Advances in Bioscience and Biotechnology
Vol.05 No.05(2014), Article ID:44865,13 pages
10.4236/abb.2014.55058
A Systems Approach for Determining Gene Expression from Experimental Observation of Compound Presence and Absence
Sangaalofa T. Clark*, Wynand S. Verwoerd
Centre for Advanced Computational Solutions (CfACS), Agriculture and Life Sciences Division, Lincoln University, Canterbury, New Zealand
Email: *sangaa@xtra.co.nz
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 1 February 2014; revised 21 March 2014; accepted 4 April 2014
ABSTRACT
Different genes are expressed in different tissues, depending on functional objectives and selection pressures. Based on complete knowledge of the structure of the metabolic network and all the reactions taking place in the cell, elementary modes (EMs) and minimal cut sets (MCSs) can relate compounds observed in a tissue, to the genes being expressed by respectively providing the full set of non-decomposable routes of reactions and compounds that lead to the synthesis of external products, and the full set of possible target genes for blocking the synthesis of external products. So, for a particular tissue, only the EMs containing the reactions that are related to the genes being expressed in those tissues, are active for the production of the corresponding compounds. This concept is used to develop an algorithm for determining a matrix of reactions (which can be related to corresponding genes) taking place in a tissue, using experimental observations of compounds in a tissue. The program is applied to the Arabidopsis flower and identified 20 core reactions occurring in all the viable EMs. They originate from the trans-cinnamate compound and lead to the formation of kaempferol and quercetin compounds and their derivatives, as well as anthocyanin compounds. Analyses of the patterns in the matrix identify reaction sets related to certain functions such as the formation of derivatives of the two anthocyanin compounds present, as well as the reactions leading from the network’s external substrate erythrose-4P to L-Phenyla- lanine, cinnamyl-alc to trans-cinnamate and so on. The program can be used to successfully determine genes taking place in a tissue, and the patterns in the resulting matrix can be analysed to determine gene sets and the state of the tissue.
Keywords:

*Corresponding author.
Metabolic Networks; Elementary Modes; Minimal Cut Sets; Gene Sets

1. Introduction
The metabolic pathway analysis concepts of elementary modes (EMs) and minimal cut sets (MCSs) represent the core concepts used in developing the method presented here. EMs are unique routes, or non-decomposable pathways, which consist of the minimal sets of reactions that allow the network to operate at steady state. Further information about EMs can be found in [1] [2] . Minimal cut sets are complementary to EMs, and consist of the minimal sets of reactions that, when blocked, will repress a certain metabolic functionality by preventing the network from achieving steady state. A review of MCSs can be found in [3] .
Although the metabolic network contains all gene-related reactions and metabolites for that organism, in real life only certain genes or enzymes are expressed in certain tissues. So, for a particular tissue, only the EMs containing the reactions that are related to the genes being expressed in those tissues, are active to produce the corresponding compounds. Consequently, different compound products are found in different tissues, depending on which genes are being expressed. In real life situations, different genes or enzymes are expressed in different tissues so certain compounds will be found in certain tissues but not others. Each compound product, however, would have the same EMs [4] and MCSs [5] . It is just a matter of determining which of these are activated for a certain cell in relation to the genes or enzymes that are expressed or suppressed.
Based on this idea, we can use experimental results of compounds that are found in a particular tissue, or are conversely absent, to identify the viable EMs [4] [6] [7] and MCSs [3] [5] [8] for certain tissues and, subsequently, genes that are being expressed in terms of the reactions that are taking place. This will be cheaper and easier to use than practical experimental methods such as microarrays which need advanced experimental technology to carry out and are hard to repeat.
The metabolic network of an organism is constructed from knowledge and information about the full genome of the organism whereby the metabolites and reactions are identified according to their corresponding genes and enzymes. Information on genes is available online at resources such as the Kyoto Encyclopaedia of Genes and Genomes (KEGG) and The Arabidopsis Information Resource (TAIR) [9] .
