Advances in Bioscience and Biotechnology
Vol.5 No.3(2014), Article ID:42707,5 pages DOI:10.4236/abb.2014.53029
Identification of a constitutional mutation in the WT1 gene in Taiwanese patients with Wilms tumor
![]()
1Department of Pediatrics, National Taiwan University Hospital, Taipei, Taiwan
2Department of Obstetrics and Gynecology, Jen-Ai Hospital, Taichung, Taiwan
3School of Medical Laboratory and Biotechnology, Chung Shan Medical University, Taichung, Taiwan
Email: lmy1079@gmail.com, leopard.wang@gmail.com, a1993129@yahoo.com.tw, *yenchein@csmu.edu.tw
Copyright © 2014 Meng-Yao Lu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Meng-Yao Lu et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 11 December 2013; revised 15 January 2014; accepted 28 January 2014
KEYWORDS: Wilms Tumor; WT1; Tumor Suppressor Gene; Nephroblastoma; Denys-Drash Syndrome
ABSTRACT
The overall frequency of WT1 gene alterations in Wilms tumor is still unclear in Taiwan. Here we conducted molecular genetic analysis of the WT1 gene in Taiwanese patients with Wilms tumor. Polymerase chain reaction and direct sequencing were performed on DNA samples from blood and paraffin-embedded tumor specimens. A constitutional mutation in the WT1 gene was found in one DNA sample from peripheral blood lymphocytes. The remaining DNA samples from peripheral blood lymphocytes and paraffin-embedded tumor specimens were tested negative for both constitutional mutations and somatic mutations. Thus, mutations at other Wilms tumor loci may play an important role in Wilms tumor development. Wilms Tumor; WT1; Tumor Suppressor Gene; Nephroblastoma; Denys-Drash Syndrome
1. INTRODUCTION
Wilms tumor, or nephroblastoma, is the most common pediatric renal malignancy [1,2]. Several genetic loci have been shown to be associated with tumor formation, including WT1 on chromosome 11p13, WT2 on chromosome 11p15, FWT1 on chromosome 17q12-q21, and FWT2 on 19q [3,4]. WT1 is the first and most important gene to be isolated [5]. The WT1 gene contains ten exons of approximately 50 kb [6,7] and encodes a zinc finger protein that binds DNA and regulates transcription of target genes [8]. Many mutations in the WT1 gene have been described (The Human Gene Mutations Database, HGMD) and WT1 gene mutations have been observed in approximately 20% of patients with Wilms tumor [9,10]. Genotype-phenotype associations at WT1 have also been demonstrated [11,12]. To date, molecular genetic studies have mainly been conducted in Western countries. The risk of Wilms tumor conferred by mutations in these genes is poorly characterized in Taiwan. In this study, we performed genetic analysis to clarify the role of the WT1 gene in the renal tumorigenesis of Wilms tumor in Tai-wan.
2. MATERIALS AND METHODS
2.1. Study Subjects
Four blood (W1 to W4) and eight paraffin-embedded tumor tissue (W5 to W12) samples from Wilms tumor patients were provided by the Department of Pediatrics of National Taiwan University Hospital. W3 and W12 were isolated from the same individual. DNA sample collection for the study protocol was approved by the Institutional Review Board of Chung Shan Medical University Hospital and informed consent was obtained from each subject.
2.2. Isolation of DNA from Blood
Genomic DNA was isolated from peripheral blood lymphocytes using the QIAamp DNA blood kit (QIAGEN GmbH., Hilden, Germany). DNA was also prepared from sections of paraffin embedded tissue using the QIAamp tissue kit (QIAGEN GmbH., Hilden, Germany). The DNA was finally dissolved in 100 μl of TE buffer (10 mM Tris-HCl, pH 8.0, and 1 mM EDTA). Concentrations of the DNA stocks were estimated by spectrophotometer. Each genomic DNA sample was adjusted to 100 ng/μl to serve as a template for subsequent analyses.
