Advances in Bioscience and Biotechnology
Vol.3 No.8(2012), Article ID:25830,9 pages DOI:10.4236/abb.2012.38141
Microarray analysis of gene expression in the liver of transgenic mouse model of HCV infection*
![]()
1Infectious Disease and Vaccine Research Centre, Children’s Hospital of Eastern Ontario Research Institute, Ottawa, Canada
2Pasteur Institute of Iran, Research and production Complex, Tehran, Iran
3Department of Microbiology, Immunology and Biochemistry, University of Ottawa, Ottawa, Canada
4Division of Virology, Children’s Hospital of Eastern Ontario (CHEO), Ottawa, Canada
5Ottawa Health Research Institute, Molecular Medicine Program, Ottawa, Canada
Email: #mghorbani@irimc.org
Received 11 September 2012; revised 12 October 2012; accepted 19 November 2012
Keywords: Microarray; HCV; Hepatocellular Carcinoma
ABSTRACT
Background: The molecular interactions of hepatitis C virus (HCV) with hepatic tissue have yet to be completely elucidated and understood. The purpose of this study was to compare differential gene expression patterns in the livers of non-transgenic and transgenic mouse model expressing HCV structural proteins Core, Envelope 1 (E1) and Envelope 2 (E2) using complementary DNA (cDNA) microarrays. Results: Total RNA extracted from the livers of HCV transgenic and non-transgenic mice was analyzed with cDNA microarray and differentially expressed genes confirmed by real-time RT-PCR. Relative expression ratios of individual genes were determined by comparing hybridization of Cy5-labelled cDNA from transgenic mouse livers and Cy3-labelled cDNA from non-transgenic mouse livers. The spot array images were quantified using QuantArray software and the outlier spots was normalized and filtered using five different criteria. 15,297 genes were analyzed using three different analytical methods. Depending on these methods, twenty-one genes were found to be differentially expressed at a statistically significant level. From these, 6 genes had a consistent differential expression. Several genes were directly involved in lipid metabolism and lipid β-oxidation. 5-azacytidine induced gene 2 (AZ2), which is involved in the methylation of genes was down regulated in HCV transgenic mice. Altered transcript levels of these 6 genes were confirmed by real-time RT-PCR analysis. Conclusion: Interactions between HCV and hepatocytes not only involve lipid metabolism and redox balance, but this interaction may also influence DNA methylation, indicating a potential association with the development of hepatocellular carcinoma.
1. BACKGROUND
Hepatitis C virus (HCV) infections represent a serious and growing worldwide threat to human health and a major etiological agent of end stage liver disease including cirrhosis and hepatocellular carcinoma [1,2]. A remarkable feature of HCV infection is the high incidence of viral persistence. More than 80% of HCV-infected individuals develop the chronic form of the disease and 20% of chronically infected patients will develop cirrhosis, and many of those will progress to hepatocellular carcinoma [3,4]. HCV infection accounts for the deaths of 8000 - 10,000 Americans each year [5,6].
Although countless efforts over the past decade have attempted to understand the pathogenesis of HCV infection, the mechanisms leading to viral persistence and disease progression are not completely understood. The core protein, which forms the viral nucleocapsid through an RNA interaction, plays an essential role in HCV pathogenesis. Studies using transgenic mice that express the core protein have been the most informative of the potential effects of the core protein on host metabolism [7]. Although no immediate pathology resulting from core expression is apparent in most core transgenic systems, in one transgenic line it was observed that a lymphocytic infiltrate with hepatocyte necrosis occurred in conjunction with an enhanced sensitivity to anti-Fas antibody [8] {Honda, 2000 871/id}. Perhaps the most significant phenotype observed in core transgenic mice was the induction of steatosis and hepatocellular carcinoma (HCC), both of which are hallmarks of chronic infections with HCV [9-11]. Increased lipid peroxidation and mitochondrial DNA damage, both indicators of increased reactive oxygen species (ROS), have been noted in some core transgenic mice [12]. Steatosis and HCC have been observed in two other transgenic lines, one expressing the structural proteins (core E1/E2 p7) and the other containing the entire HCV ORF [9]. It should be noted that not all lines of core transgenic mice develop steatosis and HCC.
In our previous study, a transgenic mouse model expressing HCV structural proteins was established [13]. This model showed severe hepatopathy characterized by the development of steatosis as well as liver and lymphoid tumors. In this study, differential gene expression in transgenic and non-transgenic mice expressing Core, E1 and E2 proteins was analyzed using cDNA microarrays. More than 15.000 genes were analyzed using three different analytical criteria. According to these criteria, 21 genes were found to be differentially expressed at a statistically significant level. These genes were directly involved in lipid metabolism and lipid β-oxidation, and DNA methylation. Changing the expression of these genes in HCV transgenic mice may help us understand HCV pathogenesis in the liver.
2. RESULT
2.1. Transgenic Mice Characteristics
In our previous study, a transgenic mouse model was established by expressing Core, E1 and E2 proteins downstream of a CMV promoter. HCV core, E1 and E2 RNA was detected in different tissues from transgenic mice, while core, E1 and E2 proteins were expressed predominantly in the transgenic liver [13]. Morphological analysis of liver cells demonstrated generalized steatosis (lipid accumulation within hepatocytes) in transgenic mice older than 3 months of age (Figure 1(a)). Both microand macrovesicular steatosis can be seen in the liver of the transgenic mice. Mice older than 10 months of age had an increased incidence of tumor formation in the livers that were mainly adenomas (Figure 1(b)). H&E staining indicates the development of hepatoma/hepatocellular carcinoma as seen by compression of ademona cells.