Knowledge about when and where a gene is expressed in a cell often provides an idea of its biological role. To get a better understanding of the possible functions of genes, DNA microarrays [10] have traditionally been used to measure the expression levels of large numbers of genes simultaneously or to study and determine the roles of genes [11] -[13] . The DNA microarrays can contain virtually all the genes in a microorganism, and thus have been used to study gene expression on a very large scale.
The pattern of genes expressed can provide information on the state of the cell, a fact that has shifted interest towards the study of gene sets from microarrays instead of individual genes, as it is a formidable task defining the role of each gene in the full genome of an organism; it is easier to biologically interpret a microarray experiment if differentially expressed genes show similarity in their functional description or occur together in a metabolic pathway to form gene sets.
A gene set is a group of functionally related genes, usually identified from a priori biological knowledge, for example, genes that are defined by functional categories or that occur in a known biological pathway.
New methods for studying gene sets have been developed, for example, [14] presents a method specifically designed for defining gene sets using pathways and taking into account the interrelationship of genes in terms of the topology of pathways. MCSs could similarly define gene sets but instead of genes, take into account the interrelationship of metabolites and reactions in the metabolic network, so would correspond to gene sets where each reaction was uniquely associated with a gene. A review of the analysis methods for gene sets can be seen in [15] [16] .
2. Methods and Approach
MatLab was used to develop the programmes (not included here) for determining gene expression as described in the methods below:
For a certain tissue at a certain metabolic state, there are sets of “present” and “absent” metabolite products. Each product contains ³ 1 EM and MCS which are complementary to each other.
The EMs are calculated in relation to compounds present in the tissue:
· EMs: E1, E2, ···, EM; different M for each product p.
Each EM forms a route consisting of a non-reducible set of reactions that lead to the formation of the compound so, in effect, form a candidate expression vector for the compound present in the tissue.
MCSs are calculated in relation to compounds absent from the tissue:
· MCSs: C1, C2, ···, CQ; Q = q(em); m = 1, 2, ···, M; different Q for each em.
Each MCS represents the minimum set of reactions that would be blocked in the tissue, based on the absent compound.
2.1. Binary Matrices for EMs and MCSs
Consider a binary matrix to represent these EMs and MCSs:
2.1.1. Binary Matrix of EMs
· A matrix indicating whether a reaction occurs or does not occur in EMs of products present in a tissue of interest:
○ a one (1) represents the presence, and
○ a zero (0) the absence, of a reaction in the corresponding EM responsible for the formation of p in a tissue:
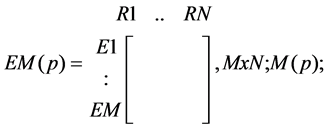 (1)
(1)
· where N is the number of reactions in the tissue- the same for all EMs;
· while M is the number (can differ for different products) of EMs for product p-; and
· 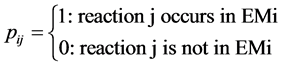 .
.
2.1.2. Binary Matrix of MCSs
· A matrix indicating whether a reaction does or does not constitute MCSs related to the absence of product p from the same tissue:
○ a one (1) in a MCS row indicates that the corresponding reaction needs to be simultaneously blocked with other ones in that MCS row, to suppress p, in order for p to be absent from the tissue;
○ a zero (0) indicates the reaction does not need to be blocked for that MCS to suppress the formation of product p.
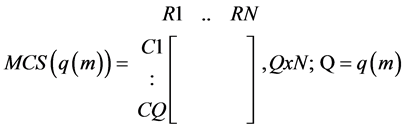 (2)
(2)
· Where N is the number of reactions in the tissue- the same for all EMs;
· Q is the number of MCSs for a particular EM, m, one of the EMs responsible for the formation (objective function) of p; and
· 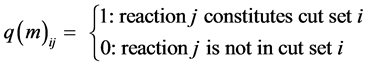 .
.
For a compound to be present in a particular tissue, at least one of the EMs responsible for producing that compound has to be active which means that the corresponding reactions/genes for that/those EM(s) would be expressed in that tissue. If products A and B are present in a tissue, it means that at least one mode of product A and one of B must be active.