2.3. Polymerase Chain Reaction
The ten exons of the WT1 gene were amplified in 10 fragments using the published primers [10]. A slight modification was made in PCR primers and experimental conditions as shown in Table 1. The PCR fragments were amplified in a Perkin-Elmer 2400 DNA thermal cycler in a final volume of 30 μl that contained one-fold Qiagen PCR buffer [Tris-HCl, KCl, (NH4)2SO4, 15 mM MgCl2; pH 8.7 at 20˚C], one-fold Q-solution, 0.015 units/μl Taq DNA polymerase supplied from Taq DNA polymerase kit (Qiagen), 500 nM for each primer, 200 μM dGTP, dATP, dCTP and dTTP (Promega) and 3 ng/μl template. The PCR conditions were initial denaturation at 95˚C for 5 min, followed by 35 cycles of 1 min at 95˚C, 1 min at annealing temperature, and 2 min at 72˚C, with final extension at 72˚C for 10 min.
2.4. Direct Sequencing
PCR products were purified using QIAquick PCR Purification kits (Qiagen GmbH., Hilden, Germany). The purified PCR products were sequenced using the cyclesequencing method with fluorescently labelled dideoxy chain terminators from ABI Prism kit (Applied Biosystems, Taipei, Taiwan) in an ABI Model 377 automated DNA sequencer, according to the distributor’s protocol.
The sequencing primers were the same as those for the preceding PCRs. When a mutation was detected, the nucleotide sequence was confirmed on both strands.