2.2. Increase of Viral RNA in Older Transgenic Mice
To investigate the pattern of expression of the viral transgene in older mice, a comparison of 4 groups of transgenic mice (between 4 and 18 months-old) was performed using real-time RT-PCR. An increased expression of the HCV Core RNA transcript was observed in older mice (Table 1). There was a 1.9, 274 and 776 fold increase in HCV transcript concentration at 6, 14 and 18 months respectively when compared to mice at 4 months of age. Moriya et al. have also demonstrated that an HCV transgenic mouse model that expressed only the core protein developed hepatic tumours and adenomas with appearance of fat droplets in cytoplasm at age of 16 months [10]. Therefore, we perofrme all microarray analysis at age 14 months when the expression of HCV core RNA reached the optimum level and animals start developing tumours.
2.3. Microarray Analysis of HCV Transgenic and Non-Transgenic Livers
There was a statistically significant difference in gene expression levels between HCV transgenic and nontransgenic livers. From the total 15,297 genes analyzed, 21 genes were selected according to their differential expression (Figure 2, Table 2). Several genes, including aldehyde dehydrogenase, Apolipoprotein A-IV, Hydroxysteroid dehydrogenase, Acetyl-CoA, Acyl CoA, and malic enzyme were upregulated in transgenic mouse livers, while tropomyosin, and 5-azacytidine induced gene 2 (AZ2), were downregulated (Figure 3). Among these
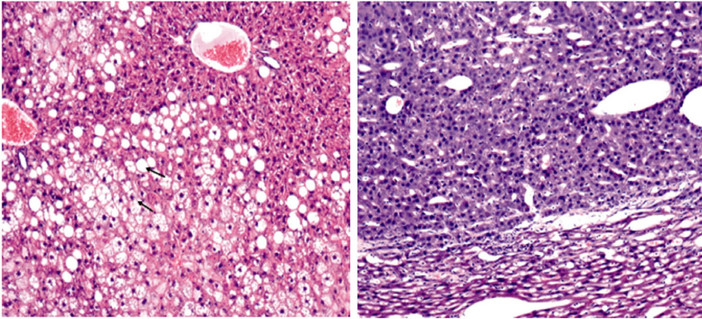 (a) (b)
(a) (b)
Figure 1. Development of steatosis and hepatocellular adenoma in transgenic mous liver.
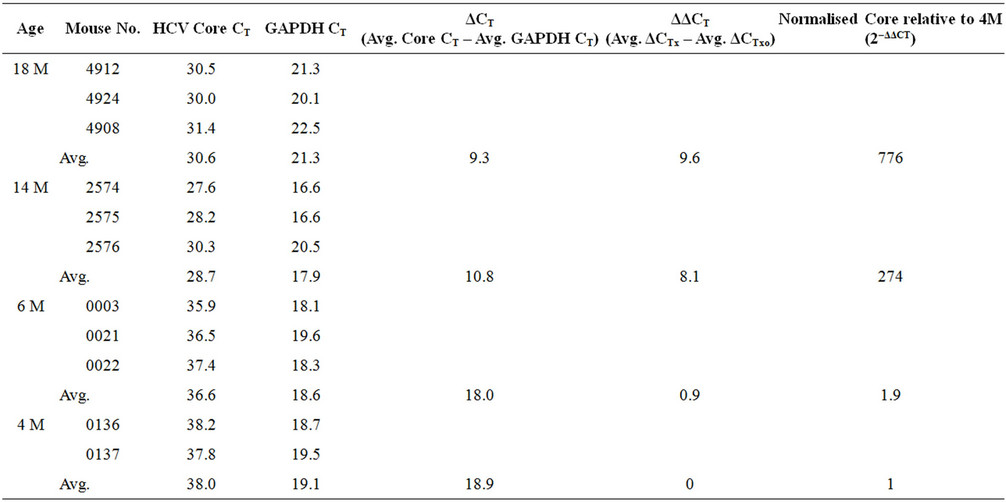
Table 1. Relative expression of RNA in the liver of transgenic mice at different ages detected by real-time RT-PCR.