For a compound to be absent, all MCSs need to be activated to block all EMs, such that the corresponding genes would not be expressed.
Based on the above idea, we could use experimental results of compounds that are found in a particular tissue and vice versa, to identify the viable EMs and MCSs and, subsequently, the genes expressed in that tissue in terms of their corresponding reactions.
The idea is to use both the EMs of compound products present in the tissue and the MCSs of compounds absent, to determine the reactions/genes for the Expression matrix X.
2.2. Compiling the Expression Matrix
The EMs of all the compounds present in the tissue of interest can be combined to form a matrix of combined EMs (cEMs) which is then reduced back to binary. Each column of this matrix would constitute EMs guaranteed to produce all the products present in the tissue.
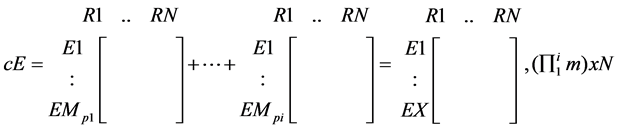 (3)
(3)
· where p is a metabolite product present in the tissue; and
· X is the product of the number of EMs for all the present products (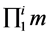 ).
).
For example, let’s say there are:
· three metabolites {A, B, C} present in a tissue; and
· two absent metabolites {D, E}.
Consider the three metabolite products A, B & C present in the tissue; product A has 3 EMs, product B has 2 EMs and C has 2 EMs. A cEM that would produce all three metabolites could be obtained by adding together an EM from each product set (note that these may overlap) as illustrated in Figure 1.
The resulting number of cEMs would be 3 × 2 × 2 = 12, so there would be 12 cEMs in the binary matrix where each row vector represents a cEM guaranteed to produce the three compounds A, B & C.
However, it is important to note that some of these cEMs would not actually be viable because their calculation does not take into account the “absent” compounds so some might actually be responsible for forming compounds that are absent in the tissue! This is also true for EMs responsible for a single compound.
In this respect, MCSs can be employed to identify viable cEMs and EMs because the reactions constituting a cEM or EM cannot be present in the tissue if they also constitute a MCS.
2.3. Use of MCSs to Verify Elementary Modes
For each “absent” compound, a MCS could be determined which contains the minimal sets of reactions to suppress in order to prevent the production of that compound. The cEMs can then be verified by multiplying its transpose by the MCSs of each absent compound:
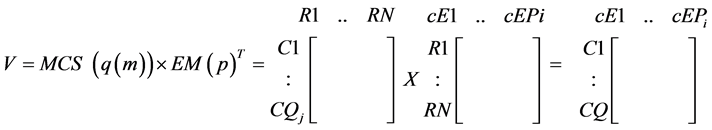 (4)
(4)
The elements of the product matrix (V) indicate the compatibility of the EMs with the MCSs:
· A “0” indicates that the MCS is compatible with the EM, i.e., the reactions taking place to produce the present compound are not the reactions being blocked to eliminate the absent compound. This means that the cEM is a valid Expression vector;
· A “1” or “non-zero” (if V is not reduced to a binary matrix) in the column indicates that the cEM is not compatible with the MCS because the reactions producing the present compounds also need to be blocked to eliminate the absent compound.
Whether the above process is practical will depend on how many EMs there are associated with each product, with the total number of combinations: multiplying all the counts could become astronomical.
2.3.1. An Example Network
To clarify and check the above processes more, we will apply them to an example network (Figure 2), that has 6 metabolites and 8 reactions:
R1: A = B; R2: B = D; R3: A = C; R4: D = C; R5: D; R6: D = E; R7: A + D = F; R8: B + D = F

Figure 1. Combining EMs of three compound products.

Figure 2. Example network, ExNet, used in developing the algorithm.
ExNet has 3 external products: C, E & F.
