Advances in Bioscience and Biotechnology
Vol.3 No.7(2012), Article ID:24838,19 pages DOI:10.4236/abb.2012.37110
Portal hypertension-related inflammatory phenotypes: From a vitelline and amniotic point of view
![]()
1Department of Surgery I, School of Medicine, Complutense University of Madrid, Madrid, Spain
2Neurosciences Laboratory, Department of Psychobiology, School of Psychology, University of Oviedo, Oviedo, Spain
3Department of Surgery, La Paz Hospital, Autonoma University of Madrid, Madrid, Spain
4Cellular Biology and Morphological Sciences Department, School of Medicine, Autonoma University of Madrid, Madrid, Spain
5Cell Engineering Laboratory, La Paz Hospital, Autonoma University of Madrid, Madrid, Spain
Email: *jariasp@med.ucm.es
Received 4 August 2012; revised 16 September 2012; accepted 14 October 2012
Keywords: Portal Hypertension; Ascites; Vitellogenic; Amniotic; Extrahepatic Cholestasis; Partial Portal Vein Ligation
ABSTRACT
Prehepatic portal hypertension induces a splanchnic low-grade inflammatory response that could switch to high-grade inflammation with the development of severe and life-threatening complications when associated with chronic liver disease. The extraembryonic origin of the portal system maybe determines the regression to an extraembryonic phenotype, i.e., vitellogenic and amniotic, during the evolution of both types of portal hypertension. Thus, prehepatic portal hypertension, or compensated hypertension by portal vein ligation in the rat, is associated with molecular mechanisms related to vitellogenesis, where hepatic steatosis and splanchnic angiogenesis stand out. In turn, extrahepatic cholestasis in the rat induces intrahepatic portal hypertension, or decompensated hypertension, with ascites and hepatorenal syndrome. The splanchnic interstitium, the mesenteric lymphatic system, and the peritoneal mesothelium seem to create an inflammatory pathway that could have a key pathophysiological relevance in the production of ascites. The hypothetical comparison between the ascitic and the amniotic fluid also allows for translational investtigation. The induced regression of the splanchnic system to extraembryonic functions by portal hypertension highlights the great relevance of the extraembryonic structures even during post-natal life.
1. INTRODUCTION
Portal hypertension is defined as a pathological increase in portal vein pressure and it is diagnosed when the hepatic venous pressure gradient is above the normal range [1]. It has been proposed that low-grade inflammation related to portal hypertension switches to high-grade inflammation with the development of severe and life-threatening complications when associated with chronic liver disease [2].
It could be considered that the underlying central theme in low-grade portal hypertensive inflammation is the disturbance in splanchnic and systemic hemodynamics [1,2]. This splanchnic and systemic hemodynamic response would be aggravated during the progression of the chronic liver disease [2,3]. Thus, a critical state is produced in which the appearance of noxious factors during the progressive evolution of chronic liver disease would favor the development of a high-grade splanchnic and systemic inflammatory response [2,4,5].
The portal system includes all veins which carry blood from the abdominal part of the alimentary tract, the spleen, pancreas and gallbladder [6]. This venous system transports the splanchnic venous flow through the portal vein to the liver [1,6]. The afferent venous circulation of the liver as well as its efferent venous system, i.e., the hepatic veins, derives from an extraembryonic venous system, the vitelline venous system, which transports blood from the yolk sac to the heart at the end of the 3rd week of gestation [7]. This extraembryonic origin of the portal system perhaps determines some of the pathological evolutive characteristics when it suffers hyperpressure.
Hence, it is important to keep in mind that a common characteristic of mammals is the development of extraembryonic supporting tissues and organs that are required for embryonic implantation, survival and development “in utero” [8]. Particularly, in the earliest stage of the embryonic development the vitelline system, as well as the amnion, stands out [7,8] (Figure 1).
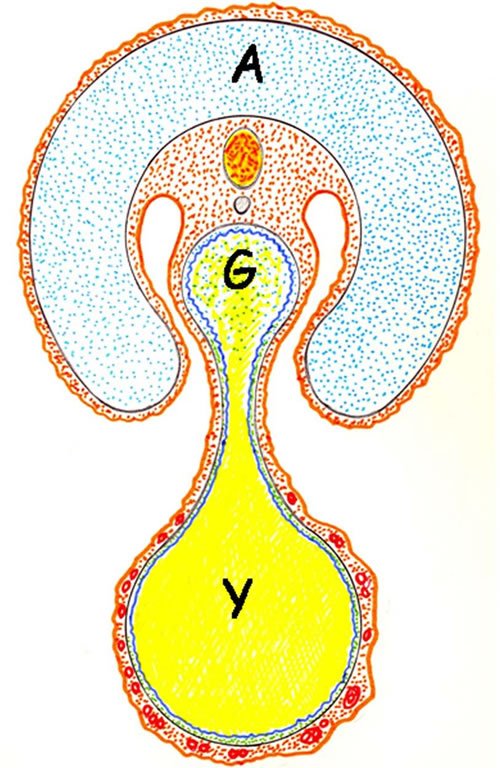
Figure 1. Schematic transverse representation of mammal embryo. The amniotic cavity surrounds the embryo and the amniotic fluid flows through the gut. The yolk sac is connected to the mid-gut by a narrow vitelline duct. Amniotic cavity; Y: yok sac; G: gut.
The portal system, derived from the extraembryonic vitelline venous system continues to perform a vital trophic role for post-natal organism survival and development, although based on its new metabolic needs [2,7]. However, when the portal system suffers an aggression, i.e., portal hypertension, perhaps a regression to the underlying or original embryonic functions could be induced by inflammatory mechanisms [9].
2. LOW-GRADE INFLAMMATORY PORTAL HYPERTENSION: THE VITELLINE CONNECTION
Prehepatic portal hypertension by partial portal vein ligation in the rat induces a splanchnic and systemic chronic low-degree inflammatory response that could be developed through the expression of three successive and overlapping phenotypes: ischemia-reperfusion phenotype, immune phenotype and angiogenic phenotype [2,3]. Thus, it has been already proposed that these phenotypes could represent the expression of trophic functional systems with increasing metabolic complexity [2,10]. Therefore, in the portal hypertensive rat it could be considered that the body adapts the support, i.e., the trophic system, to the metabolic needs characteristic of each inflammatory phenotype. In turn, the metabolic activity of each inflammatory phenotype would be determined by the mechanisms used for cellular energy production [9] (Table 1).
2.1. The Ischemia-Reperfusion Phenotype: The Hydroelectrolitic Changes Enforce the Beginning and Maintenance of a Low-Grade Inflammatory Response
The ischemia-reperfusion phenotype secondary to the hyperdynamic syndrome could represent oxidative and nitrosative stress with edema, which favors diffusion through the inflamed tissues and organs [2,10]. Hyperdynamic circulation stands out among the splanchnic and systemic alterations related to portal hypertension [1,11-15]. It has been suggested that the splanchnic and systemic vasodilation is the initial step leading to the hyperdynamic syndrome or progressive vasodilatory syndrome [12,16]. Multiple organ failure in portal hypertensive chronic liver disease is in large part attributeable to this syndrome [14,16]. Furthermore, hyperdynamic circulation could favor the initiation and mainte-

Table 1. Proposed splanchnic overlapping inflammatory phenotypes in compensated experimental portal hypertension.
nance of an inflammatory response. First, the pathological increase of the portal pressure that occurs in the hyperdynamic splanchnic circulation could favor a disturbed splanchnic venous flow with shear stress mediated by non-laminar flow [12]. The disturbed or non-laminar flow with associated reciprocating low shear stress has profound effects on the biology of the vascular wall, particularly the vascular endothelium, and could stimulate inflammation [17,18]. Second, both the increase in blood flow speed and the opening of arteriovenous shunts that induces the splanchnic hyperdynamic circulation would reduce oxygen tissue availability. This fact would produce tissue hypoxia and therefore chronicity of the inflammatory response [19].
It is widely understood that the gastrointestinal tract functions in a state of low-grade inflammation because the constant exposure to potentially inflammatory stimuli [19]. Therefore, in portal hypertension hypoxia-related with disturbed intestinal blood flow could aggravate this low-grade inflammatory state, favoring the hyperactivetion of hypoxia-inducible factor (HIF) and the IκB/nuclear factor (NF)-κB system, both endogenous alarm signals for the presence of mucosal gastrointestinal inflammatory disease [19]. In this way, the gastrointestinal tract often represents the source for the development of systemic inflammation with multiple organ dysfunction [20,21]. The association of mucosal hypoxia, splanchnic hyperemia and development of portal systemic collaterals bypassing the liver in portal hypertension also suggests that the gastrointestinal tract could play a key role in the induction and maintenance of a low-grade inflammatory response [2,3].
During the expression of the ischemia-reperfusion phenotype through the progression of the interstitial edema, the lymphatic circulation could be simultaneously activated. This circulatory switch increases mesenteric lymph flow and favors gut-derived factors that could reach the systemic circulation avoiding the liver filter [22,23].
Since the evolutionary symbiosis between gut, i.e., enteric nervous system considered as a “mini-brain” [24], and the Central Nervous System, it is possible to hypothesize the contributory role of altered bidirectional gut-brain neural, noradrenergic, cholinergic and nitrergic communications resulting from damaged intestinal mucosal integrity to the development of a systemic inflammatory response [25]. More recently, multiple studies have shown a potentially important neural pathogenic role in the portal hypertension-induced hyperdynamic circulation [1]. Vagal afferent nerves may be the signaling pathway from the hypertensive gastrointestinal tract to the central nervous system [26]. Also, although in portal hypertension there is a global overactivity of the adrenergic system, a splanchnic sympathetic nerve regression, which may contribute to aggravating splanchnic vasodilation, is produced [1].
It has been considered that homeostatic mechanisms attempt to correct the progressive hyper dynamic circulatory state of portal hypertension, including activation of the sympathetic nervous system and the renin-angiotensin-aldosterone system with renal sodium retention [27]. These homeostatic mechanisms seem to be complementary since increased sodium intake or retention can directly affect the excitability or responsiveness of the rostral ventrolateral medulla neurons to enhance sympathetic reflexes [28].