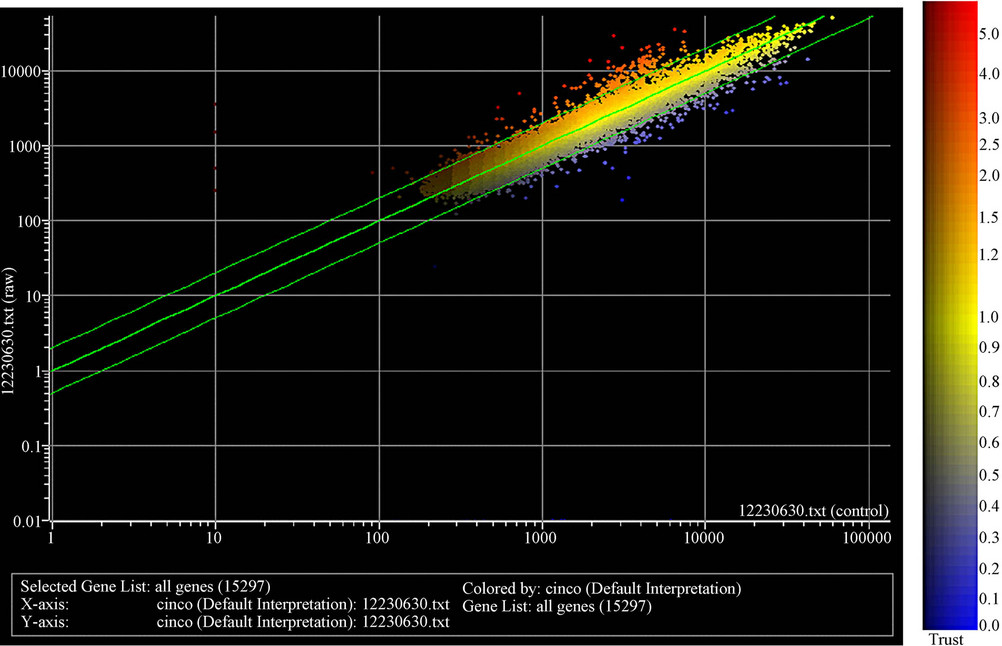
Figure 2. Scatter plot of gene expression values for 15,297 genes in an HCV transgenic mouse versus a control.
21 selected genes, 6 genes were identified as being the most differentially expressed. These genes were Apolipoprotein A IV (Apo A IV),Acetyl-Coenzyme A acyltransferase (Acaa), Tropomyosin 2 beta (Tpm2), 5-azacytidine induced gene 2 (AZ2), Acyl-Coenzyme A oxidase (AOX), and the unknown gene H3030C12. These findings suggest that HCV structural proteins induce changes in gene expression of the transgenic mice.
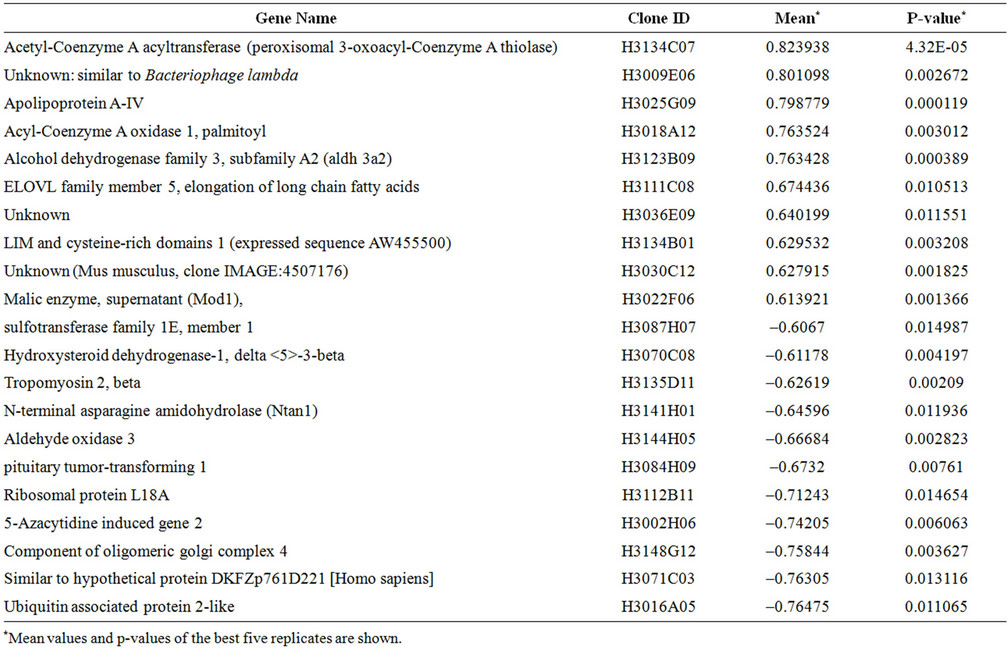
Table 2. Genes differentially regulated in HCV transgenic mice.
2.4. Validation of Microarray Data by Real Time-RT-PCR Analysis
To verify our microarray results, we performed real timePCR analysis for these 6 differentially expressed genes in the HCV transgenic mouse. Real time PCR experiments revealed that transcripts were up or down regulated in agreement with microarray data (Figure 4).
3. DISCUSSION
While younger transgenic mice have no detectable HCV proteins, at 3 months of age an increasing number of hepatocytes expressing Core, E1, E2 proteins around central veins are observed along with appearance of both severe micro and macro-vesicular steatosis [13]. HCV infection becomes persistent in 80% of affected patients. This frequently results in the development of chronic hepatitis and cirrhosis and eventually may lead to hepatocarcinoma [14]. The molecular mechanism underlying the development of HCV histopathology is still not fully understood. HCV transgenic mice expressing different viral proteins in the liver may offer insights into the pathogenesis of liver disease after HCV infection. Several transgenic HCV models have recently been described [15-18]. One of the differences between those models and the model we developed is the presence of extensive liver steatosis and the frequent formation of liver adenomas, lymphoid tumors and carcinoma in our model. In this transgenic animal model, HCV structural proteins are preferentially expressed in the liver [13]. In this study, we examined the differential gene expression profiles of hepatocytes from the Core-E1-E2 hepatitis C transgenic and non-transgenic mice. In contrast to normal livers, several genes were upregulated in transgenic livers, including genes that encode for metabolic enzymes involved in lipid metabolism, such as aldehyde dehydrogenase, apoliprotein A-IV, hydroxysteroid dehydrogenase, acetyl and acyl coenzyme A and malic enzyme.