EMs can be calculated for the three products that guarantee the production of each compound
Number of EMs calculated = 14:
· 4 EMs for C: {R5, R4}; {R3}; {R8, R2, R4}; {R1, R2, R4};
· 3 EMs for E: {R5, R6}; {R8, R2, R6}; {R1, R2, R6};
· 8 EMs for F: {R5, R7}; {R1, R5, R8}; {R2, R4, R8}; {R1, R2, R8}; {R2, R6, R8}; {R8, R2, R7}; {R1, R2, R7}; {R2, R5, R8}.
Now, say that of the 3 compound products, C & E are found in a tissue but not compound F. This indicates that the reactions needed to produce compounds C & E are taking place in the tissue so the related genes are being expressed, whilst the reactions for compound F are not taking place and the related genes are being suppressed.
2.3.2. Transpose of cEMs Matrix for Compounds C & E
The matrix of cEMs for compounds C & E is shown in Table 1. The matrix contains 12 possible combinations of EMs (cEMs) that would produce compounds C & E together:
· For each column vector of the transposed matrix (cEMT):
o the reactions with a one (1) form the set of reactions in that cEM that would lead to the formation of both compounds C & E;
o A zero (0) indicates the reactions are not involved in that cEM.
· Each column in the cEMsT is a candidate expression vector X representing an EM (route) containing candidate reactions that would guarantee the formation of both compounds C & E.
2.3.3. MCS matrix of the Absent Compound F
The absence of compound F indicates that at least one of its MCSs is activated, i.e., the reactions constituting a MCS are simultaneously blocked. As illustrated in Table 2, each MCS row in the MCS matrix contains the minimal set of reactions that would need to be simultaneously blocked to eliminate the formation of compound F.

Table 1. Matrix of cEMs for products C & E.
Table 2. MCSs for compound F where a “1” indicates the reaction is blocked.
2.3.4. Verifying Combined Elementary Modes
Multiplying Table 2 (the MCSs in for eliminating compound F) with Table 1 (the transposed matrix of combined EMs (cEMsT) responsible for producing C & E), produces a product matrix V that can be used to verify the combined modes (cEM) in terms of the compound F being absent in the tissue. The product matrix
 is shown in Table 3.
is shown in Table 3.
The resulting values in the above product matrix V indicate the following:
· A zero (0) indicates that the MCS is compatible with the cEM, e.g., for our example network it means that the reactions needed to be blocked to eliminate F are not needed for the formation of compound products C & E, and vice versa;
· A non-zero indicates that the cEM clashes with the MCS so the cEM is not viable.
In this respect, the product matrix V is also the verification matrix and can be interpreted to indicate which cEMs are valid in terms of forming compounds C & E without forming compound F.
A column vector (cEM) containing at least one 0 element in the product matrix V in Table 3, represents a viable cEM because there is a MCS for blocking compound F, that does not clash with this particular cEM for forming compounds C & E.
So, for our example network ExNet, the viable expression vectors or cEMs are those containing zeroes in Table 3:
· cEM1, cEM3, cEM4, cEM6, cEM10 & cEM12 in relation to MCS2 for which R7 & R8 are blocked;
· cEMs 1 & 4 in relation to MCS4 for which R1, 2, 7 are blocked.
The non-viable cEMs are cEM2, cEM5, cEM6, cEM8, cEM9, cEM11 so these can be deleted.
Table 3. Product matrix V indicating cEM candidates in relation to MCSs.
The resulting expression matrix X would then be as shown in Table 4.
Using the above approach, an algorithm (not included here) for determining a matrix of reactions (which can be related to corresponding genes) taking place in a tissue was developed in MatLab. The algorithm uses experimental observations of compounds in a tissue and includes verifying EMs for single compounds and eliminating duplicates and inactive reactions to address the problem posed by combinatorial explosion when combining EMs (illustrated in Figure 1).
3. Application of Algorithm to a “REAL” Example
The algorithm was applied to floral pigmentation in Arabidopsis using the flavonoid subnetwork reconstructed for earlier studies and information from experimental work on identifying flavonoids produced by Arabidopsis thaliana wild-type and flavonoid biosynthetic mutant lines [17] .