Hydration by sodium retention is a fundamental step of inflammation [29]. The increase of endothelial permeability in the tissues and organs that suffer inflammation causes interstitial edema [18]. This also allows the selective diffusion into the interstitial space of other circulating substances into the blood. Among those released as a response to the neuro-endocrine system to induced stress, in this case, by portal hypertension, the hypothalamic-pituitary-adrenal axis, the sympathetic-adrenal medullary or sympathetic nervous system and the reninangiotensin-aldosterone system stand out [27]. This selectivity in the localization of the early inflammatory response that induces interstitial edema could also favor the posterior interstitial storage of substances derived from the acute phase response. In this way, the interstitial space is shaped from the start of the portal hypertensive pathology as the main field where the battle of inflammation takes place.
2.2. The Immune Phenotype: The Pathological Management of Fat
The expression of the immune phenotype by the splanchnic system which has suffered ischemia and/or reperfusion is coupled with interstitial infiltration by inflammatory cells and by bacteria [30-32]. It has been hypothesized that the symbiosis of inflammatory cells and bacteria for extracellular (fermentation) and intracellular (phagocytosis) could favor tissue trophism [2,10]. Improper use of oxygen persists in this immune phase and it is associated with enzymatic stress. Compensation of the acute phase response includes the production of positive acute phase proteins that bind proteolytic enzymes and inhibitors of leukocyte and liposomal proteolytic enzymes [33,34]. Likewise, the natural inhibitors of matrix metalloproteinases (TIMPs) could promote anti-enzymatic stress [3].
Portal hypertension is one factor determining bacterial intestinal translocation to mesenteric lymph nodes [30, 32]. In addition, the increased presence of mast cells in the hypertrophied mesenteric lymph nodes of prehepatic portal hypertensive rats [35,36] would not only collaborate in the production of mesenteric adenitis, but also it would constitute a source of inflammatory mediators located between the intestine and systemic blood circulation [37,38]. The mesenteric lymph nodes are key structures involved in the gut-associated lymphoid tissue (GALT) [39]. GALT constitutes the largest lymphatic organ in the body and has an important function in the maintenance of the intestinal mucosal integrity, as well as in the control of mucosal inflammation [40]. GALT activation in portal hypertensive enteropathy would produce the release of inflammatory mediators. Then, these mediators would be transported by the intestinal lymph vessels to the systemic circulation aggravating the inflammatory response [9]. In different conditions related to intestinal ischemia, such as hemorrhagic shock or severe burns, mesenteric lymph node circulation has the priority over portal circulation for transporting inflammatory mediators released in the intestinal wall [41,42]. This fact suggests that in other conditions that also produce intestinal hypoxia, like portal hypertension, the mesenteric lymph is a regional pro-inflammatory mediator vehicle, that is, a splanchnic one, but with a systemic effect [9,10].
Prehepatic portal hypertension in the rat, both in the short-(1 month) and in the long-term (1 year) could produce hepatic accumulation of triglycerides and cholesterol [43-45]. The mechanisms by which portal hypertension could induce liver steatosis are not fully understood. Hepatic steatosis, as well as visceral adipose tissue, are metabolic risk factors in mesentery and omentum or visceral fat because their anatomical position drain the venous blood directly to the liver through the portal vein [46,47]. We speculate that these pathological strategies of the body inducing intraabdominal fat deposits around the portal venous system could represent ontogenic reminiscences associated with the yolk sac development, or phylogenetical, related to vitellogenesis. In the first case, the liver and particularly the omentum can be thought of as the substitutes of the yolk sac, in which the animal carries out a pathological deposit of lipids. In this hypothetical situation through the expression of inflammatory mediators, i.e., tumor necrosis factor (TNF)-α, IL-6, C-reactive protein and leptin, [47] the liver and the omentum would be able to regress to evolutive phases in which the metabolic characteristics were suitable [2]. In the second way, and from an ancestral perspective, the body would adopt the molecular mechanisms related to the vitellogenesis. This is a process by which all oviparous species provision their eggs with vitelline, as a major yolk storage glycolipoprotein, that in turn constitutes a reserve food-source for the future embryo [48] (Figure 1).
The ability to transport fat in the form of lipoprotein through the circulatory system by eukaryotes is one of their more significant functions right from the beginning of existence [49]. Thus, the evolutionary advancement of storing energy in the form of fat has provided organisms with an enormous advantage in adapting to environmental and developmental changes [49]. An isoform of apolipoprotein B, apo B48 particles, are chylomicrons, which transport dietary cholesterol and triglycerides from the intestine to sites of storage and utilization within the body, such as the adipose tissue, skeletal and cardiac muscle and the liver [50].
Hepatic steatosis results from increased uptake of free fatty acids derived mainly from the hydrolysis of adipose-tissue triglycerides, increased because of insulin resistance, but also from dietary chylomicrons and hepatic biogenesis [51]. Inflammation and the concomitant acute phase response induce marked changes in the lipoprotein profile [52]. Thus, in portal hypertensive-rat, hepatic steatosis is associated with the plasmatic increase of low density lipoprotein (LDL) and lipopolysaccharide binding protein (LBP) as well as reduction of high-density lipoproteins (HDL) [43,45]. In turn, hepatic steatosis might play a key role in the pathogenesis of cardiovascular disease through the systemic release of several inflammatory mediators and/or through the production of insulin resistance and atherogenic dyslipidemia [51,53].
The acute-phase-response is a core part of the innate immune response and produces changes in more than 200 proteins grouped as either positive or negative acute phase proteins [54,55]. Positive acute phase protein biomarkers which increase in concentration during the inflammatory response include the classic short pentraxins, i.e., C-reactive protein, serum amyloid P component and serum amyloide A [54,55]. IL-6 produced in response to tissue damage and infection is a central mediator of the immune system inducing the liver acute-phase-response [56]. IL-6 and positive acute phase proteins, particularly C-reactive protein, have been correlated to disease severity and outcome in most of the chronic low-grade inflammatory conditions like cardiovascular diseases, obesity, metabolic syndrome, type-2 diabetes and non-alcoholic fatty liver disease (NAFLD), which includes a large spectrum that ranges from fatty liver, non-alcoholic steatohepatitis and cryptogenetic cirrhosis [51,53,57-59].
However, conflicting results exist as to whether IL-6 is a beneficial or harmful cytokine [60]. There is increasing evidence to show that IL-6 has a protective role during liver injury [56]. Hence, it has been suggested that IL-6 favors hepatic secretion of very low density lipoprotein (VLDL) triglyceride by increasing the availability of apo B and thereby reducing hepatic lipid content by increased triglyceride export [60]. It is also accepted that acute phase proteins, particularly serum amyloid A, inhibits the ability of acute phase HDL to serve as an acceptor for cellular cholesterol efflux. This promotes the removal of excess cholesterol from macrophages, as well as increases the availability of cellular free cholesterol and thereby, increases the development of atherosclerotic lesions [52,61].
On the contrary, during cardiovascular inflammation cholesterol esters may be transferred from HDL to apolipoprotein B-containing particles, such as LDL or VLDL [62]. Trapping of apolipoprotein B containing particles within the arterial wall is unquestionably the essential initiating event for the development of complex atherosclerotic lesions [50]. Recent evidences support a strongly association between NAFLD and cardiovascular diseases. Particularly, NAFLD is considered a risk factor for atherosclerosis [51] and ample evidence indicates that inflammation could be the pathophysiological mechanism linking NAFLD and cardiovascular diseases [51,63]. In this context, we have found in the long-term (22 months) prehepatic portal hypertensive-rats that liver steatosis is related to an inflammatory aortic response. In particular, the increased expression of NF-κB, TNF-α, IL-1β and IL-6 in the rat’s aortic wall suggests the existence of a chronic inflammatory state [64].
2.3. The Angiogenic Phenotype: Remodeling through Extraembryonic Vitelline Functions
The remodeling by angiogenesis could characterize the third inflammatory phenotype of the portal hypertensive response. Angiogenesis is defined as the growth of new vessels from pre-existing ones [65]. Although the final objective of endothelial growth is to form new vessels for oxygen, substrates and blood cells, other functions, like antioxidative as well as antienzymatic stress properties, could be carried out before the new vessels, i.e., mature capillaries, are formed [2,9]. During the evolution of chronic liver diseases the establishment of an abnormal splanchnic and systemic angioarchitecture, including portosystemic collaterals, stand out [2,66,67].
Since in portal hypertension the basic structural alteration found in the gastrointestinal tract is vascular and consists in the increased size and the number of vessels, the very appropriate name of “hypertensive portal intestinal vasculopathy” has been proposed [68]. However, in addition to vascular alterations, histological evidence of non-specific inflammation has been described in the gastroenteropathy associated with portal hypertension [68]. Therefore, angiogenesis plays a key role in development of portal hypertension and represents a potential therapeutic target [69].
Chronic inflammatory infiltration found in the small bowel predominantly consists of mononuclear cells, and it is accompanied by atrophy, a decreased villous/crypt ratio, edema of the lamina propia, fibromuscular proliferation, and thickened muscularis mucosa [70,71]. Since most of the aforementioned characteristics can be explained on the basis of increased levels of mast cell me diators [72], these cells could be involved in the pathogenesis of portal hypertensive chronic enteropathy [35, 36]. Furthermore, in experimental portal hypertensive enteropathy, the increased degree of mast cell infiltration coexists with a higher vessel number in these intestinal layers. Indeed, the number of mast cells shows a positive and statistically significant correlation with the vascular diameter and total microvascular surface [73]. Splanchnic hyperemia, increased splanchnic vascularization, and the development of portal-systemic collateral circulation in experimental portal hypertension are all angiogenic processes that are partly vascular endothelial growth factor (VEGF) dependent [74,75]. Given that mast cells release many angiogenic factors, including VEGF [72], they possibly are involved in the promotion of the hypertensive portal vasculopathy, as well as of the portalsystemic collateral circulation.
One characteristic of mast cells is their phenotypic heterogeneity. Mast cell heterogeneity is presumably a result of micro environmental conditions that dictate gene expression and phenotype development [76]. Given the evolutive nature of portal hypertension, the splanchnic mast cell response could be influenced by the predominant phenotype in each evolutive phase.