Apolipoprotein A-IV (apo A-IV) is a protein that is associated with chylomicrons, and it is secreted only by the small intestine in humans [19]. In rodents, both the small intestine and the liver secrete apo A-IV; however, the small intestine is the major organ responsible for circulating apo A-IV [20-22]. Of all the apolipoproteins associated with chylomicrons secreted by the small intestinal epithelial cells (enterocyte), apo A-IV is the only one that is stimulated by the absorption of fat [23]. Since apo A-IV is one of the elements of chylomicrons, it is therefore important in the transport of dietary triglycerides, phospholipids, cholesterols and cholesterol esters absorbed by entercytes to other tissues such as hepatocytes [22].
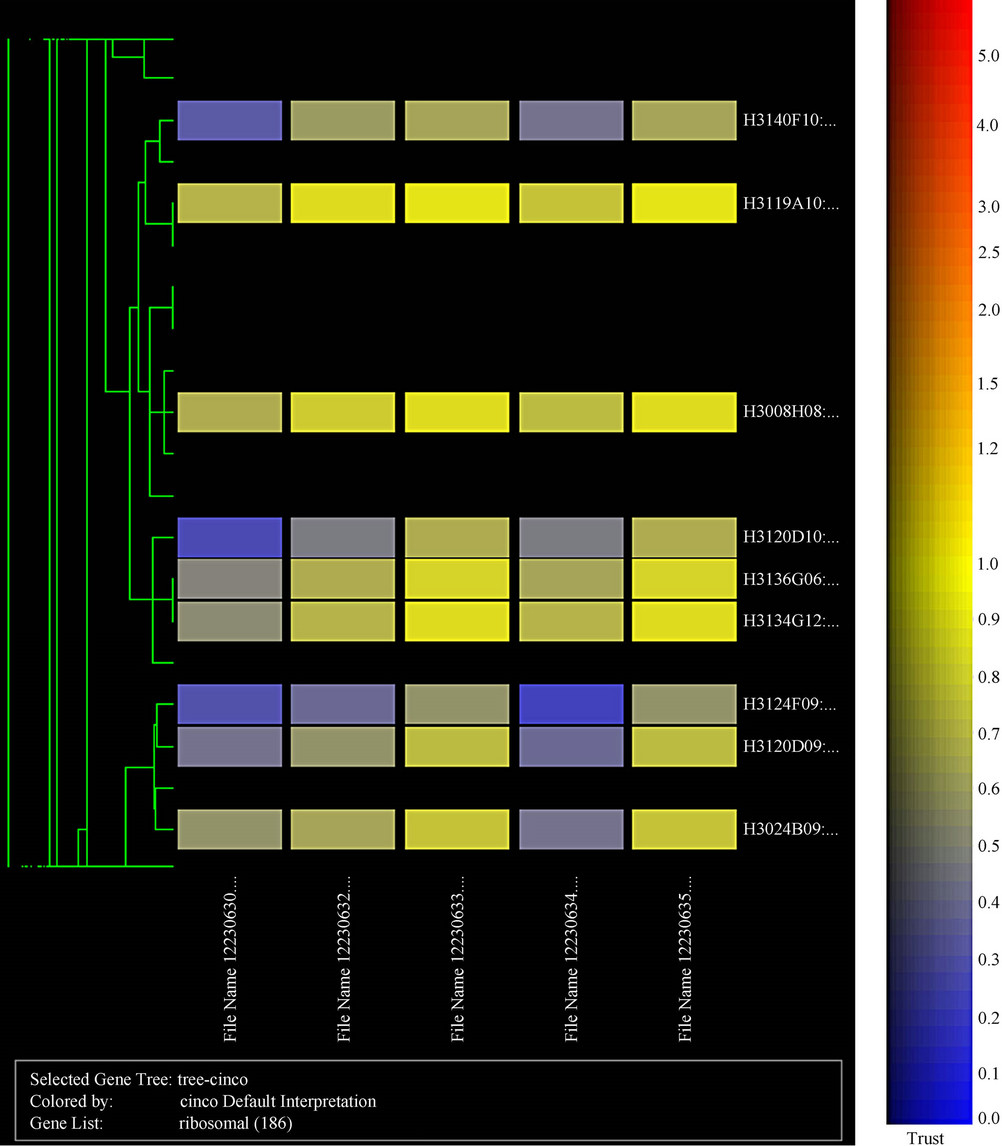
Figure 3. Cluster display of data HCV transgenic mice.
It has been shown that HCV core protein localizes to fat droplets [24-26] as well as mitochondria [27]. Since apo A-IV protein binds to lipids and renders the water soluble in the form of lipoproteins [28], it may be a likely candidate for core protein interaction. On the other hand, HCV has been described as a lipid-containing virus; in the plasma of HCV infected patients there is a heterogeneous density distribution partially due to the binding of HCV to low density lipoprotein, very low density lipoprotein, and to a minor degree to high density lipoprotein [21]. It is important, therefore, to emphasize that several in vivo observations regarding HCV infection
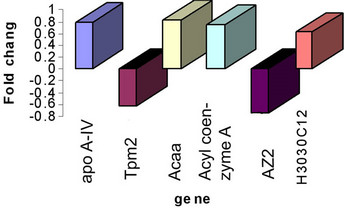
Figure 4. Gene expression by realtime.
might be related to our findings. Firstly, a characteristic of HCV infection is the presence of liver steatosis; it is plausible that the steatosis could arise, at least in part, from direct effects of HCV proteins on lipid metabolism. The importance of the association of HCV to lipids is further reinforced by blood lipid composition in patients with chronic hepatitis C treated with interferon alpha. One study found that primary responses to interferon correlated with higher apolipoprotein A1 plasma levels [29].