3. RESULTS
We found a constitutional mutation in the WT1 gene in one DNA sample from peripheral blood lymphocytes. Wilms tumor W2 showed a deletion of one base pair at codon 115 CCT in exon 1 (Figure 1(A)), designated 359delC according to the Reference Sequence (RefSeq) of GenBank (accession number M80217). This mutation causes a reading frame shift after codon 115 and premature chain termination at codon 217.
W2 also showed a homozygous point mutation at codon 323 in exon 7 (Figure 2(B)), 71A > G (GenBank M80229). This is a silent mutation, without a change in the amino acid Arg. The remaining samples also showed this silent mutation, 71A > G in exon 7 (Figure 2(A)), however in heterozygous form.
Except for W2, the DNA samples from peripheral blood lymphocytes and paraffin-embedded tumor specimens tested negative for both constitutional mutations and somatic mutations in the exon regions. There were nucleotide alterations in the intron regions in almost all samples (Table 2). However, splicing status was not af-
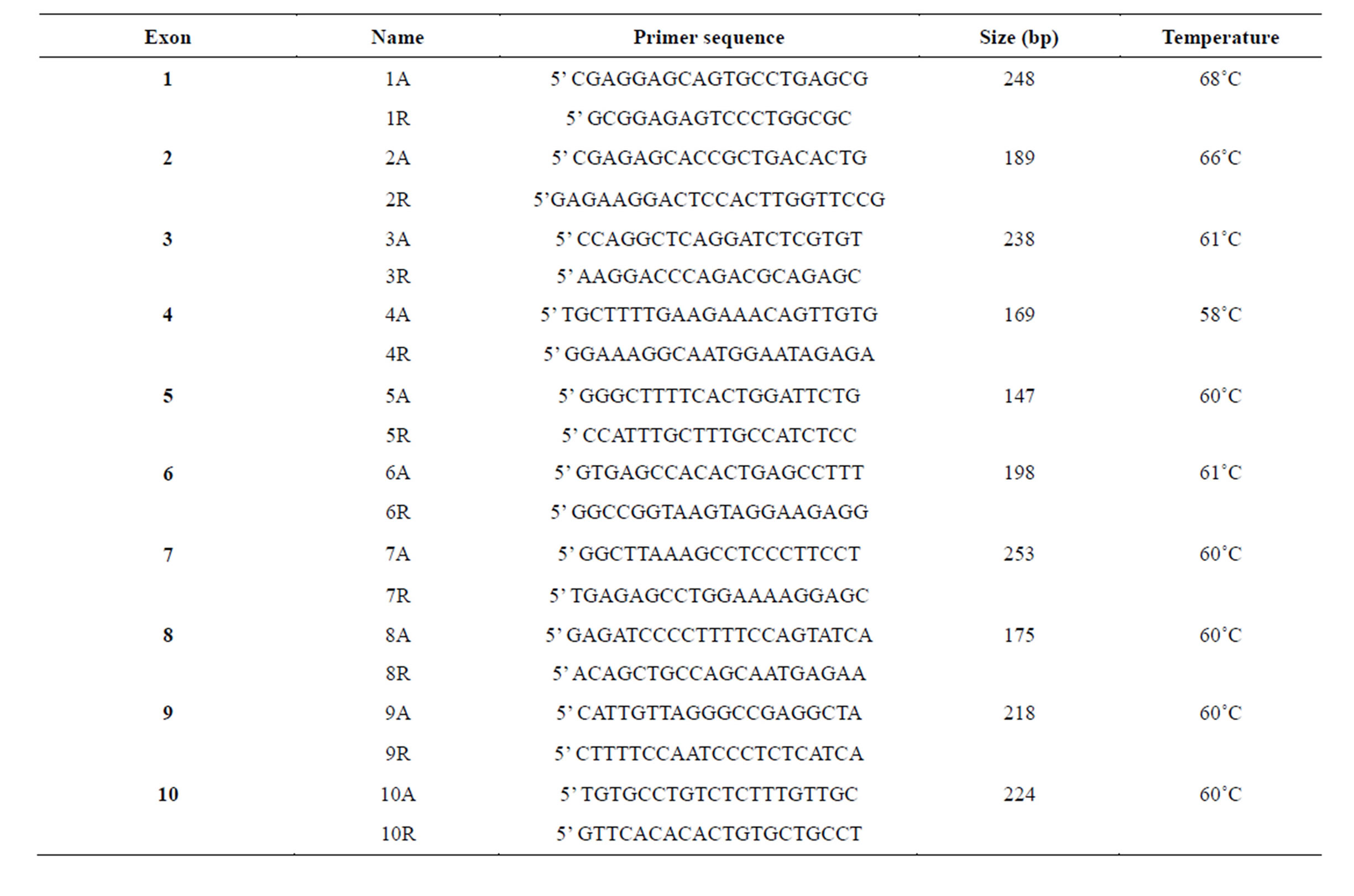
Table 1. Primers for the amplification of WT1 exons.
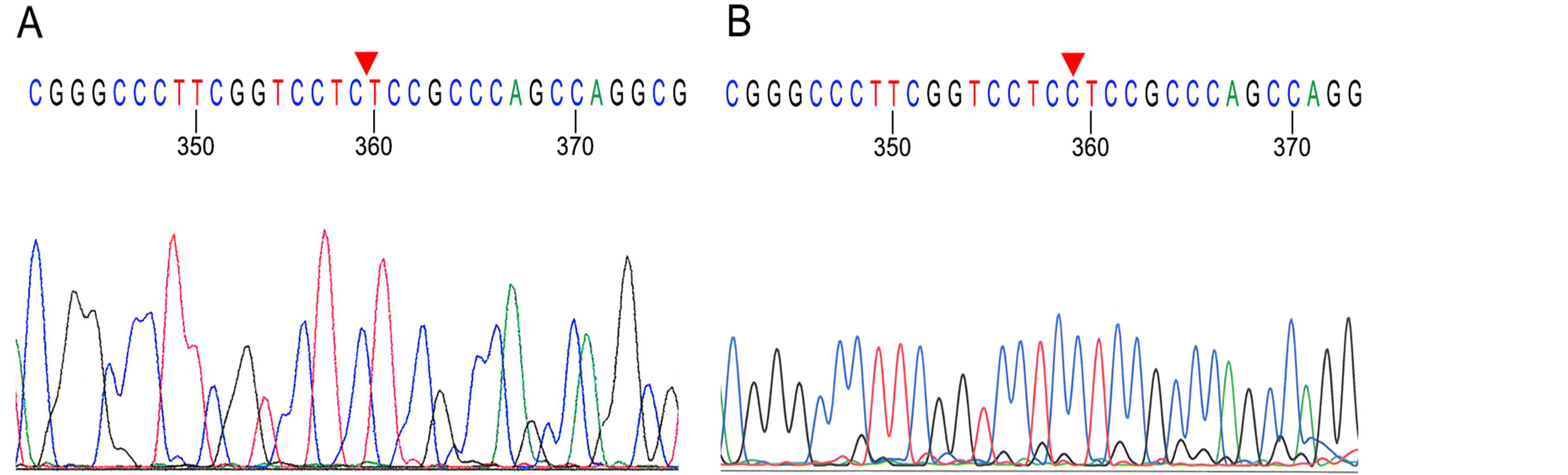
Figure 1. Partial sequencing chromatograms of Wilms tumor W2 show a base pair deletion 359delC in exon 1 (A). W7 does not show this deletion (B). Red triangles indicate the positions of mutations.
