AS2 high pressure autosampler (32 µl loop installed). Peptides were trapped on an in-house fabricated monolithic (100 µm × 250 mm) trap column (20% of both divinyl benzene & polystyrene, 60% dodecanol as porogen, 1% w/w azobisisobutyronitrile initiator) [29] installed onto the gradient 1 switching valve of an Eksigent™ Nano LC-Ultra 2D system. The analytical column used was a PicroFrit (75 µm × 150 mm, 15 µm tip) packed in-house with Phenomenex C18 Kinetex™ packing (2.6 µm). The sample was transferred from the autosampler loop using the loading pump flowing at 5 µl/min (5% methanol in water) for 8 minutes. Thereafter the valve on channel 1 was switched and the starting mobile phase 200 nl/min with a composition of 96% A (0.15% formic acid water) and 4% B (acetonitrile) was changed to 50% A at 15 minutes. Heavily bound compounds were flushed from the LC column by modifying the gradient to 100% B at 15.1 and holding this until 25 minutes.
The LC was interfaced to a Thermo Scientific LTQ Orbitrap Discovery™ operated with New Objective (USA) PicoView™ mounting system that was modified to allow the sheath gas from the instrument to be connected to the PicoFrit™ column via a T-piece. A co-axial gas flow of 20 (arbitrary units) was used to dislodge droplets forming on the tip when there was high aqueous content and to assist the ionization process. The linear ion trap in the MS/MS mode m/z 402.2, 449.7 and 462.3 precursor ions were selected for the analysis of the three peptide fragments generated from the EPO and a collision energy of 35 with wide band activation selected. The mass range for the product ions was 120 - 1000.
3. Results & Discussion
A commonly used approach for improving the limit of detection (LOD) for an analysis is to introduce steps to increase the selectivity of the extraction methodology, as enhancing the recovery of the target analyte whilst concurrently reducing the chemical background gives a better signal to noise ratio and this makes detection easier. The most effective way of achieving this goal is to employ a highly selective affinity ligand attached to a solid support, since this is able to extract the analyte by specifically interacting with a moiety or a part of the tertiary structure that is unique to the compound that you are isolating from the matrix. Even though there are many ligands available from both from biological and non-biological sources, IAC using the appropriate antibody is probably the most widely used implementation of this approach.
Hence it is no surprise that IAC is the cornerstone of many of the methods [4] -[7] [10] [20] [21] [30] that have been published for the detection of EPO & DPO in equine samples. However, with the exception of one paper [4] that reported 77% efficiency for EPO isolation, all of the recoveries reported (see Table 1) in these publications fell within the 40% to 60% range, a figure that is only marginally acceptable. We have used three ([4] [10] [21] ) of the methods to isolate recombinant human EPO from aliquots taken from a single plasma sample that had

Table 1 . Recovery of recombinant human EPO from equine plasma.
aPublished result was ≤70%.
been spiked at 1.5 ng/ml. We then estimated the recovery of the EPO using an immunochromatographic assay (ICA) and applying a simple calculation based upon the ratio of a post-elution spiking of the extract from a blank plasma sample to obtain the results shown in Table1
It was interesting that the calculated recoveries were higher than those reported for two of the isolation methods that we used, whereas were only able to obtain an average recovery of 66% for the one developed by Guan & co-workers [4] . Our preference was for the approach used in the Maiia EPO Purification Kit®, as this gave the highest % recovery in the smallest elution volume (~55 μl) and was also much simpler to implement and less time consuming to use than either of the two other methods that were evaluated.
Aside from the issue of lower than optimal recovery, the intra-sample variability for several aliquots of a single sample extracted in triplicate using the same batch of immunoaffinity media can also be noticeably higher than when the extraction is performed on aliquots of spiked buffer. This may be due to the fact that there are some intermittent/unpredictable interactions between the highly abundant plasma proteins and these epogens, which may interfere with the ability of the anti-EPO antibody to capture the antigen. As a result, some of the analyte could have been lost during the loading and/or washing steps used. For this reason, the reduction of protein-protein interaction was attempted [9] through the application of an albumin immunodepletion column prior to IAC, but this also appeared to trap a large proportion of the EPO and it was reported to cause a lower (25% - 40%) recovery. Based upon this observation, Singh and coworkers proposed that the sample should rather be purified with either a hydroxyapatite (HTP-Progel®) or an immobilized lectin concavalin A (ConA) column before IAC.