The splanchnic dependent angiogenic process in portal hypertension, could also represent a reminiscence of the extraembryonic vitelline functions. The yolk sac is an extraembryonic vitelline structure not only related with lipid storage, metabolism and transport [48,49], but also with angiogenesis and hematopoiesis [7,77]. Endothelial cells and hematopoietic stem cells simultaneously differentiate within the blood islands in the yolk sac [77]. Indeed, because hematopoietic stem cells always emerge in close physical association with endothelial cells, a common origin for the two cell types has been proposed [78]. It is generally accepted that hematopoietic stem cells generated in yolk sac blood islands sequentially colonize the embryonic liver, then the aorta-gonads-mesonephros and finally the bone marrow [7]. If so, the excessive splanchnic angiogenic response induced by portal hypertension could be related to the embrionary colonization process by cells bearing a dual hemato-endothelial phenotype. In addition, there is evidence of the association between liver cirrhosis and bone marrow alterations in both rats and patients [79]. Inflammatory mediators may be responsible for increased expression of adhesion molecules in the bone marrow sinusoidal endothelium. However, more investigation is needed to clarify the precise mechanism of how sinusoidal endothelium changes affect the hematopoietic cells [79]. Greater understanding is required pertaining to the relationship of portal hypertension with both processes, i.e., angiogenic and hematopoietic. Thus, the mast cell, a plurifunctional bone-marrow-derived cell, could emulate a rudimentary hematopoietic response in the gastrointestinal tract.
3. HIGH-GRADE INFLAMMATORY PORTAL HYPERTENSION: THE AMNIOTIC CONNECTION
Liver disease could be the most frequent factor for worsening portal hypertensive syndrome. Particularly, chronic liver disease and cirrhosis aggravate this syndrome exceedingly [80]. Hepatic dysfunction related to fibrosis or cirrhosis would aggravate the grade of systemic inflammation characteristic of the chronic portal hypertensive syndrome. Consequently, the vascular dysfunction with both blood and lymphatic hyperdynamic circulation would be favored [9].
3.1. Ascites: The Portal Hypertensive Peritoneum
Increased lymph flow is known to occur in diffuse abnormalities of liver architecture such as fibrosis and cirrhosis [81,82]. The size and number of lymphatics are increased due to the increased hepatic lymph production which is caused by disturbance of the microcirculation typical of portal hypertension [82]. Expansion of lymphatics is also a prominent feature of gastrointestinal inflammation. The dysregulation of lymphatics exacerbates, in turn, gastrointestinal disease [83]. During cirrhotic portal hypertension dilation of esophageal and gastric lymph vessels may be related to the absorption of excess interstitial fluid [84]. However, the main features of liver decompensation in cirrhosis are ascites [85], hepatorenal syndrome [80,86] and hepatic encephalopathy [80].
Ascites and hepatorenal syndrome are the major challenging complications of cirrhosis and portal hypertension that significantly affect the course of the disease [87].
Hepatorenal syndrome is a serious complication of end-stage disease occurring mainly in patients with advanced cirrhosis and ascites, who have marked circulatory dysfunction [86]. Although ascites can be observed in multiple diseases [88], it is most frequent due to cirrhosis with portal hypertension.
The three major factors involved in the pathogenesis of ascites are portal hypertension, arterial vasodilation and neurohormonal activation, all of them leading to sodium and water retention [87]. Arteriolar vasodilation causes underfilling of systemic arterial vascular space with a decrease in the effective arterial blood volume. Consequently, baroreceptor-mediated activation of reninangiotensin-aldosterone system, sympathetic and parasympathetic nervous systems and non-osmotic release of antidiuretic hormone occur to restore normal blood homeostasis [87,89-91]. On the other hand, splanchnic vasodilation increases splanchnic lymph production exceeding the lymph transportation system capacity and leads to lymph leakage into the peritoneal cavity [92]. Persistent renal sodium and water retention, alongside increased splanchnic vascular permeability in addition to lymph leakage into peritoneal cavity play the major role in a sustained ascites formation [90,92]. Overtime the stasis of lymph flow in lymphatic channels of the intestine could lead to lymphangiectasia with an accompanying loss of proteins and lymphocytes [93].
In the past, it was considered that ascites treatment depends on a reduction of lymph formation by indirect i.e. dietary restriction of salt and water, and diuretic drugs, or direct, i.e. portosystemic shunt, portal decompression or, alternatively, and acceleration of an already rapid lymph return (peritoneovenous shunt) to match the high rate of lymph production. Conversely, factors that favor lymph formation i.e. mineralocorticoids, exogenous salt, or inhibit lymph return i.e. impaired lymphatic contractility or central venous hypertension, intensify lymph imbalance and worsen ascites [92,94].
Other mechanisms proposed to be involved in ascites formation are based on hepatorenal reflex, secondary to a rapid increase in sinusoidal hepatic pressure [92,95] and the splenorenal reflex-mediated reduction in renal vascular conductance, which exacerbates sodium and water retention in the kidneys and may eventually contribute to renal dysfunction and, consequently, ascites formation [96].
The development of ascites is a major complication of cirrhosis that induces an impaired quality of life and decreases survival. The most difficult patients to treat are those with refractory ascites, which are characterized by a lack of response to diuretic treatment [97]. Spontaneous bacterial peritonitis is a frequent and severe complication of decompensated cirrhosis [98]. Bacterial translocation is the key mechanism in its pathogenesis and the common causative microorganisms are gram-negative bacteria such as Escherichia coli and Klebsiella pneumoniae [99]. The clinical manifestations of spontaneous bacterial peritonitis are subtle and require a high index of suspicion and almost always occur in a large volume of ascites in patients with liver cirrhosis. Abdominal pain can be continuous and is different from tense ascites. Ascitic polymorphonuclear leukocyte cell count is essential for diagnosis and management [98,99]. However, by means of a polymerase chain reaction (PCR)-based method and automated nucleotide sequencing is possible to detect and identify the presence of fragments of bacterial DNA in patients with culture-negative, non-neutrocytic ascites, indicating the existence of bacterial DNA translocation [100]. Paralysis ileus, hypotension and hypothermia are seen in advanced illness, where prognosis may be death and one third of patients will develop renal failure [90,99].
Secondary bacterial peritonitis is an infrequent complication in cirrhotic patients and presents a significant more severe local inflammatory response than in patients with spontaneous bacterial peritonitis [100,101]. On the contrary, “bacteriascites” is the term used to describe the colonization of ascitic fluid by bacteria in the absence of a local inflammatory reaction, which suggests the concurrent failure of defensive mechanisms [99].
3.2. Splanchnic Interstitium, Mesenteric Lymphatics and Peritoneal Mesothelium: A Hypothetically Inflammatory Continuum in the Portal Hypertensive Syndrome
The standard view of inflammation as a reaction to injury or infection might need to be expanded to account for the inflammatory processes induced by other types of adverse conditions [102]. Therefore, the splanchnic and systemic impairments that are produced during the evolution of the syndrome associated with portal hypertension could be considered of an inflammatory nature. If so, and similar to other type of inflammatory response, it would begin in the interstitial space [2,3,29] (Table 2).

Table 2. The hypothetical continuum inflammatory splanchnic pathway in decompensated portal hypertension.
The early inflammatory response in portal hypertension could be associated with abnormal ion transport. There is increasing evidence that the conditions characterized by an inflammatory response have alterations in cellular membrane potential with depolarization and abnormal ion transport [103]. In addition, disturbances of ion transport are produced in intraand extracellular edema. It has been stated that small fluctuations in cell hydration or cell volume act as a potent signal for cellular metabolism and gene expression [104]. Oxidative and nitrosative stress increase degradation of extracellular matrix and causes edema [105]. The accumulation of glycosaminoglycans fragments has been proposed as an important mechanism for edema formation due to its hydrophilic properties [106]. Likewise, while the progression of interstitial edema reduces the blood capillary function, it simultaneously enhances lymphatic circulation, thus producing a circulatory switch in the inflamed tissues and organs.
Furthermore, splanchnic interstitial flow could be relevant for lymphangiogenesis [107]. The interstitial fluid flow associated with edema, even though it can be extremely slow, can have important effects on tissue morphogenesis and function, cell migration and differentiation and matrix remodeling, among other processes [108]. Abnormally increased interstitial flow rates can occur during low-grade inflammation and can also trigger fibroblasts to differentiate or remodel the extracellular matrix, contributing thus to the development of tissue fibrosis [107,109-112]. Interstitial flow may significantly alter the distribution of metalloproteinases and lymphatic growth factors, inducing lymphatic endothelial cell migration and capillary morphogenesis [107,110]. Mast cells and lymphatics could be important players in the development of the splanchnic inflammatory process [9]. Thus, it has been shown that in vitro mast cell degranulation impairs lymphatic contractile activity, probably through activation of H1 receptors by histamine. It has been suggested that this action could interfere with the expected ability of lymphatic vessels to reduce edema during inflammation [113].
Two of the principal functions of intestinal lymphatics are to assist in the maintenance of interstitial volume within relatively normal limits during alterations in capillary filtration, i.e. acute portal hypertension, and the removal of absorbed water and chylomicrons [114]. Unidirectional fluid transport into the initial lymphatics from the splanchnic interstitial space could be facilitated by the primary and secondary valve systems and then by contractile lymphatics [115,116]. During inflammation the elevation of lymphatic endothelial permeability in an outward direction has two major effects. First, fluid is being cleared from the tissue less efficiently. With the increased permeability of the blood vessels already producing more fluid in the tissue than normal, the decreased transport by the lymphatics causes this edema to increase even more [116,117]. Second, the leaking lym phatics allow inflammatory mediators to remain in the tissue longer increasing thus the inflammatory intestinal response [116] (Figure 2).
Mesenteric lymph transports cells (lymphocytes), lipids (chylomicrons), proteins (plasma proteins, immunoglobulins), enzymes (alkaline phosphatase, amylase), hormones (insulin, incretins) and electrolytes (chloride and bicarbonate) to the systemic circulation [115,118]. The biological role of the mesenteric lymph in the pathogenesis of splanchnic and systemic inflammation is not fully clarified up to date. However, there is now evidence to show that mesenteric lymph plays a key role in the pathogenesis of multiple organ dysfunction in trauma/ hemorrhagic shock, burns, reperfusion injury and surgical stress [41,42,115,119-121].