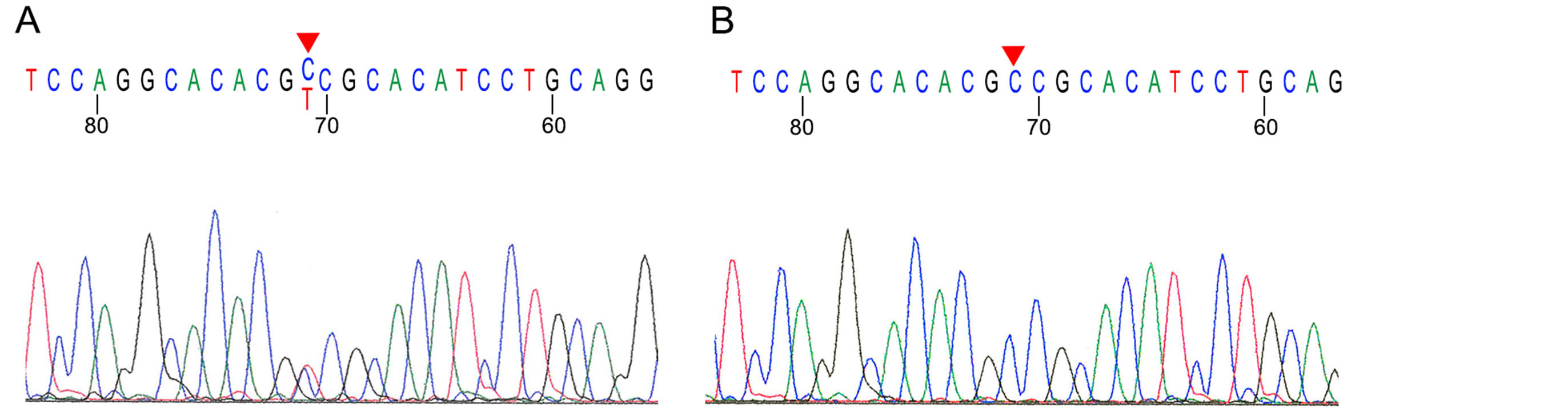
Figure 2. Partial sequencing chromatograms of Wilms tumor W3 show a heterozygous 71A > G point mutation in exon 7 (A). W2 shows a homozygous 71A > G point mutation (B) in reverse direction. Red triangles indicate the positions of mutations.