Acetyl-Coenzyme A acyltransferase (Acaa) is an enzyme specific for the peroxisomal breakdown of fatty acids producing acetyl-CoA. Acaa plays an integral role in fatty acid metabolism. Similarly, acyl-Coenzyme A oxidase is involved in the oxidation of free fatty acids in peroxisomes [30]. Our HCV transgenic model demonstrated proliferation of peroxisomes, which in conjunction with the upregulation of these peroxisomal enzymes, suggests that HCV alters the lipid metabolism and perhaps the redox balance through these interactions.
Tropomyosin 2 beta (Tpm2) and 5-azacytidine induced gene 2 (AZ2), were downregulated in our transgenic mice livers. The tropomyosin (Tm) family of actin-binding proteins plays a pivotal role in regulating the function of actin filaments in both muscle and nonmuscle cells. The tropomyosin 2 beta (Tpm2) gene encodes the fibroblast protein tropomyosin, for which many isoforms exist as a multi-gene family of actin-binding proteins. Tropomyosin stabilizes the actin filaments by modulating the interaction with proteins that are responsible for the regulation of actin dynamics. The tropomyosin proteins are expressed in tissues examined including skeletal muscle, stomach, heart, liver, kidney, uterus, spleen, brain, and diaphragm [31]. Previous study performed proteomic analysis of cancerous and noncancerous tissues from HCC patients with HCV infection determined that tropomyosin β chain proteins were decreased [32]. The 5-azacytidine induced gene 2 (AZ2) plays a role in gene regulation through methylation of CpG islands. The downregulation of this gene product observed in our HCV transgenic mice, suggests that hypomethylation which may be a required step in cancer promotion. Cancerous cell genomes are dysregulated due to global hypomethylation [33,34]. Recently, rats fed a methyl-deficient diet demonstrated extensive DNA methylation in the liver, which was associated with expansion of tumor foci suggesting that DNA hypomethylation is a promoting factor for clonal expansion of parenchymal cells. This evidence provides a mechanistic basis to explain hepatocarcinogenesis [35]. Finally, an unknown gene named H3030C12 was studied (NCBI), though its molecular function is yet to be discovered.
4. CONCLUSION
Using the microarray technique, we demonstrated differential gene expression profiles in the livers of HCV transgenic mice. The interaction between the HCV structural proteins influences the lipid metabolism, redox balance and DNA methylation in the transgenic mice, indicating an association with the development of hepatocellular carcinoma in this HCV model. These results provide a window into understanding the global changes in gene expression that occur in vivo during chronic HCV infection. This study provides insight into the molecular mechanisms leading to HCV pathogenesis and HCC development.
5. MATERIALS AND METHODS
5.1. Production of Transgenic Mice
All animal protocols were reviewed and approved by the University of Ottawa Animal Care Protocol Review Committee.
A transgenic mouse model expressing HCV core, E1 and E2 under the control of a CMV promoter was previously described [13]. Briefly, Total RNA was extracted from the plasma of a patient infected with HCV genotype 1a. The RNA was used as a template to amplify core, E1 and E2 genes. The HCV fragment containing core, E1 and truncated E2 genes, encoding for amino acid residues 1-683 (2049 nt), was constructed by RT-PCR using the forward primer 5’-ACCATGAGCACGAATCCTAAACCTC- 3’ and the reverse primer 5’-TGGTAGGGTTGTGAAGGAACACG-3’. The amplified fragment was cloned into the EcoRI sites of pCR 2.1 vector using the TOPO-TA cloning kit (Invitrogen). The nucleotide sequence was verified by DNA sequencing using the University of Ottawa DNA sequencing facility. The core, E1 and E2 fragments were subsequently subcloned into pVAX (Invitrogen) downstream of a cytomegalovirus (CMV) promoter [13]. The pVAX-HCV (core, E1, E2) was linearized by MluI restriction enzyme digestion. The linearized plasmid containing HCV genes encoding Core, E1 and E2 proteins was microinjected into the pro-nuclei of fertilized eggs from a B6C3F1 murine strain (Charles River Laboratories, Wilmington, MA). The injected eggs were surgically implanted into the oviducts of pseudopregnant mothers. Founder mice harboring HCV fragments were identified as transgenic via PCR using DNA isolated from tail biopsies using the Dneasy kit (Qiagen, Mississauga, ON). Viral protein expression and liver histological changes in this model were previously described.