The hemodynamic alterations that portal hypertension imposes on the splanchnic circulation [16,80] allows considering that changes in the mesenteric lymph flow and composition are also produced [9]. Therefore, the mesenteric lymph could play an etiopathogenic role in

Figure 2. Schematic view of the evolutive phases in the formation of the interstitial-lymphatic-peritoneal mesothelium inflammatory axis in portal hypertensive ascites: a) Splanchnic lymphatic flow resulting from the gastrointestinal tract (GIT) and the hepatic (H) interstitium drained through the mesenteric lymph node (MLN) in physiological situations; b) Chronic-lowgrade inflammation in the compensated portal hypertensive syndrome increases mesenteric lymphatic flow; c) Acute-on-chronic splanchnic inflammation in the decompensated portal hypertensive syndrome increases the ascitic fluid.
the multiple organ dysfunction developed in the portal hypertensive syndrome [16]. Particularly, bacteria and toxins, like bacterial lipopolysaccharide (LPS), and inflammatory mediators with high affinity for chylomicrons, that is lipids, could use the mesenteric lymph vessels to bypass the liver and induce a systemic response [122].
Lymphangiogenesis, the formation of new lymphatic vessels, occurs in several pathological conditions ssociated with chronic inflammation [123,124], including portal hypertensive enteropathy [84] and liver cirrhosis [81,82]. Inflammatory cells, through the secretion of stimulatory factor such as VEGF-C and TNF-α, can stimulate lymphatic endothelial cells [125-127] and consequently regulate gut lymphatic remodeling in portal hypertensive enteropathy. Lymphangiogenesis generally accompanies angiogenesis [128], the basic structural alteration of the hypertensive portal gastroenteropathy [68]. However, pathological lymphangiogenesis may occur in absence of blood vessels [127,129].
The biological role of lymphangiogenesis in the pathogenesis of gastrointestinal chronic inflammation needs further clarification. Inflammation triggers gut lymphangiogenesis [130-133], and this may be beneficial for the resolution of chronic inflammation since lymphatic vessels remove inflammatory cells and mediators from the inflammation sites, favoring resolution [133-135]. Lymphovenous communications located both in lymphatic vessels and mesenteric lymph nodes [115] could also have a pathophysiological significance in the setting of chronic intestinal inflammation that need to be explored. Furthermore, one highly specialized form of tissue remodeling in chronic inflammation is lymphoid neogenesis, that is, the development of new lymphoid tissue in inflammatory sites [134,136]. It has been proposed that during chronic inflammation lymphangio-genesis and lymphoid neogenesis would be synergistic processes that mutually amplify each other [136].
Severe hepatic dysfunction related to fibrosis or cirrhosis would aggravate the grade of splanchnic inflammation in portal hypertension, and as a result would increase the incidence of complications. Consequently, the vascular splanchnic dysfunction, with increased blood flow and portal pressure, would get worse and the interstitial hepato-intestinal-lymph flow would be favored. Splanchnic ischemia-reperfusion injury secondary to the acute-on-chronic vascular dysfunction could result in higher reninangiotensin-aldosterone system activity and excessive interstitial edema and lymph flow [137,138]. The liver has been assumed to be the likely source of ascites in patients with liver disease [81]. Nevertheless, the cirrhotic liver is not the sole or even the major source of ascites in most patients [89,94]. Weeping of fluid in great excess from the peritoneal lining and serosal surfaces of the bowel is often striking in portal hypertension as are dilated lymphatics on the surface of the small intestine, in the mesentery and within the retroperitoneal space [94]. Therefore, it is accepted that when lymphatic drainage mechanisms are overwhelmed, excess lymph collects in the peritoneal cavity, thus causing ascites [90, 139,140] (Figure 2).
Nevertheless, peritoneal mesothelial cells would not be considered as a passive barrier for the lymph leakage of splanchnic origin in ascites formation. In the normal peritoneal cavity mesothelial cells play an important role as a source of intraperitoneal phospholipid lubricant [141]. Surfactant proteins have been found to be associated with the phospholipid-containing surfactant-like material that line mesothelial cells and exert anti-inflammatory and immunomodulatory functions [142]. In addition, the overlapped peritoneal mesothelial cells have a surface covered with great number of microvilli and a cytoplasm filled with a greater number of ribosomes, mitochondria, rough endoplasmic reticulum and Golgi apparatus [143]. Particularly, the numerous pinocytic vesicles in the membrane and the cytoplasm indicate active endoand exo-transcytosis in the process of secretion and reabsorption of peritoneal fluid [143]. Peritoneal mesothelial cells by secreting chemokines, cytokines and growth factors such as VEGF [144] could have an active participation in the inflammatory response as a developer in the intestinal wall in portal hypertensive syndrome and, therefore, in ascites formation.
3.3. Obstructive Cholestasis in the Rat: A Model of Decompensated Portal Hypertensive Syndrome
Obstructive cholestasis is characterized by jaundice, discolored urine, pale stools and pruritus [145]. The serious repercussions of cholestasis on the liver and on the systemic level have led to the creation of many experimental models so as to better understand its pathogenesis, prophylaxis and treatment [146,147].
Obstructive cholestasis causes cirrhotic chronic hepatic insufficiency and portal hypertension. Several surgical techniques for developing obstructive cholestasis have been described, especially in the rat. These techniques could be divided into two groups, macrosurgical and microsurgical. Macrosurgical techniques are based on the section of the common bile duct between ligatures. These macrosurgical techniques of obstructive extrahepatic cholestasis are called “bile duct ligation” (BDL) and cause the development of infected biliary pseudocysts by dilation of the bile duct proximal end. As a result an important number of the animals die during the first two weeks of the postoperative period because of sepsis [147,148]. To avoid these infectious complications we have proposed performing a microsurgical technique which consists in the resection of the extrahepatic biliary tract [146,147,149]. The use of broad-spectrum antibiotic and vitamin K allows the long-term evolution of the obstructive cholestatic-rats [146,147].
In the long-term evolution, between 8 to 10 weeks, microsurgical extrahepatic cholestatic-rats develop hepatomegaly with a marked ductular proliferation and fibrosis [150]. It has been suggested that liver fibrogenesis resembles a wound-healing process leading to scar formation [151-153]. In relation to extrahepatic alterations, jaundice, choluria, portal hypertension with an enlarged spleen and collateral portosystemic circulation, hepatic encephalopathy and ascites stand out [147,154,155]. Therefore, experimental extrahepatic cholestasis is not only a good model for studying chronic hepatic disease related to biliary obstruction, but also for studying extrahepatic complications, particularly ascites.
In the rat, chronic liver disease due to obstructive cholestasis produces progressive hemodynamic dysfunction with ascites and hepatorenal syndrome. BDL rats after four weeks of biliary obstruction present an initial disturbance in renal function associated with ascites. Two weeks later, rats with obstructive cholestasis clearly developed hepatorenal syndrome with ascites [156]. In these chronic phases of macrosurgical obstructive cholestasis, high rates of bacterial translocation, with endotoxemia and consecutive systemic inflammatory response, also exist [157,158].
In rats with obstructive cholestasis, the portal hypertensive syndrome with low-degree splanchnic and systemic inflammation can progress to severe systemic inflammatory response syndrome leading to multiple organ failure. After extrahepatic cholestasis, the rat can suffer the effects of generalized ischemia/reperfusion injury and exacerbation of oxidative and nitrosative stress. It is accepted that there is a strong correlation between experimental obstructive jaundice and oxidative stress [159]. BDL mainly impairs the liver rat ability of antioxidant regeneration especially at the mitochondria level [160]. Thus, it has been demonstrated that treatment with antioxidants improves the hepatic cellular redox status [161, 162].
Long-term (6 weeks) microsurgical cholestatic rats show a splanchnic redistribution of cytokines, with an increase of Th1 (TNFα and IL-1β) and Th2 (IL-4 and IL-10) production in the small bowel and in the mesenteric lymph nodes [163]. It has been proposed that this splanchnic inflammatory response associated with ascites could be mediated, among others factors, by mast cells. Degranulation of splanchnic mast cells, including the mast cells present in the peritoneum, could result in the release of mediators with a potent vasodilator and exudative actions such as histamine, proteases and other enzymes, arachidonic acid metabolites and reactive oxygen and nitrogen species [76] which could cause intense exudation related to an endothelial permeability increase, which could be in turn the cause of swelling, increased splanchnic lymphatic flow and production of peritoneal exudate [9].
The suggested pathophysiological mediator role of the mesenteric lymph in worsening the splanchnic portal hypertensive inflammatory response induced by obstructtive cholestasis can be studied by the cannulation of the rat mesenteric lymph duct [164-166]. The mesenteric lymph duct in the rat is observed as a white dyed duct, approximately 10 mm above the origin of the left adrenal vein, and is accompanied by the mesenteric artery. Using an operating microscope, the connective tissue surrounding the duct could be carefully cleared. In brief, the cannulation technique is made using a heparinized polyethylene catheter with an outer diameter of 1 mm. Then, a ligature is placed around the duct and the catheter. Lastly a small drop of cyanoacrylate glue or tissue adhesive is applied to the duct at the point of entry of the catheter to prevent lymph leakage [164,166].
The relative distribution profiles of protein functional classes in normal rodent mesenteric lymph differ signifycantly from that reported for the plasma. The most abundant protein classes in mesenteric lymph are protease inhibitors, immune-related proteins, particularly those implicated in innate immunity, and carrier proteins [167]. Therefore, mesenteric lymph has a unique profile compared with plasma and thus represents more than a simple filtrate [167]. Recent proteomic analyses of posthemorrhagic shock mesenteric lymph have documented the increase of proteins functionally involved in tissue inflammation [168]. These results provide a starting point for the in-depth investigation of the pathophysiology role of mesenteric lymph in other conditions in which it is considered that the gastrointestinal tract is the driving force behind multiple organ dysfunctions, like the portal hypertensive syndrome. In rats with long-term surgical cholestasis we have shown using the transcription factor LYVE-1, as a marker of lymphangiogenesis [169], the existence of intestinal lymphatic hyperplasia. In this experimental model subperitoneal lymphangiogenesis associated with ascites is also developed.
Lastly, ascites produces intra-abdominal hypertension. Elevated intraabdominal pressure could compresses thin walled mesenteric veins promoting venous hypertension, intestinal interstitial edema, bacterial translocation, sepsis and multiple organ failure [170-172].