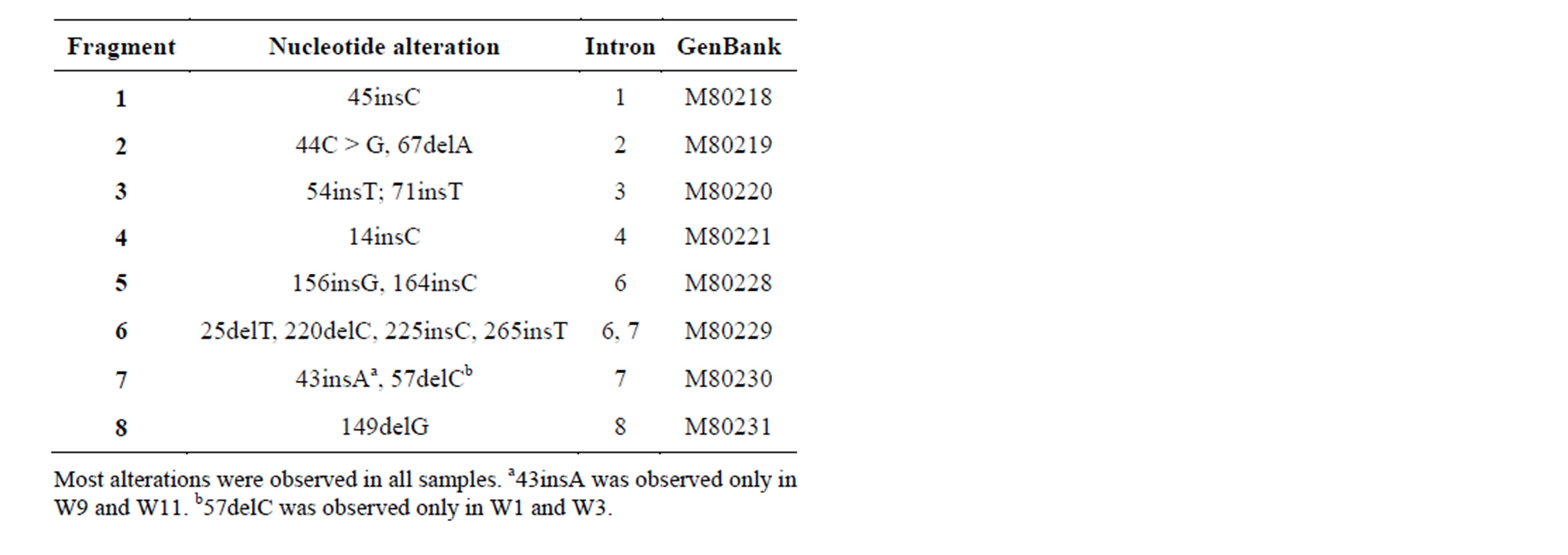
Table 2. Nucleotide alterations in the intron regions of WT1 gene.
fected. These alterations may be attributed to ethnic effects as they were also observed in the DNA from a healthy individual (data not shown).
4. DISCUSSION
According to statistics compiled by the Childhood Cancer Foundation, about 10 new cases of Wilms tumor are reported in Taiwan every year. This accounts for 3.4% of total childhood malignancies (www.ccfroc.org.tw). Breslow et al. have shown that 1% - 3% of cases of Wilms tumor are familial and that a predisposition to Wilms tumor is most likely caused by rare germline mutations acting in a dominant fashion [13]. Constitutional mutations in the WT1 gene on chromosome 11p13 predispose an individual to Wilms tumor and are associated with genitourinary abnormalities [4]. Several genetic syndromes (e.g., Denys-Drash syndrome) have been correlated with intragenic WT1 mutations [12]. In contrast to other WT1 mutations, these mutations are dominant and mainly found in the zinc finger region encoded by exon 7 through exon 10 of WT1 [5].
None of the patients in this study were diagnosed with Denys-Drash syndrome or Beckwith-Wiedemann syndrome. In addition, none presented with familial Wilms tumor. Evidence of WT1 inactivating mutations was apparent in only one of the 11 cases on mutational analysis, suggesting that direct alterations of the WT1 gene are present in only a very small fraction of Wilms tumor patients in Taiwan. This result is consistent with the findings of a previous study [14]. Less than 10 percent of sporadic Wilms tumors are associated with WT1 mutations. Thus, mutations at other Wilms tumor loci may play an important role in Wilms tumor development.