The hydroxyapatite option appears, at face value, to be unsatisfactory as it produced a large volume of highly diluted EPO isolate and thus it would be inconvenient to combine this process with the subsequent IAC extraction step. Whilst their proposal of using ConA as a pre-extraction step seemed to present a viable option, unfortunately, our experiment with this lectin showed a large proportion of the EPO was not retained on the affinity medium. We concluded that it did not exhibit a strong affinity for the analyte and this is consistent with the findings of Spivak et al. [23] -[25] as well as that of Franco Fraguas et al. [31] . The same studies had also shown that WGA coupled to Sepharose gel had exhibited good affinity for our target analytes and thus we switched to the use of this media to determine whether this would provide a satisfactory extraction when used by itself or in tandem with an IAC extraction.
In order to assess the amount of the affinity media that would be required to give efficient isolation of the analytes, the WGA 4B-CL Sepharose slurry was transferred into tubes until beds with a volume of approximately 0.1, 0.5 & 1 ml were formed on settling. Thereafter these aliquots were mixed with 5 ml of plasma samples that had been spiked with a high (50 ng/ml) or medium concentration (2 ng/ml) of one of the epoetins (EPO, CERA and DPO) before being diluted with an equal volume of the loading buffer. Interaction between the sample and gel was facilitated by slowly tumbling the tubes on a rotator for 4 hours before the gel was transferred into a fritted column. After washing and subsequent elution (2 × 1 ml) with 0.5 M GlucNAc in buffer B, the quantification of the epoetin concentration was performed using ICA. Our results demonstrated that even 0.1 ml of the WGA gel was effective in isolating the DPO (approximately 100% recovery). Not unexpectedly, there was little or no affinity for CERA and using the MAIIA purification kit® we were able to recover this protein from the flow through from the loading/washing steps. Since this was approximately the same % recovery as achieved when extracting the unprocessed sample, coming in contact with the WGA gel appears to neither advantage nor disadvantage the later IAC extraction of this substance from the un-retained fractions.
The result for the isolation of recombinant human EPO fell in between these two extremes and it could be seen that a minimum of 0.5 ml of WGA gel would be required to provide satisfactory isolation of this protein. The requirement for a larger amount of media to extract EPO can be attributed to the lower sugar content of the substance in comparison to DPO. The loss of the analyte that was observed when using the lower bed volume column was also not greatly diminished by a further 12 hours of contact of the sample with the gel, as EPO was still found in the un-retained fraction from the loading step at approximately the same level as seen after 4 hours of incubation. Therefore increasing the gel volume to >0.5 ml for 5 ml of plasma is the only way to achieve satisfactory isolation of recombinant human EPO during the loading stage.
In order to establish the optimal elution conditions for the WGA, we loaded a number of 0.5 ml gel portions with 5 ml of 1.5 ng/ml of EPO spiked plasma that had been diluted with buffer (1:1). Then after washing had been completed, we used a step elution (100 µl) of either a 0.2 or 0.5 M GlucNAc buffer solution and collected the fractions for ICA quantification. We also evaluated 0.2 M NANA as a substitute for GlucNac, as it has previously been reported [25] to elute EPO from WGA Sepharose.
However, the recovery (see Figure 1(a) & Graph 1) with NANA was lower than the equivalent result achieved with GlucNAc and due to this poor outcome and the high cost of the sugar; this experiment was not pursued any further. Our other results, as shown in Figure 1(b) & Figure 1(c), demonstrated that, even with the more concentrated sugar solution, a minimum of 600 µl was required to recover the majority of spiked materials from the gel.
So in all cases the elution volume far exceeded the 50 to 100 µl range that is optimal to achieve satisfactory sensitivity from the trypsin digestion and subsequent LC/MS/MS analysis step, which means that an additional step to reduce the volume would be required. However, since we have had mixed success with either molecular weight cutoff filtration or evaporation for the purposes of concentrating solutions containing these epogens, we abandoned the idea of a lectin only extraction procedure. Instead we proceeded to examine the potential for using the WGA as the pre-extraction for IAC to improve the recovery of the analyte and also increase the robustness of the method.
The WGA extraction was introduced as a preliminary preparation step before proceeding on with extractions using all of the three methods that were described previously. Using aliquots taken from the spiked plasma sample that was used for the IAC alone extractions, we noted an improvement in the concentration of EPO detected using the ICA. Overall, the addition of this step raised the percentage recovery (Table 1) by around 15%.