3.1. Method of Application
Information from [17] was used in conjunction with the flavonoid subnetwork, FlavNet, used in [18] , to compile a product binary vector for an Arabidopsis flower, specifying which flavonoids are present and which are not.
It is important to note that the compounds found present in the Arabidopsis flower from the results of [17] , are, in effect, cast in stone and definite. However, compounds that are absent are not quite definite because it might be that they were just not tested for and/or reported on, and by applying the algorithm, we are assuming that the compounds are definitely absent. This case would also apply to any tissue and any other experimental results that are used, unless the experiment specifically sets out to check that compounds are actually absent from the tissue.
The CellNetAnalyzer (CNA) program [19] was used to calculate the corresponding EMs needed to produce the compounds present in the flower product vector and the MCSs for compounds absent from the product vector.
The CNA EMs and MCSs outputs were reformatted and, along with the product vector, form the inputs entered into the algorithm to determine the Expression vector X containing a set of valid EMs for producing all the compounds present in the tissue.
3.2. Results and Discussions
The product vector compiled for an Arabidopsis flower using FlavNet [18] and information from [17] is shown in Table 5 where a one (1) represents the presence, and a zero (0) the absence, of the compound in the flower:
Table 4. The resulting expression matrix X.
Table 5. Product vector for the Arabidopsis flower.
3.2.1. The Expression Matrix X
Applying the first 6 steps of the algorithm produces the matrix X containing eligible EMs (columns) with their corresponding reactions (rows) as shown in Figure 3.
As predicted earlier, applying the algorithm to the flavonoid subnetwork produces an Expression matrix X containing a large number of eligible EMs such that it is not easy to define the role of each gene and is better to analyse X in terms of patterns in the matrix that could be used to determine different gene sets.
There are 378 eligible EMs and 165 reactions in the Expression matrix X. The reactions are not included in the above tables but are kept track of during the manipulation and analysis of matrix X.
3.2.2. Analysing the Expression Matrix X
To prepare for analysis, matrix X was cleaned up of reactions that were not needed. The eligible EMs, collectively, don’t use a lot of the reactions (yellow cells), as shown in Figure 4, where matrix X is sorted in increasing order of the total number of EMs a reaction is involved in.
The green part corresponds to reactions not participating in any EMs so they can be taken out of the matrix, leaving a matrix containing only those reactions that constitute an eligible EM, as shown in Figure 5.
The matrix in Figure 5 shows that 48 reactions occur in at least one EM of which 20 are ubiquitous reactions occurring in all of the eligible EMs (plain yellow part) for the flower tissue. This means that, based on the information used, these 20 reactions are vital for metabolism in the Arabidopsis flower as they are required for all the viable EMs. These core reactions are shown in Table 6.
The above 20 reactions form the core reactions occurring in all viable EMs of the Arabidopsis flower tissue.

Figure 3. Matrix X containing eligible EMs (columns) and their reactions (rows).

Figure 4. Sorting by total reactions (rows) in EMs.

Figure 5. Without reactions involved in no EM.
Table 6. The 20 ubiquitous reactions.
Looking at their position in the network, the reactions originate from the external trans-cinnamate substrate (refer to Figure 1 and Figure 3 of [18] ) and lead to the formation of kaempferol and quercetin compounds and their derivatives as well as anthocyanin compounds although not the anthocyanin derivatives.
If the yellow part containing the 20 reactions in the above Table 6 is taken out, the remaining matrix is as shown in Figure 6 with the reactions on the vertical axis and the viable EMs on the horizontal axis.
A yellow area in Figure 6 indicates a reaction is involved in the corresponding viable EM while a green one means the reaction is not involved in the corresponding EM. The 28 reactions illustrated in the matrix are shown in Table 7.
These 28 reactions occur in some EMs but not others. The different patterns this creates in the matrix could be analysed for any relationship it could have to gene sets or information about the corresponding reactions and their related genes as well as the EMs and whether different patterns in the EMs could be related to certain functions or the state of the cell.
Grouping the reactions in Table 7 according to the different patterns discernible by eye in Figure 6, is provided in Table 8.