5.2. Microarray Experiments
Three transgenic and three non-transgenic mice were used in doing microarray experiments. Total RNA was isolated from frozen liver tissues of HCV transgenic and non-transgenic mice using an RNeasy isolation kit (Qiagen, Mississauga, ON) and used as a template to generate cDNA labeled with Cyanine dye-conjugated (Cy3 or Cy5) dUTP (Amersham Pharmacia, Baie d’Urfe, QC). Labeling reactions contained 10 µg of total RNA, 4 µg Oligo (dT)-18, 3 µg random hexamer primers and 400 units of Superscript II reverse transcriptase (Invitrogen, Burlington, ON). Reactions were incubated at 42˚C for 1.5 h, concentrated to 10 µl in a Microcon-30 filter (Amicon, Nepean, Ontario). Non-transgenic RNA was labeled with Cy3-green, and the transgenic RNA with Cy5-red. Labeled cDNAs were combined in 26 µl of 3 × SSC, 0.2% SDS, and were competitively hybridized to an arrayed surface under a coverslip for 16 h at 37˚C. Slides were washed in 1 × SSC and 0.03% SDS (1 min), 1 × SSC (1 min), 0.2 × SSC (20 min), and 0.05 × SSC (10 min) and dried by centrifugation.
More than 15,297 mouse genes and expressed sequence tags (ESTs), from the array Microarray Center (Ontario Cancer Institute, Toronto, ON) were examined. Array images were collected for both Cy3 and Cy5 by using a Scan Array XL 4000 fluorescent scanner (Packard Biochip, Montreal, QC) with a 10-µm resolution to detect Cy3 and Cy5 fluorescence. The image intensity data were extracted and analyzed using the QuantArray 3.0 (Packard Bio-chip, CA) software.
5.3. Data Filtering and Normalization
The data consisted of seven microarray experiments (biological repeats). Every data set (array) consisted of 30,622 rows of reading of mouse genes, and 578 rows of controls. There was one pair of duplicate readings of each gene or control. Columns specify readings from a nontransgenic mouse (Cy3, channel) and Cy5, Channel 2), respectively. Microarray raw data from QuantaArray was pre-processed using an add-in package for Microsoft Excel 2000 specifically designed to perform background subtraction, normalization and filtering. Briefly, after background subtraction the data preprocessed using five filtering criteria. The first criteria involved filtering spots with intensity in the 5th percentile or lower in either channel. The second is filtering spots with intensity in the 98th percentile or higher in either channel. Spots with intensity/background less than 2.5 were also dismissed. These data are close to background and as such yield unreliable ratio values. The 4th criterion applied was an “ignore” filter, to remove data that was identified as poor quality by visual inspection of the images during the quantization step. Finally, entire subarrays were filtered in cases where more than 75% of the spots were already filtered. Then the intensity data were converted to logarithmic scale (log2). Channel 2 intensities were normalized using a linear regression correction of the Channel 2 vs. Channel 1 intensity data. This transformation was used in order to balance the differential expression of the genes around a log2 ratio of 0. The method applies the assumptions that most of the genes do not differentially express and that there is no bias towards upor downregulation of expression in the system. The resulting Ch2 vs. Ch1 intensity scatter plot has a slope of 1 and an intercept of 0.
5.4. Data Analysis
Three analytical methods were used to study the differentially expressed genes detected by computing p-values using a paired t-test in Microsoft Excel. The mean log ratio of intensity for each gene across either all 7, or the best 5, replicates was tested against (i.e. ratios significantly different from 0). Thresholds were also applied to the genes with both a p-value of 0.01 or less and a minimum absolute log2 ratio of 0.6 (which is approximately 1.5-fold). An outlier detection strategy was applied to the data set to remove data points that were significantly different from the mean of all replicates for a particular gene. First, the standard deviation from the mean of all ratios on the array was computed. This value was then used as the threshold for outlier detection. Two rounds of outlier detection using the same threshold value were applied to the data set. A minimum of 4 remaining replicates was required following the filtering for a gene to be further considered. P-value and ratio thresholds were then applied as previously mentioned using Excel. Second, a Cyber T with Bayesian analysis was performed using the application Cyber T. Bayesian correction factors were determined using the method Significance Analysis of Microarrays (SAM).
RT-PCR: Total RNA was extracted from different mouse tissues (liver, spleen, kidney, brain, lung, and heart) using the Rneasy Mini Kit (Qiagen, Mississauga, ON). Reverse transcription of total RNA was performed with Mul.V reverse transcriptase (Applied Biosystems, Foster City, CA, USA) and random hexamers (Applied Biosystems, Foster City, CA, USA) to generate cDNA, according to the manufacturer’s instructions. The cDNA was amplified by PCR using HCV forward primer (5’ ACC ATG AGC ACG AAT CCT AAA CCTC 3’) and reverse primer (5’ TGG TAG GGT TGT GAA GGA ACA CG 3’). RNA extract from the liver of a non-transgenic littermate was used as a negative control.
5.5. Real-Time RT-PCR Assay
Total RNA was extracted from mouse liver using the RNeasy Mini Kit (Qiagen, Mississauga, ON). The RNA was reverse transcribed using oligo (dT) 20-mer and Superscript II reverse transcriptase (InVitrogen), according to the manufacturer’s instructions. RT was performed for 80 min at 42˚C using 500 ng of RNA for all samples. In order to prevent any potential contamination by protein, the samples were incubated for 10 min at 95˚C. The real time semi-quantitative PCR was performed in special optical tubes in a 96-in well microtiter plate (PerkinElmer/Applied Biosystems) with an iCycler (Bio-Rad, Hercules, CA). Fluorescent signals were generated using the Quantitect SYBR Green PCR kit (Qiagen).