The WT1 gene is located at chromosome 11p13 [6,7]. It was discovered as a part of the WAGR syndrome (Wilms tumor, Aniridia, Genitourinary anomaly, and mental Retardation). Those who present with this syndrome have germline deletion on chromosome 11p13 [5]. This gene has 10 exons and encodes a protein called WT1 which has tumor suppression function [15]. High expression of this protein on developing kidney results in suppression of growth-related gene [16]. Tumor suppression gene is usually involved in a two-hit mechanism [17]. In our study, there was only one case with one mutant allele from blood sample. This may be explained by WT1 product acting as dimeric or multimeric protein [18]. When one allele produces an abnormal product, it presents as negative dominant event with loss of function. When there is mutation on only one allele, there are normal polypeptides with preserved function. The phenotype of Denys-Drash syndrome involves mutations in the zinc finger domain [19], so does that of Frasier syndrome [20], neither of which was observed in this study.
In conclusion, our data showed a low frequency of constitutional and somatic WT1 mutations in children with nonsyndromatic Wilms tumor. This is the first study to detect WT1 mutation in children with Wilms tumor in Taiwan.
ACKNOWLEDGEMENTS
Direct sequencing in an ABI Model 377 automated DNA sequencer was performed in the Instrument Center of Chung Shan Medical University, which is supported by the National Science Council, Ministry of Education and Chung Shan Medical University. The authors would like to thank the Gene Codes Corporation for providing evaluation license of Sequencher® version 5.2 sequence analysis software (http://www.genecodes.com) to analyze the DNA sequencing data.
REFERENCES
- Brown, K.W. and Malik, K.T. (2001) The molecular biology of Wilms tumour. Expert Reviews in Molecular Medicine, 2001, 1-16. http://dx.doi.org/10.1017/S1462399401003027
- Pastore, G., Znaor, A., Spreafico, F., Graf, N., PritchardJones, K. and Steliarova-Foucher, E. (2006) Malignant renal tumours incidence and survival in European children (1978-1997): Report from the Automated Childhood Cancer Information System project. European Journal of Cancer, 42, 2103-2114. http://dx.doi.org/10.1016/j.ejca.2006.05.010
- Coppes, M.J. and Egeler, R.M. (1999) Genetics of Wilms’ tumor. Seminars in Urologic Oncology, 17, 2-10.
- Rahman, N., Arbour, L., Houlston, R., Bonaiti-Pellie, C., Abidi, F., Tranchemontagne, J., Ford, D., Narod, S., Pritchard-Jones, K., Foulkes, W.D., Schwartz, C. and Stratton, M.R. (2000) Penetrance of mutations in the familial Wilms tumor gene FWT1. Journal of the National Cancer Institute, 92, 650-652. http://dx.doi.org/10.1093/jnci/92.8.650
- Menke, A., van der Eb, A.J. and Jochemsen, A.G. (1998) The Wilms’ tumor suppressor WT1: Approaches to gene function. Kidney International, 53, 1512-1518. http://dx.doi.org/10.1046/j.1523-1755.1998.00935.x
- Call, K.M., Glaser, T., Ito, C.Y., Buckler, A.J., Pelletier, J., Haber, D.A., Rose, E.A., Kral, A., Yeger, H., Lewis, W.H., Jones, C. and Housman, D.E. (1990) Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell, 60, 509-520. http://dx.doi.org/10.1016/0092-8674(90)90601-A
- Gessler, M., Konig, A. and Bruns, G.A. (1992) The genomic organization and expression of the WT1 gene. Genomics, 12, 807-813. http://dx.doi.org/10.1016/0888-7543(92)90313-H