3.4. Portal Hypertensive Ascites and the Amniotic Way of Life
Ascites is a common manifestation of liver failure, being one of the cardinal signs of portal hypertension [173]. Ascitic fluid formation is a not well known pathogenic mechanism. However, ascitic fluid is a bioactive medium containing electrolytes, with high levels of sodium, proteins including albumin and enzymes, as well as cells, including leukocytes [174]. Some of these characteristics make it similar to another bioactive medium, the amniotic fluid [175-177]. Amniotic fluid, the protecting liquid contained in the amnion cavity, is an essential extraembryonic component for fetal development and maturation during pregnancy [176,178].
Body fluid is distributed among three major fluid spaces: intracellular fluid, interstitial fluid and plasma. Nevertheless, the distribution of fluid in each of these compartments is dramatically different in the fetus compared with the adult [179]. Particularly, the amniotic fluid that surrounds the fetus may be considered an extension of the extracellular space of the fetus [178,179]. Thus, the lymphatic system plays an essential role in the regulation of fluid distribution between the plasma and the interstitial fluid and probably with the amniotic fluid [179]. In patients with portal hypertension and ascites, it could also be hypothesized that the lymphatic splanchnic system plays a key role in the fluid distribution between the plasma and the ascitic fluid. If so, a pathological pathway of increasingly complex structures with a similar function to the management of the extracellular fluid would be formed [9] (Figure 3).

Figure 3. Comparative and schematic representation of the fetal (left) and portal hypertensive (right) gastrointestinal tract. Amniotic fluid through gastrointestinal tract of the embryo favors trophism and maturation. On the contrary, the proposed equivalent fluid, i.e. ascitic fluid, in the adult organism is confined within the peritoneal mesothelial cavity. Am: amniotic fluid; As: ascitic fluid; Y: yolk sac; L-O: liver and great omentum; GIT: gastrointestinal tract.
In the early stages of pregnancy, amniotic fluid consists of a filtrate of maternal blood [177,180]. That is why in this period drugs taken by the mother can enter amniotic fluid by diffusion across the placenta [177,181]. However, its composition is known to change as pregnancy proceeds [175]. At these stages amniotic fluid is a bioactive medium actively secreted by the cells lining the amniotic cavity and as gestation progresses it includes significant volume of fetal urine [182].
The hypothetical comparison of the characteristics of amniotic and ascitic fluids would oblige raising again the role of peritoneal mesothelial cells in the etiopathogeny of ascites that occurs in the decompensated portal hypertensive syndrome.
The first fluid to enter the gastrointestinal system is amniotic fluid and contributes to fetal nutritional requirements and plays a significant role in gut development and maturation [182]. Thus, growth-promoting effects of amniotic fluid are equivalent to human milk [182]. The trophic effect of orally consumed amniotic fluid is attributed in part to its content in growth factors, including epidermal growth factor (EGF), hepatocyte growth factor (HGF), transforming growth factor-alpha (TGF-α), fibroblast growth factor (FGF), insulin-like growth factor (IGF-s) and VEGF [182,183].
The functional comparison of amniotic and ascitic fluids would imply that in the decompensated portal hypertensive syndrome the abdominal mesothelium acquires properties of the amniotic membrane or amnion. In essence, the body acquires the ability to express functions that correspond to a structure of extra-embrionary origin, the amnion. This hypothesis would imply several suppositions or suggestions. For example, the intestine in the case of portal hypertensive ascites could not benefit from the supposed trophic properties of the ascitic fluid given that the peritoneal cavity-gastrointestinal pathway doesn’t exist in post-natal life. Likewise, the prenatal interruption of the amniotic fluid transit in cases of prenatal intestinal obstruction prevents the fetus from benefiting from its trophic properties and it has been suggested that it contributes to fetal undergrowth [184].
The amniotic membrane is a tissue of particular interest because it possesses cells characteristic of stem cells with multipotent differentiation ability [185,186]. Therefore, to explore the possibilities that the “ascitic peritoneum” could be a source of stem cells and growth factors could open new paths of knowledge for the study of its pathogeny. Moreover, the ascitic fluid could be a source of powerful therapies for using its supposedly beneficial properties. The embryonic regression of the peritoneum that induces the inflammatory response induced by portal hypertension also could favor the presence of active antimicrobial components as those described in the amniotic fluid [187]. Thus, the resistance to infection of the ascites would be explained, with usual subtle clinical manifestations in cases of secondary bacterial peritonitis [97,100,101].
The hypothetical existence of a regression to the prenatal functional mechanisms of the abdominal interstitium-lymphatic-mesothelium axis could serve to integrate the existing knowledge of the decompensated portal hypertensive syndrome. In this hypothesis is considered that the main objective of this de-repression of “dormant” biochemical functions is the recuperation of the amnion. The major phylogenetic importance that this structure has perhaps lies in that during development it protectively surrounds the embryo and creates a fullfilled cavity in which the embryo develops [178]. Its acquisition, that is, the “amniotic egg” was one of the main causes that enabled amniotes to escape the bonds that confined their ancestors to aquatic environments [178,188]. If so, it could be proposed that there this is no better method to escape the sea than taking the sea with us, as an amniotic-mesothelial structure full of saline trophic fluid [188,189]. Since this ability would be associated in the post-natal life with mesothelial cells, its major spread through the body is not surprising, coating cavities, i.e. aracnoides, sinovial, pleura, testicular and vaginal, to create virtual spaces that facing traumatic, infectious or tumoral injury, quickly carry out an exudative response that is simultaneously protecting and trophic. Therefore, although the mesothelial exudative inflammatory response is considered pathological, its proven evolutive and beneficial characteristics should not be forgotten both from the ontogenic and phylogenetic points of view [9].
4. CONCLUSIONS
In summary, the extraembryonic origin of the portal system seems to determine some of its evolutive pathological properties when it suffers hypertension, both compensated, i.e. prehepatic, and decompensated, i.e. intrahepatic. The partial portal vein ligation, or compensated, and the extrahepatic cholestasis or decompensated, are the experimental models most frequently used to study both types of portal hypertension [190]. Prehepatic portal hypertension by partial portal vein ligation in the rat induces the expression of three overlapping phenotypes, whose pathophysiological mechanisms recall those that are associated with the yolk sac development or vitallogenics. In turn, in obstructive cholestatic-rats the decompensation induced by the severe liver disease would produce ascites. The functional comparison between the ascitic and amniotic fluids would imply that in the decompensated portal hypertensive syndrome the abdominal mesothelium acquires properties of the amnion.
The expression of these extraembryonic functions during the evolution of these two types of experimental portal hypertension can be subject to diverse interpretations. Regardless of its meaning, it is important that the re-expression of these extraembryonic functions induced by portal hypertension might be considered today in inflammation. This consideration suggests that the hypothetical comparison between the inflammatory portal hypertensive evolutive types and the evolutive development of extraembryonic systems, i.e. vitelline and amniotic, could allow for translational research.
5. ACKNOWLEDGEMENTS
The authors are indebted to Maria Elena Vicente for preparing the manuscript and Elisabeth Mascola for translating it into English. This study was supported in part by grants from the Mutua Madrileña Foundation (FMM. Ref. n AP 69772009), and from the National Department of Science and Innovation (MICINN Ref. n PSIC2010- 19348).
![]()
![]()
REFERENCES
- Martell, M., Coll, M., Ezkurda, N., Raurell, I. and Genesca, J. (2010) Physiopathology of splanchnic vasodilation in portal hypertension. World Journal of Hepatology, 2, 208-220. doi:10.4254/wjh.v2.i6.208
- Aller, M.A., Arias, J.L., Cruz, A. and Arias, J. (2007) Inflammation: A way to understanding the evolution of portal hypertension. Theoretical Biology and Medical Modeling, 4, 44. doi:10.1186/1742-4682-4-44
- Aller, M.A., Arias, J.L. and Arias, J. (2007) The mast cell integrates the splanchnic and systemic inflammatory response in portal hypertension. Journal of Translational Medicine, 5, 44. doi:10.1186/1479-5876-5-44
- Cazzaniga, M., Dionigi, E., Gobbo, G., Fioretti, A., Monti, V. and Salerno, F. (2009) The systemic inflammatory response syndrome in cirrhotic patients: Relationship with their in-hospital outcome. Journal of Hepatology, 51, 475- 482. doi:10.1016/j.jhep.2009.04.017
- Malik, R., Mookerjee, R.P. and Jalan, R. (2009) Infection and inflammation in liver failure: Two sides of the same coin. Journal of Hepatology, 51, 426-429. doi:10.1016/j.jhep.2009.06.013
- Sherlock, S. (1989) The portal venous system and portal hypertension. In: Sherlock, S., Ed., Diseases of the liver and biliary system, Blackwell Scientific Publications, London, 151-207.
- Collardeu-Frachon, S. and Scoazec, J.Y. (2008) Vascular development and differentiation during human liver organogenesis. The Anatomical Record, 291,614-621. doi:10.1002/ar.20679
- Dobreva, M.P., Pereira, P.N.G., Deprest, J. and Zwijsen, A. (2010) On the origin of amniotic stem cells: Of mice and men. The International Journal of Developmental Biology, 54, 761-777. doi:10.1387/ijdb.092935md
- Aller, M.A., Prieto, I., Argudo, S., de Vicente, F., Santamaria, L., de Miguel, M.P., Arias, J.L. and Arias, J. (2010) The interstitial lymphatic peritoneal mesothelium axis in portal hypertensive ascites: When in danger, go back to the sea. International Journal of Inflammation, 2010, 18 p. doi:10.4061/2010/148689
- Aller, M.A., Nava, M.P., Duran, M., Chivato, T., Arias, J.L., Sánchez-Patan, F., de Vicente, F., Alvárez, E. and Arias, J. (2007) Evolutive phases of the experimental prehepatic portal hypertension. Journal of Gastroenterology & Hepatology, 22, 1127-1133. doi:10.1111/j.1440-1746.2007.04876.x
- Groszmann, R.J. (1994) Hyperdinamic circulation of liver disease forty years later: Pathophysiology and clinical consequence. Hepatology, 20, 1359-1363. doi:10.1002/hep.1840200538
- Iwakiri, Y. and Groszmann, R.J. (2006) The hyperdynamic circulation of chronic liver diseases: From the patient to the molecule. Hepatology, 43, S121-131. doi:10.1002/hep.20993
- Moreau, R. and Lebrec, D. (2006) Molecular and structural basis of portal hypertension. Clinics in Liver Disease, 10, 445-457. doi:10.1016/j.cld.2006.08.011
- Rodriguez-Vilarrupla, A., Fernandez, M., Bosch, J. and Garcia-Pagan, J.C. (2007) Current concepts on the pathophysiology of portal hypertension. Annals of Hepatology, 6, 28-36.