The following differentially expressed genes were analyzed using oligonucleotide primers (300 nM): AOX (Acyl-CoA oxidase) sense 5’ GAA CTC CAG ATA ATT GGC ACC TA 3’ and antisense 5’ AGT GGT TTC CAA GCC TCG AA 3’, Tpm-2 (tropomyosin 2, beta) sense 5’ ACA AGG AGA ATG CCA TCG AC 3’ and antisense 5’ CTT TTC CAC CTC GTC CTC TG 3’, Acaa (acetyl-Coenzyme A acyltransferase) sense 5’ CCT GAC TCC TAT GGG GAT GA 3’ and antisense 5’ AAT CCC AGC ACG TAA GCA TG 3’, AZ-2 (5-azacytidine induced gene 2) sense 5’ CTG GTG ACT CAA GTG CAA GC 3’ and antisense 5’ GTG CTG TCT CTC CCA AGG TC 3’, Apo A-4 (apolipoprotein A-IV) sense 5’ CTG GTG GCC ATC ACC GGC AC 3’ and antisense 5’ AGT CCT GGA AGA GGG TAC TGA GC 3’, and gene H3030C12 sense 5’ AGT AGT GAG AGG TTC TCG 3’ and antisense 5’ GTA AGG GAG TTC AGT AGC 3’. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal control gene with the following sense and antisense primer sequences: 5’ ATGTGTCCGTCGTGGATCTGA 3’ and 5’ TTGAAGTCGCAGGAGACAACCT 3’ respectively. As negative control, a no template and double distilled water was used in a total volume of 50 µl. For each amplification using real time PCR, the protocol consisted of 15 min at 95˚C and 40 cycles of 15 s at 95˚C and 30 s at 63˚C. The iCycler software detected the CT for each amplicon. Normalization was performed using the 2–∆∆CT method [36].
6. ACKNOWLEDGEMENTS
The authors acknowledge Rita Frost towards her assistance with the RNA extraction and microarray technique optimization. Great thanks to Katrina Gee and Nicole D. Scherling for critical revising the manuscript. This work was supported by health Canada and Canadian Institute of Health Research.
REFERENCES
- Cohen, J. (1999) The scientific challenge of hepatitis C. Science, 285, 26-30. doi:10.1126/science.285.5424.26
- Rosen, H.R. and Gretch, D.R. (1999) Hepatitis C virus: Current understanding and prospects for future therapies. Molecular Medicine Today, 5, 393-399. doi:10.1016/S1357-4310(99)01523-3
- Colombo, M. and Covini, G. (1995) Hepatitis C virus and hepatocellular carcinoma. Clinical and Experimental Rheumatology, 13, S23-S27.
- Liang, T.J. and Heller, T. (2004) Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology, 127, S62-S71. doi:10.1053/j.gastro.2004.09.017
- Alter, M.J. (1997) Epidemiology of hepatitis C. Hepatology, 26, 62S-65S. doi:10.1002/hep.510260711
- Gasiorowicz, M., Hurie, M., Russell, A., Hoxie, N. and Vergeront, J. (2006) Epidemiologic trends in infection, mortality, and transplants related to hepatitis C in Wisconsin. WMJ, 105, 34-39.
- Ray, R.B., Lagging, L.M., Meyer, K. and Ray, R. (1996) Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. Journal of Virology, 70, 4438-4443.
- Honda, A., Arai, Y., Hirota, N., Sato, T., Ikegaki, J., Koizumi, T., et al. (1999) Hepatitis C virus structural proteins induce liver cell injury in transgenic mice. Journal of Medical Virology, 59, 281-289. doi:10.1002/(SICI)1096-9071(199911)59:3<281::AID-JMV4>3.0.CO;2-S
- Lerat, H., Honda, M., Beard, M.R., Loesch, K., Sun, J., Yang, Y., et al. (2002) Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology, 122, 352- 365. doi:10.1053/gast.2002.31001
- Moriya, K., Fujie, H., Yotsuyanagi, H., Shintani, Y., Tsutsumi, T., Matsuura, Y., et al. (1997) Subcellular localization of hepatitis C virus structural proteins in the liver of transgenic mice. Japanese Journal of Medical Science & Biology, 50, 169-177.
- Moriya, K., Fujie H, Shintani, Y., Yotsuyanagi, H., Tsutsumi, T., Ishibashi, K., et al. (1998) The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nature Medicine, 4, 1065-1067. doi:10.1038/2053
- Moriya, K., Nakagawa, K., Santa, T., Shintani, Y., Fujie, H., Miyoshi, H., et al. (2001) Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Research, 61, 4365-4370.
- Naas, T., Ghorbani, M., Alvarez-Maya, I., Lapner, M., Kothary, R., De, R.Y., et al. (2005) Characterization of liver histopathology in a transgenic mouse model expressing genotype 1a hepatitis C virus core and envelope proteins 1 and 2. Journal of General Virology, 86, 2185- 2196. doi:10.1099/vir.0.80969-0
- Sola, R., Alvarez, M.A., Balleste, B., Montoliu, S., Rivera, M., Miquel, M., et al. (2006) Probability of liver cancer and survival in HCV-related or alcoholic-decompensated cirrhosis. A study of 377 patients. Liver International, 26, 62-72. doi:10.1111/j.1478-3231.2005.01181.x
- Alonzi, T., Agrati, C., Costabile, B., Cicchini, C., Amicone, L., Cavallari, C., et al. (2004) Steatosis and intrahepatic lymphocyte recruitment in hepatitis C virus transgenic mice. Journal of General Virology, 85, 1509-1520. doi:10.1099/vir.0.19724-0
- Feitelson, M.A. and Larkin, J.D. (2001) New animal models of hepatitis B and C. ILAR Journal, 42, 127-138.