Figure 6. The cleaned Expression matrix X: Reactions (y-axis) by viable EMs (x-axis).
3.2.3. Analysis of Grouped Reactions
The resulting groups in Table 8 show that reactions sharing the same pattern actually share a related function, so their corresponding genes would form gene sets related to a certain function e.g., those corresponding to Group 1 would form the set of genes related to the formation of derivatives of the anthocyanin cyanidin-3-0-glucoside.
Some reactions do not quite fit into their group (in relation to Figure 6), either because their patterns are a bit different or they are not involved in a relatively similar number of EMs, e.g., Reaction 27 from Group 1 and Reaction 28 from Group 7 which would fit in with Group 4. These reactions and their corresponding genes could be studied further to see if they are actually different in some way.
3.2.4. Analysis of Grouped EMs
Looking at vertical patterns in Figure 6 related to EMs, there are four obvious patterns which could be used to group the EMs:
Group 1: [EM1 - EM54], Group 2: [EM55 - EM162], Group 3: [EM163 - EM270] and Group 4: [EM271 - EM378].
The most obvious difference in these groups is in whether the reactions leading from Erythrose-4P to L- Phenylalanine are used (Grp 2 & 4) or not (Grp 1 & 3) but the four groups of EMs could be analysed in detail, e.g., to see which reactions are involved and what products they form, to determine if there is a relationship between them, such as what kind of metabolite products are being synthesised and through which EMs? This could provide an idea of the state of the Arabidopsis flower and the biological processes in the tissue. This would involve extensive analysis of the 378 EMs which could form a basis for future work.
3.2.5. Next Steps
To obtain further and improved information from the Expression matrix the next steps to pursue include:
· Making use of tools, such as clustering methods, that could further analyse the Expression matrix in detail and elucidate gene sets and related biological processes in the tissue;
· Improving the completeness of experimental information being used. The information used in this study was from experimental work that did not specifically look at compounds that were present and absent in the Arabidopsis flower, so the fact that a compound was absent could have been just because the study was concentrating on other compounds. Having more complete information would improve the accuracy of the results.
4. Conclusions
The approach would work best in collaboration with experimental scientists to specifically look at compounds present and absent in a tissue of interest. However, the fact that useful information can already be obtained from a limited set of data, provides credence for the use and further development of this systems approach method for determining and studying gene expression.
The study shows that the developed algorithm and programmes, work to determine an Expression matrix containing reactions expressed in the tissue, from experimental information about compounds that are present.
Table 7. The reactions corresponding to the matrix in Figure 6.
Table 8. Reaction groups and related functions.
The 20 core reactions that occur in all the viable EMs of the Arabidopsis flower tissue originate from the trans- cinnamate compound and lead to the formation of kaempferol and quercetin compounds and their derivatives as well as anthocyanin compounds although not the anthocyanin derivatives.
Analyses of the matrix patterns show that these correspond to reaction sets related to certain functions such as the formation of derivatives of the two anthocyanin compounds present, as well as the reactions leading from the network’s external substrate erythrose-4P to L-Phenylalanine, cinnamyl-alc to trans-cinnamate and so on. The results provide credibility to the program being able to determine reaction sets, from which their corresponding gene sets can be established.
Acknowledgements
The authors wish to thank Lincoln University for supporting the research.
References
- Klamt, S. and Stelling, J. (2003) Two Approaches for Metabolic Pathway Analysis? Trends in Biotechnology, 21, 64- 69. http://dx.doi.org/10.1016/S0167-7799(02)00034-3
- Papin, J.A. (2004) Comparison of Network-Based Pathway Analysis Methods. Trends in Biotechnology, 22, 400-405. http://dx.doi.org/10.1016/j.tibtech.2004.06.010
- Clark, S.T. and Verwoerd, W.S. (2012) Minimal Cut Sets and the Use of Failure Modes in Metabolic Networks. Me- tabolites, 2, 567-595.