- La Villa, G. and Gentilini, P. (2008) Hemodynamic alterations in liver cirrhosis. Molecular Aspects of Medicine, 29, 112-118. doi:10.1016/j.mam.2007.09.010
- Iwakiri, Y. and Groszmann, R.J. (2007) Vascular endothelial dysfunction in cirrhosis. Journal of Hepatology, 46, 927-934. doi:10.1016/j.jhep.2007.02.006
- Harrison, D.G., Widder, J., Grumbach, I., Chen, W., Weber, M. and Searles, C. (2006) Endothelial mechanotransduction, nitric oxide and vascular inflammation. Journal of Internal Medicine, 259, 351-363. doi:10.1111/j.1365-2796.2006.01621.x
- Chiu, J.-J. and Chien, S. (2011) Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiological Reviews, 91, 327-387. doi:10.1152/physrev.00047.2009
- Colgan, S.P. and Taylor, C.T. (2010) Hypoxia: An alarm signal during intestinal inflammation. Nature Reviews. Gastroenterol & Hepatology, 7, 281-287.
- Carrico, C.J., Meakins, J.L., Marshall, J.C., Fry, D. and Maier, R.V. (1986) Multiple-organ-failure-syndrome: The gastrointestinal tract. The “motor” of MOF. Archives of Surgery, 121, 196-208. doi:10.1001/archsurg.1986.01400020082010
- Suliburk, J., Helmer, K., Moore, F. and Mercer, D. (2008) The gut in systemic inflammatory response syndrome and sepsis. European Surgical Research, 40,184-189. doi:10.1159/000110859
- Deitch, E.A. (2002) Bacterial translocation or lymphatic drainage of toxic products from the gut: What is important in human beings? Surgery, 131, 241-244. doi:10.1067/msy.2002.116408
- Alexander, J.S., Ganta, V.C., Jordan, P.A. and Witte, M.H. (2010) Gastrointestinal lymphatics in health and disease. Pathophysiology, 17, 315-335. doi:10.1016/j.pathophys.2009.09.003
- Gershon, M.D. (2010) Developmental determinants of the independence and complexity of the enteric nervous system. Trends in Neuroscience, 33, 446-454. doi:10.1016/j.tins.2010.06.002
- Lyte, M. (2009) Reciprocal gut-brain evolutionary symbiosis provokes and amplifies the post injury systemic inflammatory response syndrome. Surgery, 146, 950-954. doi:10.1016/j.surg.2009.06.002
- Liu, H., Schuelert, N., McDougall, J.J. and Lee, S.S. (2008) Central neural activation of hyperdynamic circulation in portal hypertensive rats depends on vagal afferent nerves. Gut, 57, 966-973. doi:10.1136/gut.2007.135020
- Schrier, R.W., Arroyo, V., Bernardi, M., Epstein, M., Henriksen, J.H. and Rodes, J. (1988) Peripheral arterial vasodilatation hypothesis: A proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology, 8, 1151-1157. doi:10.1002/hep.1840080532
- Stocker, S.D., Madden, C.J. and Sved, A.F. (2010) Excess dietary salt intake alters the excitabiligy of central sympathetic networks. Physiology & Behavior, 100, 519- 524. doi:10.1016/j.physbeh.2010.04.024
- Aller, M.A., Arias, J.L., Nava, M.P. and Arias, J. (2004) Post-traumatic inflammation is a complex response based on the pathological expression of the nervous, immune and endocrine functional systems. Experimental Biology and Medicine, 229, 170-181.
- Garcia-Tsao, G., Albillos, A., Barden, G.E. and West, A.B. (1993) Bacterial translocation in acute and chronic portal hypertension. Hepatology, 17, 1081-1085. doi:10.1002/hep.1840170622
- Garcia-Tsao, G. and Wiest, R. (2004) Gut microflora in the pathogenesis of the complications of cirrhosis. Best Practice & Research Clinical Gastroenterology, 18, 353- 372. doi:10.1016/j.bpg.2003.10.005
- Llamas, M.A., Aller, M.A., Marquina, D., Nava, M.P. and Arias, J. (2010) Bacterial translocation to mesenteric lymph nodes increases in chronic portal hypertensive rats. Digestive Diseases and Sciences, 55, 224-254. doi:10.1007/s10620-009-1001-3
- Gabay, C. and Kushner, I. (1999) Acute-phase proteins and other systemic responses to inflammation. The New England Journal of Medicine, 340, 448-454. doi:10.1056/NEJM199902113400607
- Cray, C., Zaias, J. and Altman, N.H. (2009) Acute phase response in animals: A review. Comparative Medicine, 59, 517-526.
- Prieto, I., Aller, M.A., Santamaria, L., Nava, M.P., Madero, R., Perez-Robledo, J.P. and Arias, J. (2005) Prehepatic portal hypertension produces increased mast cell density in the small bowel and in mesenteric lymph nodes in the rat. Journal of Gastroenterology and Hepatology, 20, 1025-1031. doi:10.1111/j.1440-1746.2005.03831.x
- Moquillaza, L.M., Aller, M.A., Nava, M.P., Santamaria, L., Vergara, P. and Arias, J. (2010) Partial hepatectomy, partial portal vein stenosis and mesenteric lymphadenectomy increase splanchnic mast cell infiltration in the rat. Acta Histochemica, 112, 372-382. doi:10.1016/j.acthis.2009.03.002
- McLachlan, J.B., Hart, J.P., Pizzo, S.V., Shelburne, C.P., Shelburne, C.P., Staats, H.F., Gunn, M.D. and Abraham, S.N. (2003) Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nature Immunology, 4, 1199-1205. doi:10.1038/ni1005
- Bischoff, S.C. (2009) Physiological and pathophysiological functions of intestinal mast cells. Semininars in Immunopathology, 31, 185-205. doi:10.1007/s00281-009-0165-4
- Cäruntu, F.A. and Benea, L. (2006) Spontaneous, bacterial peritonitis: Pathogenesis, diagnosis, treatment. Journal of Gastrointestinal and Liver Disease, 15, 51-56.
- Jung, C., Hugot, J.-P. and Barreau, F. (2010) Peyer’s patches: The immune sensors of the intestine. International Journal of Inflammation, 2010, Article ID: 823710. doi:10.4061/2010/823710
- Magnotti, L.J. and Deitch, E.A. (2005) Burns, bacterial translocation, gut barrier function, and failure. Journal of Burn Care Rehabilitation, 26, 383-391. doi:10.1097/01.bcr.0000176878.79267.e8
- Damie, S.S., Moore, E.E., Nydam, T.L., Banerjee, M., Gamboni-Robertson, F., Su, X., and Banerjee, A. (2007) Post-shock mesenteric lymph induces endothelial NF-κB activation. Journal of Surgical Research, 143, 136-140. doi:10.1016/j.jss.2007.04.016
- Aller, M.A., Vara, E., Garcia, C., Nava, M.P., Angulo, A., Sanchez-Patan, F., Calderon, A., Vergara, P. and Arias, J. (2006) Hepatic lipid metabolism changes in shortand long-term prehepatic portal hypertensive rats. World Journal of Gastroenterology, 12, 6828-6834.
- Alonso, M.J., Aller, M.A., Corcuera, M.T., Nava, M.P., Gomez, F., Angulo, A. and Arias J. (2005) Progressive hepatocytic fatty infiltration in rats with prehepatic portal hypertension. Hepato-Gastroenterology, 52, 541-546.
- Sanchez-Patan, F., Anchuelo, R., Aller, M.A., Vara, E., García, C., Nava, M.P. and Arias, J. (2008) Chronic prehepatic portal hypertension in the rat: Is it a type of metabolic inflammatory síndrome? Lipids in Health and Disease, 7, 4. doi:10.1186/1476-511X-7-4
- Kim, L.J., Nalls, M.A., Eiriksdottir, G., Sigurdsson, S., Launer, L.J., Koster, A., Chaves, P.H., Jonsdottir, B., Garcia, M., Gudnason, V., Harris, T.B. and AGES-Reykjavik Study Investigators (2011) Association of visceral and liver fat with the metabolic syndrome across the spectrum of obesity: The AGES-Reykjavik study. Obesity, 19, 1265- 1271. doi:10.1038/oby.2010.291
- Ibrahim, M.M. (2010) Subcutaneous and visceral adipose tissue: Structural and functional differences. Obesity Reviews, 11, 11-18. doi:10.1111/j.1467-789X.2009.00623.x
- Tofail, M. and Takeda, M. (2008) Molecular characteristics of insect vitellogenins. Journal of Insect Physiology, 54, 144714-144758.