- Ren, J.Y., Cheng, G.X., Kong, X.F., Chen, J.Q., Zhou, R.J. and Lu, Z.M. (2005) Establish a transgenic mice model harboring structural genes of hepatitis C virus. Chinese Journal of Hepatology, 13, 501-504.
- Wakita, T. (1998) Construction of HCV transgenic mice with Cre/loxP switching expression system. Journal of Virology, 48, 9-18.
- Elshourbagy, N.A., Walker, D.W., Paik, Y.K., Boguski, M.S., Freeman, M., Gordon, J.I., et al. (1987) Structure and expression of the human apolipoprotein A-IV gene. The Journal of Biological Chemistry, 262, 7973-7981.
- Windmueller, H.G. and Spaeth, A.E. (1985) Regulated biosynthesis and divergent metabolism of three forms of hepatic apolipoprotein B in the rat. The Journal of Lipid Research, 26, 70-81.
- Kono, Y., Hayashida, K., Tanaka, H., Ishibashi, H. and Harada, M. (2003) High-density lipoprotein binding rate differs greatly between genotypes 1b and 2a/2b of hepatitis C virus. Journal of Medical Virology, 70, 42-48. doi:10.1002/jmv.10372
- Wu, A.L. and Windmueller, H.G. (1981) Variant forms of plasma apolipoprotein B. Hepatic and intestinal biosynthesis and heterogeneous metabolism in the rat. The Journal of Biological Chemistry, 256, 3615-3618.
- Wu, A.L. and Windmueller, H.G. (1978) Identification of circulating apolipoproteins synthesized by rat small intestine in vivo. The Journal of Biological Chemistry, 253, 2525-2528.
- Barba, G., Harper, F., Harada, T., Kohara, M., Goulinet, S., Matsuura, Y., et al. (1997) Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proceedings of the National Academy of Sciences USA, 94, 1200-1205. doi:10.1073/pnas.94.4.1200
- Perlemuter, G., Sabile, A., Letteron, P., Vona, G., Topilco, A., Chretien, Y., et al. (2002) Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: A model of viral-related steatosis. FASEB Journal, 16, 185- 194. doi:10.1096/fj.01-0396com
- Sabile, A., Perlemuter, G., Bono, F., Kohara, K., Demaugre, F., Kohara, M., et al. (1999) Hepatitis C virus core protein binds to apolipoprotein AII and its secretion is modulated by fibrates. Hepatology, 30, 1064-1076. doi:10.1002/hep.510300429
- Korenaga, M., Wang, T., Li, Y., Showalter, L.A., Chan, T., Sun, J., et al. (2005) Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. The Journal of Biological Chemistry, 280, 37481-37488. doi:10.1074/jbc.M506412200
- Tso, P., Chen, Q., Fujimoto, K., Fukagawa, K. and Sakata, T. (1995) Apolipoprotein A-IV: A circulating satiety signal produced by the small intestine. Obesity Research, 3, 689S-695S. doi:10.1002/j.1550-8528.1995.tb00487.x
- Soardo, G., Pirisi, M., Fonda, M., Fabris, C., Falleti, E., Toniutto, P., et al. (1995) Changes in blood lipid composition and response to interferon treatment in chronic hepatitis C. Journal of Interferon & Cytokine Research, 15, 705-712. doi:10.1089/jir.1995.15.705
- Cheng, Y., Dharancy, S., Malapel, M. and Desreumaux, P. (2005) Hepatitis C virus infection down-regulates the expression of peroxisome proliferator-activated receptor alpha and carnitine palmitoyl acyl-CoA transferase 1A. World Journal of Gastroenterology, 11, 7591-7596.
- Schevzov, G., Vrhovski, B., Bryce, N.S., Elmir, S., Qiu, M.R., O’neill, G.M., et al. (2005) Tissue-specific tropomyosin isoform composition. Journal of Histochemistry & Cytochemistry, 53, 557-570. doi:10.1369/jhc.4A6505.2005
- Kuramitsu, Y. and Nakamura, K. (2005) Current progress in proteomic study of hepatitis C virus-related human hepatocellular carcinoma. Expert Review of Proteomics, 2, 589-601. doi:10.1586/14789450.2.4.589
- Chekhun, V.F., Kulik, G.I., Yurchenko, O.V., Tryndyak, V.P., Todor, I.N., Luniv, L.S., et al. (2006) Role of DNA hypomethylation in the development of the resistance to doxorubicin in human MCF-7 breast adenocarcinoma cells. Cancer Letters, 231, 87-93. doi:10.1016/j.canlet.2005.01.038
- Tryndyak, V.P., Kovalchuk, O. and Pogribny, I.P. (2006) Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferase 1, Suv4-20h2 histone methyltransferase and methyl-binding proteins. Cancer Biology & Therapy, 5, 65-70. doi:10.4161/cbt.5.1.2288
- Pogribny, I.P., Ross, S.A., Wise, C., Pogribna, M., Jones, E.A., Tryndyak, V.P., et al. (2006) Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutation Research, 593, 80-87. doi:10.1016/j.mrfmmm.2005.06.028
- Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402-408.
NOTES
*The author(s) declare that they have no competing interests.
#Corresponding author.