The most dramatic improvement in the overall recovery was found with the samples spiked with the DPO,
 (a)
(a) (b)
(b) (c)
(c)
Figure 1. (a) Lane 10-blank, 11 to 20 are 100 μl aliquots of 200 mM GlucNAc, (b) lane 30-blank, 31 to 40 are 100 μl aliquots of 500 mM GlucNAc & (c) lane W-blank, 1 to 10 are 100 μl aliquots of 200 mM NANA.
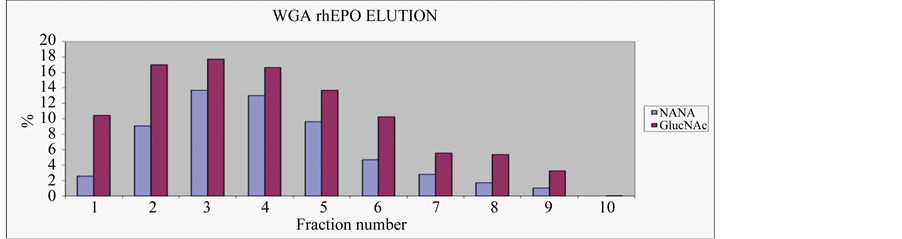
Graph 1. The concentration of EPO detected by ICA in fractions (100 μl) eluted with either 0.2 M NANA or GlucNAc.
which is reported [21] to be extracted from equine plasma with a 47% recovery when using the monolithic support based IAC. This relatively poor result may be due to the hyperglycosylation and/or the modifications to the protein chain leading to a reduction of the strength of the antibody-antigen interaction. However, by purifying the sample first, we appear to have reduced the competition from other weak antigens, which increased the total amount of DPO that was able to bind to the IAC column and the recovery of DPO was on par with our result for EPO.
The intra-batch reproducibility was evaluated by spiking plasma with EPO and DPO at the 1.5 ng/ml concentration level and performing a triplicate extraction of two samples of each of the epoetins. The concentration of the epoetin in the eluent was estimated using ICA after the elution of the WGA gel with 0.5 m GlucNAc and also after the IAC was completed. After both stages of the extraction the standard error for the measured results was less than 10%, which was considered as satisfactory for our purposes.
To test the suitability of this approach when used in conjunction with LC/MS/MS analysis, we used the WGA pre-extraction combined with the MAIIA purification kit to extract 5 ml of blank equine plasma that was spiked with EPO at a medium concentration (2 ng/ml). The samples were eluted in a total volume of 55 μl and were then digested with trypsin and subjected to LC/MS/MS. The T17 peptide was detected and the ion trap spectrum (Figure 2) closely matches that from a synthetic peptide with the amino acid composition VYSNFLR. Hence the combination of the two affinity extractions was demonstrated to have been effective in isolating this epogen from the spiked equine plasma.
An additional benefit of using the WGA pre-extraction is that it is also relatively simple to increase the total volume of sample that can be extracted and this tactic can be used to lower the LOD of the method. Scaling up is simply a matter of using a larger volume of plasma and proportionally increasing the amount of affinity matrix
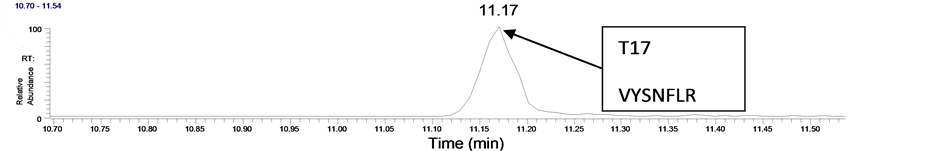 (a)
(a) (b)
(b)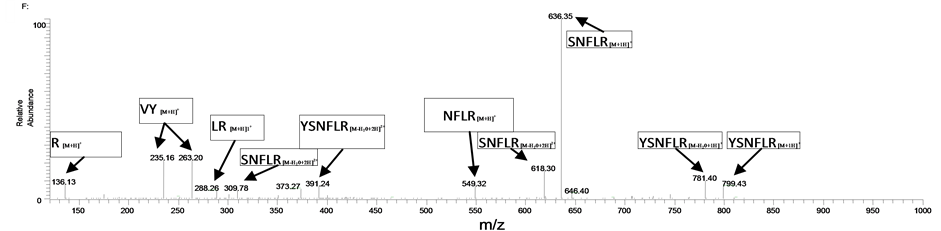 (c)
(c)
Figure 2. (a) XIC 636.3 (MS/MS of the m/z 449.7 ion) from the analysis of a WGA + IAC extract of an equine plasma sample spiked with EPO at 2 ng/ml, (b) Product ion spectrum of T17 from the extract and (c) Product ion spectrum from the VYSNFLR synthetic peptide.