- Arukwe, A. and Goksφyr, A. (2003) Eggshell and egg yolk proteins in fish; hepatic proteins for the next generation: Oogenetic, population, and evolutionary implications of endocrine disruption. Comparative Hepatology, 2, 4. doi:10.1186/1476-5926-2-4
- Sniderman, A., Couture, P. and De Graaf, J. (2010) Diagnosis and treatment of apolipoprotein B dyslipoproteinemias. Nature Reviews. Endocrinology, 6, 335-346. doi:10.1038/nrendo.2010.50
- Targher, G., Day, C.P. and Bonora, E. (2010) Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. New England Journal of Medicine, 363, 1341-1350. doi:10.1056/NEJMra0912063
- Jahangiri, A. (2010) High-density lipoprotein and the acute phase response. Current Opinion in Endocrinology, Diabetes, and Obesity, 17, 156-260. doi:10.1097/MED.0b013e328337278b
- Tarantino, G., Savastano, S. and Colao, A. (2010) Hepatic steatosis, low-grade chronic inflammation and hormona/growth factor/adipokine imbalance. World Journal of Gastroenterology, 16, 4773-4783. doi:10.3748/wjg.v16.i38.4773
- Eckersall, P.D. and Bell, R. (2010) Acute phase proteins: Biomarkers of infection and inflammation in veterinary. Veterinary Journal, 185, 23-27. doi:10.1016/j.tvjl.2010.04.009
- Schreiber, G., Tsykin, A., Aldred, A.R., Thomas, T., Fung, W.P., Dickson, P.W., Cole, T., Birch, H., De Jong, F.A. and Milland, J. (1989) The acute phase response in the rodent. Annals of the New York Academy of Sciences, 557, 61-85. doi:10.1111/j.1749-6632.1989.tb24000.x
- Kopf, M., Bachmann, M.F. and Marsland, B.J. (2010) Averting inflammation by targeting the cytokine environment. Nature Reviews Drug Discovery, 9, 703-718. doi:10.1038/nrd2805
- Tordjman, J., Poitou, C., Hugol, D., Bouillot, J.L., Basdevant, A., Bedossa, P., Guerre-Millo, M. and Clement, Kl. (2009) Association between adipose tissue macrophages and liver histopathology in morbid obesity: Influence of glycemic status. Journal of Hepatology, 51, 354- 362. doi:10.1016/j.jhep.2009.02.031
- Marsland, A.L., McCaffery, J.M., Muldoon, M.F. and Manuck, S.B. (2010) Systemic inflammation and the metabolic syndrome among middle-aged community volunteers. Metabolism, 59, 1801-1088. doi:10.1016/j.metabol.2010.05.015
- Solinas, G. and Karin, M. (2010) JNK1 and IKKβ: Molecular links between obesity and metabolic dysfunction. FASEB Journal, 24, 2596-2609. doi:10.1096/fj.09-151340
- [61] Sparks, J.D., Cianci, J., Jokinen, J., Chen, L.S. and Sparks, C.E. (2010) Interleukin-6 mediates hepatic hypersecretion of apolipodrotein B. American Journal of Physiology, Gastrointestinal and Liver Physiology, 299, G980-G989. doi:10.1152/ajpgi.00080.2010
- [62] Van der Westhuyzen, D.R., De Beer, F.C. and Webb, N.R. (2007) HDL cholesterol transport during inflammation. Current Opinion in Lipidology, 18, 147-151. doi:10.1097/MOL.0b013e328051b4fe
- [63] Natarajan, P., Ray, K.K. and Cannon, C.P. (2010) Highdensity lipoprotein and coronary heart disease. Journal of the American College of Cardiology, 55, 1283-1239. doi:10.1016/j.jacc.2010.01.008
- [64] Loria, P., Lonardo, A. and Targher, G. (2008) Is liver fat detrimental to vessels: Intersections in the pathogenesis of NAFLD and atherosclerosis. Clinical Science, 115, 1- 12. doi:10.1042/CS20070311
- [65] de Las Heras, N., Aller, M.A., Martín-Fernández, B., Miana, M., Ballesteros, S., Regadera, J., Cachofeiro, V., Arias, J. and Lahera, V. (2012) A wound-like inflammatory aortic response in chronic portal hypertensive rats. Molecular Immunology, 51, 177-187. doi:10.1016/j.molimm.2012.03.016
- [66] Puxeddu, I., Ribatti, D., Crivallato, E. and Levi-Schaffer, F. (2005) Mast cells and eosinophils: A novel link between inflammation and angiogenesis in allergic diseases. Journal of Allergy and Clinical Immunology, 116, 531- 536. doi:10.1016/j.jaci.2005.06.007
- [67] Coulon, S., Heindryckx, F., Geerts, A., Van Steenkiste, C., Colle, I. and Van Vlierberghe, H. (2011) Angiogenesis in chronic liver disease and its complications. Liver International, 31, 146-162. doi:10.1111/j.1478-3231.2010.02369.x
- [68] Thabut, D. and Shah, V. (2010) Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: New targets for the treatment of portal hypertension? Journal of Hepatology, 53, 976-980. doi:10.1016/j.jhep.2010.07.004
- [69] Viggiano, T.R. and Gostout, C.J. (1992) Portal hypertensive intestinal vasculopathy: A review of the clinical endoscopic and histopathological features. American Journal of Gastroenterology, 87, 944-954.
- [70] Rondonotti, E., Villa, F., Signorelli, C. and De Francis, R. (2006) Portal hypertensive enteropathy. Gastrointestinal Endoscopy Clinics of North America, 16, 277-286. doi:10.1016/j.giec.2006.01.019
- [71] Nagral, A.S., Joshi, A.S., Bhatia, S.J., Abraham, F.P. and Vora, I.M. (1993) Congestive jejunopathy in portal hypertension. Gut, 34, 694-697. doi:10.1136/gut.34.5.694
- [72] Misra, V., Misra, S.P., Dwivedi, M. and Gupta, S.C. (1997) Histomorphometric study of portal hypertensive enteropathy. American Journal of Clinical Pathology, 108, 625- 657.
- [73] Galli, S.J., Kalesnikoff, J., Grimbaldeston, M.A., Piliponsky, A.M., Williams, C.M. and Tsai, M. (2005) Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annual Review of Immunology, 23, 749- 786. doi:10.1146/annurev.immunol.21.120601.141025
- [74] Diez-Arias, J.A., Aller, M.A., Palma, M.D., Arias, J.L., Arias, J.L., Muñiz, E., Sánchez, M. and Arias, J. (2001) Increased duodenal mucosa infiltration by mast cells in rats with portal hypertension. Digestive Surgery, 18, 34- 40. doi:10.1159/000050094
- [75] Fernandez, M., Mejias, M., Angermayr, B., Garcia-Pagan, J.C., Rodés, J., and Bosch J. . (2005) Inhibition of VEGF receptor-2 decreases the development of hyperdynamic splanchnic circulation and porta-systemic collateral vessels in portal hypertensive rats. Journal of Hepatology, 43, 98-103. doi:10.1016/j.jhep.2005.02.022
- [76] Angermayr, B., Mejias, M., Gracia-Sancho, J., GarciaPagan, J.C., Bosch, J. and Fernandez, M. (2006) Hemeoxigenase attenuates oxidative stress and inflammation, and increases VEGF expresión in portal hypertensive rats. Journal of Hepatology, 44, 1033-1039. doi:10.1016/j.jhep.2005.09.021
- [77] Moon, T.C., St. Lauren, C.D., Morris, K.E., Marcet, C., Yoshimura, T., Sekar, Y. and Befus, A.D. (2010) Advances in mast cell biology: New understanding of heterogeneity and function. Nature Immunology, 3, 111-128.
- [78] Bielinska, M., Narita, N. and Wilson, D.B. (1999) Distinct roles of visceral endoderm during embryonic mouse development. The International Journal of Developmental Biology, 43, 183-205.
- [79] Oberlin, E., El Hafny, B., Petit-Cocault, L. and Souyri, M. (2010) Definitive human and mouse hematopoiesis originates from the embryonic endothelium: A new class of HSCs based on VE-cadherin expression. The International Journal of Developmental Biology, 54, 1165-1173. doi:10.1387/ijdb.103121eo
- [80] Zhao, S., Fu, Y.-M., Li, X.-F., Zhao, R.B., Huang, Q., Zhang, F.M. and Zhang, W.H. (2010) Alterations of bone marrow sinusoidal endothelium in rat and patients with liver cirrhosis. Digestive Disease and Science, 55, 654- 661. doi:10.1007/s10620-009-0785-5
- [81] Sanyal, A.J., Bosch, J., Blei, A. and Arroyo, V. (2008) Portal hypertension and its complications. Gastroenterology, 134, 1715-1728. doi:10.1053/j.gastro.2008.03.007
- [82] Vollmar, B., Wolf, B., Siegmund, S., Katsen, A.D. and Menger, M.D. (1997) Lymph vessel expansion and function in the development of hepatic fibrosis and cirrhosis. American Journal of Pathology, 151, 169-175.
- [83] Yamauchi, Y., Ikeda, R., Michitaka, K., Hiasa, Y., Horiike, N. and Onji, M. (2002) Morphometric analysis of lymphatic vessels in primary biliary cirrhosis. Hepatology Research, 24, 107-113. doi:10.1016/S1386-6346(02)00019-0
- [84] Alexander, J.S., Ganta, V.C., Jordan, P.A. and Witte, M.H. (2010) Gastrointestinal lymphatics in health and disease. Pathophysiology, 17, 315-335. doi:10.1016/j.pathophys.2009.09.003
- [85] Ikeda, R., Michitaka, K., Yamauchi, Y., Matsui, H. and Onji, M. (2001) Changes in gastrointestinal lymph and blood vessels in patients with cirrhotic portal hypertension. Journal of Gastroenterology, 36, 689-695. doi:10.1007/s005350170032
- [86] Cardenas, A. and Arroyo, V. (2007) Management of ascites and hepatic hydrothorax. Best Practice & Research Clinical Gastroenterology, 21, 55-75. doi:10.1016/j.bpg.2006.07.012
- [87] Arroyo, V., Fernandez, J. and Gines, P. (2008) Pathogenesis and treatment of hepatorenal syndrome. Seminars in Liver Disease, 28, 81-95. doi:10.1055/s-2008-1040323
- [88] Mφller, S., Henriksen, J.H., and Bendtsen, F. (2008) Pathogenetic background for treatment of ascites and hepatorenal syndrome. Hepatology International, 2, 416-428.
- [89] Mahmood, G., Debnath, C.R. and Mandal, A.K. (2009) Evaluation of 100 cases of ascites. Mymensingh Medical Journal, 18, 62-66.
- [90] Schrier, R.W., Arroyo, V., Bernardi, M., Epstein, M., Henriksen, J.H. and Rodes, J. (1988) Peripheral arterial vasodilation hypothesis: A proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology, 8, 1151-1157. doi:10.1002/hep.1840080532
- [91] Kashani, A., Landaverde, C., Medici, V. and Rossaro, L. (2008) Fluid retention in cirrhosis: Pathophysiology and management. The Quarterly Journal of Medicine, 101, 71-85. doi:10.1093/qjmed/hcm121
- [92] Salerno, F. and Cazzaniga, M. (2009) Autonomic dysfunction: Often present but usually ignored in patients with liver disease. Liver International, 29, 1451-1453. doi:10.1111/j.1478-3231.2009.02141.x
- [93] Arroyo, V. and Gines, P. (1993) Mechanism of sodium retention and ascites formation in cirrhosis. Journal of Hepatology, 17, 24-28. doi:10.1016/S0168-8278(05)80451-9
- [94] Paulus, B.M., Ali, S., Zia, A.A., Munir, A., Davis Jr., R.C., Mansbach, C.M, Smith, W.C. and Weber, K.T. (2008) Causes and consequences of systemic venous hypertension. American Journal of Medical Sciences, 336, 489- 497. doi:10.1097/MAJ.0b013e318176abe9
- [95] Witte, C.L. and Witte, M.H. (1983) Splanchnic circulatory and tissue fluid dynamics in portal hypertension. Federation Proceedings, 42, 1685-1689.