- Gagneur, J. and Klamt, S. (2004) Computation of Elementary Modes: A Unifying Framework and the New Binary Approach. BMC Bioinformatics, 5, 175.
- Klamt, S. (2006) Generalized Concept of Minimal Cut Sets in Biochemical Networks. Biosystems, 83, 233-247. http://dx.doi.org/10.1016/j.biosystems.2005.04.009
- Schuster, S., Dandekar, T. and Fell, D.A. (1999) Detection of Elementary Flux Modes in Biochemical Networks: A Promising Tool for Pathway Analysis and Metabolic Engineering. Trends in Biotechnology, 17, 53-60. http://dx.doi.org/10.1016/S0167-7799(98)01290-6
- Trinh, C.T., Wlaschin, A. and Srienc, F. (2009) Elementary Mode Analysis: A Useful Metabolic Pathway Analysis Tool for Characterizing Cellular Metabolism. Applied Microbiology and Biotechnology, 81, 813-826. http://dx.doi.org/10.1007/s00253-008-1770-1
- Klamt, S. and Gilles, E. (2004) Minimal Cut Sets in Biochemical Reaction Networks. Bioinformatics, 20, 226-234. http://dx.doi.org/10.1093/bioinformatics/btg395
- (2011) The Arabidopsis Information Resource (TAIR).
- Roberts, P.C. (2008) Gene Expression Microarray Data Analysis Demystified. Biotechnology annual Review, 14, 29- 61. http://dx.doi.org/10.1016/S1387-2656(08)00002-1
- Casati, P. and Walbot, V. (2003) Gene Expression Profiling in Response to Ultraviolet Radiation in Maize Genotypes with Varying Flavonoid Content. Plant Physiology, 132, 1739-1754. http://dx.doi.org/10.1104/pp.103.022871
- DeRisi, J.L., Iyer, V.R. and Brown, P.O. (1997) Exploring the Metabolic and Genetic Control of Gene Expression on a Genomic Scale. Science, 278, 680-686. http://dx.doi.org/10.1126/science.278.5338.680
- Díaz, H., Andrews, B.A., Hayes, A., Castrillo, J., Oliver, S.G. and Asenjo, J.A. (2009) Global Gene Expression in Recombinant and Non-Recombinant Yeast Saccharomyces cerevisiae in Three Different Metabolic States. Biotechnology Advances, 27, 1092-1117. http://dx.doi.org/10.1016/j.biotechadv.2009.05.015
- Massa, M.S., Chiogna, M. and Romualdi, C. (2010) Gene Set Analysis Exploiting the Topology of a Pathway. BMC Systems Biology, 4, 121.
- Ackermann, M. and Strimmer, K. (2009) A General Modular Framework for Gene Set Enrichment Analysis. BMC Bioinformatics, 10, 47. http://dx.doi.org/10.1186/1471-2105-10-47
- Nam, D. and Kim, S.-Y. (2008) Gene-Set Approach for Expression Pattern Analysis. Briefings in Bioinformatics, 9, 189-197. http://dx.doi.org/10.1093/bib/bbn001
- Yonekura-Sakakibara, K., Tanaka, Y., Fukuchi-Mizutani, M., Fujiwara, H., Fukui, Y., Ashikari, T., Murakami, Y., Yamaguchi, M. and Kusumi, T. (2000) Molecular and Biochemical Characterization of a Novel Hydroxycinnamoyl-CoA: Anthocyanin 3-O-Glucoside-6”-O-acyltransferase from Perilla frutescens. Plant and Cell Physiology, 41, 495-502. http://dx.doi.org/10.1093/pcp/41.4.495
- Clark, S. and Verwoerd, W. (2011) A Systems Approach to Identifying Correlated Gene Targets for the Loss of Colour Pigmentation in Plants. BMC Bioinformatics, 12, 343. http://dx.doi.org/10.1186/1471-2105-12-343
- Klamt, S., Sae-Rodriguez, J. and Gilles, E.D. (2007) Structural and Functional Analysis of Cellular Networks with CellNetAnalyzer. BMC Systems Biology, 1, 2.