that is added. Our experiments have shown that reliable recovery can be achieved from a 5 ml affinity gel bed with 2 × 1 ml of the 0.5 M GlucNAc containing buffer. This is a particularly useful attribute, as it is difficult to scale up the IAC step because there are limitations on the capacity of the extraction media and other practical difficulties, such as the lack of equipment capable of handling larger quantities of magnetic beads when contained in a larger volume.
Many of the blood collection tubes used to collect samples from racehorses were designed for human use and hence they collect a relatively small volume. This can be offset by taking more tubes, but this time-consuming and for practicality we are currently limited to 4 × 10 ml gel separator blood tubes and this provides approximately 15 ml of plasma in total. However, for the collection of OOCT blood samples we are currently trialing the implementation of a larger volume collection device that was designed for use in veterinary medicine. Each of the 60 ml collections provides sufficient blood for an aliquot of at least 20 ml of plasma to be used for the purposes of confirming any suspected epogens detected during ELISA screening. We have tested this approach on 20 ml of a 0.1 ng/ml EPO spiked plasma sample and the results were in line with data from extraction of 2 ml of a 1 ng/ml sample.
4. Conclusion
We have concluded from these results that the isolation of EPO or DPO from equine plasma is optimized by combining WGA affinity chromatography with IAC. This will improve the probability of confirming the presence of these ESAs if and when they are detected during the screening. The retention of the load and subsequent wash steps from the WGA extraction step will allow for the same aliquot from the sample to be used to confirm the presence of CERA, which does not bind to the lectin. Consequently, a lack of response in the WGA & IAC extraction, coupled with the detection of the EPO peptides in the un-retained fraction after an IAC extraction could be used to alert the laboratory to the likelihood that the sample may contain this ESA. Hence, any further confirmatory work can be tailored towards detecting the characteristic elements found in this substance. The addition of the WGA extraction step also offers the advantage that it can be easily scaled up to handle a large volume of plasma and this provides a simple way of increasing the sensitivity of the method.
References
- Stanley, S.M. and Poljak, A. (2003) Matrix-Assisted Laser-Desorption Time-of Flight Ionisation and High-Performance Liquid Chromatography—Electrospray Ionisation Mass Spectral Analyses of Two Glycosylated Recombinant Epoetins. Journal of Chromatography B, 785, 205-218. http://dx.doi.org/10.1016/S1570-0232(02)00824-3
- Wang, Y.J., Hao, S.J., Liu, Y.D., Hu, T., Zhang, G.F., X. Zhang, Qi, Q.S., Ma, G.H. and Su, Z.G. (2010) PEGylation Markedly Enhances the in Vivo Potency of Recombinant Human Non-Glycosylated Erythropoietin: A Comparison with Glycosylated Erythropoietin. Journal of Controlled Release, 145, 306-313. http://dx.doi.org/10.1016/j.jconrel.2010.04.021
- Macdougall, I.C. (2008) Hematide, a Novel Peptide-Based Erythropoiesis-Stimulating Agent for the Treatment of Anemia. Current Opinion in Investigational Drugs, 9, 1034-1047.