- Madden, S.L., Cook, D.M., Morris, J.F., Gashler, A., Sukhatme, V.P. and Rauscher 3rd, F.J. (1991) Transcriptional repression mediated by the WT1 Wilms tumor gene product. Science, 253, 1550-1553. http://dx.doi.org/10.1126/science.1654597
- Huff, V. (1998) Wilms tumor genetics. American Journal of Medical Genetics, 79, 260-267. http://dx.doi.org/10.1002/(SICI)1096-8628(19981002)79:4<260::AID-AJMG6>3.0.CO;2-Q
- Gessler, M., Konig, A., Arden, K., Grundy, P., Orkin, S., Sallan, S., Peters, C., Ruyle, S., Mandell, J., Li, F., Cavenee, W. and Bruns, G. (1994) Infrequent mutation of the WT1 gene in 77 Wilms’ Tumors. Human Mutation, 3, 212-222. http://dx.doi.org/10.1002/humu.1380030307
- Segers, H., Kersseboom, R., Alders, M., Pieters, R., Wagner, A. and van den Heuvel-Eibrink, M.M. (2012) Frequency of WT1 and 11p15 constitutional aberrations and phenotypic correlation in childhood Wilms tumour patients. European Journal of Cancer, 48, 3249-3256. http://dx.doi.org/10.1016/j.ejca.2012.06.008
- Little, M., and Wells, C. (1997) A clinical overview of WT1 gene mutations. Human Mutation, 9, 209-225. http://dx.doi.org/10.1002/(SICI)1098-1004(1997)9:3<209::AID-HUMU2>3.0.CO;2-2
- Breslow, N.E., Olson, J., Moksness, J., Beckwith, J.B. and Grundy, P. (1996) Familial Wilms’ tumor: A descriptive study. Medical and Pediatric Oncology, 27, 398-403. http://dx.doi.org/10.1002/(SICI)1096-911X(199611)27:5<398::AID-MPO2>3.0.CO;2-H
- Varanasi, R., Bardeesy, N., Ghahremani, M., Petruzzi, M.J., Nowak, N., Adam, M.A., Grundy, P., Shows, T.B. and Pelletier, J. (1994) Fine structure analysis of the WT1 gene in sporadic Wilms tumors. Proceedings of the National Academy of Sciences of the United States of America, 91, 3554-3558. http://dx.doi.org/10.1073/pnas.91.9.3554
- Menke, A.L., van der Eb, A.J. and Jochemsen, A.G. (1998) The Wilms’ tumor 1 gene: Oncogene or tumor suppressor gene? International Review of Cytology, 181, 151-212. http://dx.doi.org/10.1016/S0074-7696(08)60418-0
- Rauscher 3rd, F.J. (1993) The WT1 Wilms tumor gene product: A developmentally regulated transcription factor in the kidney that functions as a tumor suppressor. FASEB Journal, 7, 896-903.
- Berger, A.H., Knudson, A.G. and Pandolfi, P.P. (2011) A continuum model for tumour suppression. Nature, 476, 163-169. http://dx.doi.org/10.1038/nature10275
- Moffett, P., Bruening, W., Nakagama, H., Bardeesy, N., Housman, D., Housman, D.E. and Pelletier, J. (1995) Antagonism of WT1 activity by protein self-association. Proceedings of the National Academy of Sciences of the United States of America, 92, 11105-11109. http://dx.doi.org/10.1073/pnas.92.24.11105
- Pelletier, J., Bruening, W., Kashtan, C.E., Mauer, S.M., Manivel, J.C., Striegel, J.E., Houghton, D.C., Junien, C., Habib, R., Fouser, L., Fine, R.N., Silverman, B.L., Haber1, D.A. and David Housman, D. (1991) Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in DenysDrash syndrome. Cell, 67, 437-447. http://dx.doi.org/10.1016/0092-8674(91)90194-4
- Klamt, B., Koziell, A., Poulat, F., Wieacker, P., Scambler, P., Berta, P. and Gessler, M. (1998) Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/-KTS splice isoforms. Human Molecular Genetics, 7, 709-714. http://dx.doi.org/10.1093/hmg/7.4.709
NOTES
*Corresponding author.