- Guan, F., Uboh, C.E., Soma, L.R., Birks, E., Chen, J., Mitchell, J., You, Y., Rudy, J., Xu, F., Li, X. and Mbuy, G. (2007) LC-MS/MS Method for Confirmation of Recombinant Human Erythropoietin and Darbepoetin α in Equine Plasma. Analytical Chemistry, 79, 4627-4635. http://dx.doi.org/10.1021/ac070135o
- Guan, F., Uboh, C.E., Soma, L.R., Birks, E., Chen, J., You, Y., Rudy, J. and Li, X. (2008) Differentiation and Identification of Recombinant Human Erythropoietin and Darbepoetin Alfa in Equine Plasma by LC-MS/MS for Doping Control. Analytical Chemistry, 80, 3811-3817. http://dx.doi.org/10.1021/ac800054t
- Guan, F., Uboh, C.E., Soma, L.R., Birksz, E. and Chen, J. (2009) Identification of Darbepoetin Alfa in Human Plasma by Liquid Chromatography Coupled to Mass Spectrometry for Doping Control. International Journal of Sports Medicine, 30, 80-86. http://dx.doi.org/10.1055/s-0028-1103283
- Guan, F., Uboh, C.E., Soma, L.R., Maylin, G., Jiang, Z. and Chen, J. (2010) Confirmatory Analysis of Continuous Erythropoietin Receptor Activator and Erythropoietin Analogues in Equine Plasma by LC-MS for Doping Control. Analytical Chemistry, 82, 9074-9081. http://dx.doi.org/10.1021/ac102031w
- Kawasaki, N., Haishima, Y., Ohta, M., Itoh, S., Hyuga, M., Hyuga,S. and Hayakawa, T. (2001) Structural Analysis of Sulfated N-Linked Oligosaccharides in Erythropoietin. Glycobiology, 11, 1043-1049. http://dx.doi.org/10.1093/glycob/11.12.1043
- Singh, A.K. and Gupta, S. (2007) Analysis of Recombinant Human Erythropoietin and Darbepoietin in Spiked Plasma. Proteomics—Clinical Applications, 1, 626-639. http://dx.doi.org/10.1002/prca.200600972
- Yu, N.H., Ho, E.N., Wan, T.S. and Wong, A.S. (2010) Doping Control Analysis of Recombinant Human Erythropoietin, Darbepoetin Alfa and Methoxy Polyethylene Glycol-Epoetin Beta in Equine Plasma by Nano-Liquid Chromatography—Tandem Mass Spectrometry. Aalytical and Bioanalytical Chemistry, 396, 2513-2521. http://dx.doi.org/10.1007/s00216-010-3455-8
- Bartlett, C., Clancy, G.J., Cowan, D.A. and Healy, J.F. (2006) Detection of the Administration of Human Erythropoietin (HuEPO) to Canines. Journal of Analytical Toxicology, 30, 663-669. http://dx.doi.org/10.1093/jat/30.9.663
- Belalcazar, V., Gutierrez Gallego, R., Llop, E., Segura, J. and Pascual, J.A. (2006) Assessing the Instability of the Isoelectric Focusing Patterns of Erythropoietin in Urine. Electrophoresis, 27, 4387-4395. http://dx.doi.org/10.1002/elps.200500891
- Breidbach, A., Catlin, D.H., Green, G.A., Tregub, I., Truong, H. and Gorzek, J. (2003) Detection of Recombinant Human Erythropoietin in Urine by Isoelectric Focusing. Clinical Chemistry, 49, 901-907. http://dx.doi.org/10.1373/49.6.901
- Caldini, A., Moneti, G., Fanelli, A., Bruschettini, A., Mercurio, S., Pieraccini, G., Cini, E., Ognibene, A., Luceri, F. and Messeri, G. (2003) Epoetin Alpha, Epoetin Beta and Darbepoetin Alfa: Two-Dimensional Gel Electrophoresis Isoforms Characterization and Mass Spectrometry Analysis. Proteomics, 3, 937-941. http://dx.doi.org/10.1002/pmic.200300405
- Catlin, D.H., Breidbach, A., Elliott, S. and Glaspy, J. (2002) Comparison of the Isoelectric Focusing Patterns of Darbepoetin Alfa, Recombinant Human Erythropoietin, and Endogenous Erythropoietin from Human. Clinical Chemistry, 48, 2057-2059.
- Kohler, M., Ayotte, C., Desharnais, P., Flenker, U., Lüdke, S., Thevis, M., Völker-Schänzer, E. and Schänzer, W. (2008) Discrimination of Recombinant and Endogenous Urinary Erythropoietin by Calculating Relative Mobility Values from SDS Gels. International Journal of Sports Medicine, 29, 1-6. http://dx.doi.org/10.1055/s-2007-989369
- Lamon, S., Robinson, N., Mangin, P. and Saugy, M. (2007) Detection Window of Darbepoetin-α Following One Single Subcutaneous Injection. Clinica Chimica Acta, 379, 145-149. http://dx.doi.org/10.1016/j.cca.2007.01.014
- Reichel, C., Abzieher, F. and Geisendorfer, T. (2009) SARCOSYL-PAGE: A New Method for the Detection of MIRCERAand EPO-Doping in Blood. Drug Testing and Analysis, 1, 494-504. http://dx.doi.org/10.1002/dta.97
- AORC Guidelines for the Minimum Criteria for Identification by Chromatography and Mass Spectrometry (2003). http://www.aorc-online.org/documents/aorc-chromatographic-and-mass-spectral-criteria/
- Dehnes, Y., Lamon, S. and Lönnberg, M. (2010) Erythropoietin (EPO) Immunoaffinity Columns—A Powerful Tool for Purifying EPO and Its Recombinant Analogues. Journal of Pharmaceutical and Biomedical Analysis, 53, 1028-1032. http://dx.doi.org/10.1016/j.jpba.2010.06.017
- Lönnberg, M., Dehnes, Y., Drevin, M., Garle, M., Lamon, S., Leuenberger, N., Quach, T. and Carlsson, J. (2010) Rapid Affinity Purification of Erythropoietin from Biological Samples Using Disposable Monoliths. Journal of Chromatography A, 1217, 7031-7037. http://dx.doi.org/10.1016/j.chroma.2010.09.034
- Mi, J.B., Wang, S., Ding, X.J., Guo, Z.Q., Zhao, M.P. and Chang, W.B. (2006) Efficient Purification and Preconcentration of Erythropoietin in Human Urine by Reusable Immunoaffinity Column. Journal of Chromatography B, 843, 125-130. http://dx.doi.org/10.1016/j.jchromb.2006.05.037
- Spivak, J.L. and Hollenberg, M.D. (1979) Affinity Chromatography with Agarose-Lectin Derivatives: Separation of Human Glycoproteins and Application to Erythropoietin Purification. Progress in Clinical and Biological Research, 29, 641-653.
- Spivak, J.L., Small, D. and Hollenberg, M.D. (1977) Erythropoietin: Isolation by Affinity Chromatography with Lectin-Agarose Derivatives. Proceedings of the National Academy of Sciences of the United States of America, 74, 4633-4635. http://dx.doi.org/10.1073/pnas.74.10.4633
- Spivak, J.L., Small, D., Shaper, J.H. and Hollenberg, M.D. (1978) Use of Immobilized Lectins and Other Ligands for the Partial Purification of Erythropoietin. Blood, 52, 1178-1188.
- Kawasaki, N., Ohta, M., Hyuga, S., Hashimoto, O. and Hayakawa, T. (1999) Analysis of Carbohydrate Heterogeneity in a Glycoprotein Using Liquid Chromatography/Mass Spectrometry and Liquid Chromatography with Tandem Mass Spectrometry. Analytical Biochemistry, 269, 297-303. http://dx.doi.org/10.1006/abio.1999.4026
- Kawasaki, N., Ohta, M., Hyuga, S., Hyuga, M. and Hayakawa, T. (2000) Application of Liquid Chromatography/Mass Spectrometry and Liquid Chromatography with Tandem Mass Spectrometry to the Analysis of the Site-Specific Carbohydrate Heterogeneity in Erythropoietin. Analytical Biochemistry, 285, 82-91. http://dx.doi.org/10.1006/abio.2000.4739
- Lasne, F., Popot, M.A., Varlet-Marie, E., Martin, L., Martin, J.A., Bonnaire, Y., Audran, M. and de Ceaurriz, J. (2005) Detection of Recombinant Epoetin and Darbepoetin Alpha after Subcutaneous Administration in the Horse. Journal of Analytical Toxicology, 29, 835-837. http://dx.doi.org/10.1093/jat/29.8.835
- Schley, C., Swart, R. and Huber, C.G. (2006) Capillary Scale Monolithic Trap Column for Desalting and Preconcentration of Peptides and Proteins in Oneand Two-Dimensional Separations. Journal of Chromatography A, 1136, 210-220. http://dx.doi.org/10.1016/j.chroma.2006.09.072
- Gupta, S., Sage, A. and Singh, A. (2005) Screening and Confirmation of Recombinant Human Erythropoietin and Darbepoietin-α in Spiked Plasma Samples from Drug-Free Horses. Analytica Chimica Acta, 552, 96-109. http://dx.doi.org/10.1016/j.aca.2005.07.065
- Franco Fraguas, L., Carlsson, J. and Lönnberg, M. (2008) Lectin Affinity Chromatography as a Tool to Differentiate Endogenous and Recombinant Erythropoietins. Journal of Chromatography A, 1212, 82-88. http://dx.doi.org/10.1016/j.chroma.2008.10.036
NOTES

*Corresponding author.