- [96] Jimenez-Saenz, M., Soria, I.C., Bernardez, J.R. and Gutierrez, J.M. (2003) Renal sodium retention in portal hypertensión and hepatorenal reflex: From practice to science. Hepatology, 37, 1494. doi:10.1053/jhep.2003.50226
- [97] Hamza, S.M. and Kaufman, S. (2009) Role of spleen in integrated control of splanchnic vascular tone: Physiology and pathophysiology. Canadian Journal of Physiology and Pharmacology, 87, 1-7. doi:10.1139/Y08-103
- [98] Cardenas, A. and Gines, P. (2009) Portal hypertension. Current Opinion in Gastroenterology, 25, 195-201. doi:10.1097/MOG.0b013e328329e154
- [99] Riggio, O. and Angeloni, S. (2009) Ascitic fluid analysis for diagnosis and monitoring of spontaneous bacterial peritonitis. World Journal of Gastroenterology, 15, 3845- 3850. doi:10.3748/wjg.15.3845
- [100] Koulaouzidis, A., Bhat, S. and Saeed, A.A. (2009) Spontaneous bacterial peritonitis. World Journal of Gastroenterology, 15, 1042-1049. doi:10.3748/wjg.15.1042
- [101] Soriano, G., Castellote, J., Alvarez, C., Girbau, A., Gordillo, J., Baliellas, C., Casas, M., Pons, C., Román, E.M., Maisterra, S., Xiol, X. and Guarner, C. (2010) Secondary bacterial peritonitis in cirrhosis: A retrospective study of clinical and analytical characteristics, diagnosis and management. Journal of Hepatology, 52, 39-44. doi:10.1016/j.jhep.2009.10.012
- [102] Bernardi, M. (2010) Spontaneous bacterial peritonitis: From pathophysiology to prevention. Internal and Emergency Medicine, 5, S37-S44. doi:10.1007/s11739-010-0446-x
- [103] Medzhitov, R. (2008) Origin and physiological roles of inflammation. Nature, 454, 428-435. doi:10.1038/nature07201
- [104] Eisenhut, M. (2006) Changes in ion transport in inflammatory disease. Journal of Inflammation, 3, 5. doi:10.1186/1476-9255-3-5
- [105] Häussinger, D. (1996) The role of cellular hydration in the regulation of cell function. Biochemical Journal, 331, 697-710.
- [106] Kennett, E.C. and Davis, M.J. (2007) Degradation of matrix glycosaminoglycans by peroxynitrite/peroxynitrous acid: Evidence of a hydroxyl-radical-like mechanism. Free Radical Biology & Medicine, 42, 1278-1289. doi:10.1016/j.freeradbiomed.2007.01.030
- [107] Jiang, D., Liang, J. and Noble, P.W. (2007) Hyaluronan in tissue injury and repair. Annals Review Cell Development and Biology, 23, 435-461. doi:10.1146/annurev.cellbio.23.090506.123337
- [108] Ng, Ch.P., Helm, C.-L.E. and Swartz, M.A. (2004) Interstitial flow differentially stimulates blood and lymphatic endothelial cell morphogenesis in vitro. Microvascular Research, 68, 258-264. doi:10.1016/j.mvr.2004.08.002
- [109] Rutowski, J.M. and Swartz, M.A. (2006) A driving force for change: Interstitial flow as a morphoregulator. Trends in Cell Biology, 17, 44-50. doi:10.1016/j.tcb.2006.11.007
- [110] Ng, C.P., Hinz, B. and Swartz, M.A. (2005) Interstitial fluid flow induces myofibroblast differentiation and collagen alignement in vitro. Journal of Cell Science, 118, 4731-4739. doi:10.1242/jcs.02605
- [111] Helm, C.-L.E., Fleury, M.E., Zisch, A.H., Boschetti, F. and Swartz, M.A. (2005) Synergy between interstitial flow and VEGF directs capillary morphogenesis in vitro through a gradient amplification mechanism. Proceedings of National Academy Science, 102, 15779-15784. doi:10.1073/pnas.0503681102
- [112] Ng, C.P. and Swartz, M.A. (2006) Mechanisms of interstitial flow-induced remodeling of fibroblast-collagen cultures. Annals of Biomedical Engineering, 34, 446-454. doi:10.1007/s10439-005-9067-3.
- [113] Goldman, J., Conley, K.A., Raehl, A., Bondy, D.M., Pytowski, B., Swartz, M.A., Rutkowski, J.M., Jaroch, D.B. and Ongstad, E.L. (2007) Regulation of lymphatic capillary regeneration by interstitial flow in skin. American Journal of Physiology Heart and Circulatory Physiology, 292, H2176-H2183. doi:10.1152/ajpheart.01011.2006
- [114] Plaku, K.J. and Von der Weid, P.Y. (2006) Mast cell degranulation alters lymphatic contractile activity through action of histamine. Microcirculation, 13, 219-227. doi:10.1080/10739680600556902
- [115] Kvietys, P.R. and Granger, D.N. (2010) Role of intestinal lymphatics in interstitial volume regulation and transmucosal water transport. Annals of the New York Academy of Sciences, 1207, E29-E43. doi:10.1111/j.1749-6632.2010.05709.x
- [116] Fanous, M.Y.Z., Phillips, A.J. and Windson, J.A. (2007) Mesenteric lymph: The bridge to future management of critical illness. Journal of the Pancreas, 8, 374-399.
- [117] Lynch, P.M., Deland, F.A. and Schmid-Schönbein, G.W. (2007) The primary valves in the initial lymphatics during inflammation. Lymphatic Research and Biology, 5, 3-10. doi:10.1089/lrb.2007.5102
- [118] Von der Weid, P.Y. and Rehal, S. (2010) Lymphatic pump function in the inflammed gut. Annals of the New York Academy of Sciences, 1207, E69-E74. doi:10.1111/j.1749-6632.2010.05715.x
- [119] Kohan, A., Yoder, S. and Tso, P. (2010) Lymphatics in intestinal transport of nutrients and gastrointestinal hormones. Annals of the New York Academy of Sciences, 1207, E44-E51. doi:10.1111/j.1749-6632.2010.05753.x
- [120] Magnotti, L.J., Xu, D.-Z., Lu, Q. and Deitch, E.A. (1999) Gut-derived mesenteric lymph. Archives of Surgery, 134, 1333-1341. doi:10.1001/archsurg.134.12.1333
- [121] Kaiser, V.L., Sifri, Z.C., Diksan, G.S., Berezina, T., Zaets, S., Lu, Q., Xu, D.Z. and Deitch, E.A. (2005) Traumahemorrhagic shock mesenteric lymph from rat contains a modified form of albumin that is implicated in endothelial cell toxicity. Shock, 23, 417-425. doi:10.1097/01.shk.0000160524.14235.6c
- [122] Watkins, A.C., Caputo, F.J., Badami, C., Barlos, D., da Xu, Z., Lu, Q., Feketeova, E. and Deitch, E.A. (2008) Mesenteric lymph duct ligation attenuates lung injury and neutrophil activation after intraperitoneal injection of endotoxin in rats. Journal of Trauma, 64, 126-130. doi:10.1097/TA.0b013e3181574a8a
- [123] Wang, Y., Ghoshal, S., Ward, M., De Villiers, W., Woodward, J. and Eckhardt, E. (2009) Chylomicrons promote intestinal absorption and systemic dissemination of dietary antigen (ovalbumin) in mice. PLoS One, 4, Article ID: e8441.
- [124] Wilting, J., Becker, J., Buttler, K. and Weich, H.A. (2009) Lymphatics and inflammation. Current Medical Chemistry, 16, 4581-4592. doi:10.2174/092986709789760751
- [125] Bruyere, F. and Noël, A. (2010) Lymphangiogenesis: In vitro and in vivo models. FASEB Journal, 24, 8-21. doi:10.1096/fj.09-132852
- [126] Mouta, C. and Heroult, M. (2003) Inflammatory triggers of lymphangiogenesis. Lymphatics Research Biology, 1, 201-218. doi:10.1089/153968503768330247
- [127] Baluk, P., Yao, L.-C., Feng, J., Romano, T., Jung, S.S., Schreiter, J.L., Yan, L., Shealy, D.J. and McDonald, D.M. (2009) TNF-α drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. Journal of Clinical Investigation, 119, 2954-2964.
- [128] Jurisic, G. and Detmar, M. (2009) Lymphatic endothelium in health and disease. Cell Tissue Research, 335, 97-108. doi:10.1007/s00441-008-0644-2
- [129] Nakao, S., Maruyama, K., Zandis, S., Melhorn, M.I, Taher, M., Noda, K., Nusayr, E., Doetschman, T. and HafeziMoghadam, A. (2010) Lymphangiogenesis and angiogenesis: concurence and/or dependence? Studies in inbred mouse strains. FASEB Journal, 24, 504-513. doi:10.1096/fj.09-134056
- [130] Ji, R.C. (2009) Lymph node lymphangiogenesis: A new concept for modulating tumor metastasis and inflammatory process. Histology and Histopathology, 24, 377-384.
- [131] Liersch, R. and Detmar, M. (2007) Lymphangiogenesis in development and disease. Thrombosis and Haemostasis, 98, 304-310.
- [132] Cueni, L.N. and Detmar, M. (2008) The lymphatic system in health and disease. Lymphatics Research and Biology, 6, 109-122. doi:10.1089/lrb.2008.1008
- [133] Vetrano, S., Borroni, E.M., Sarukhan, A., Savino, B., Bonecchi, R., Correale, C., Arena, V., Fantini, M., Roncalli, M., Malesci, A., Mantovani, A., Locati, M. and Danese, S. (2010) The lymphatic system controls intestinal inflammation and inflammation-associated colon cancer through the chemokine decoy receptor D6. Gut, 59, 197-206. doi:10.1136/gut.2009.183772
- [134] Alexander, J.S., Chaitanya, G.V., Grisham, M.B. and Boktor, M. (2010) Emerging roles of lymphatics in inflammatory bowel disease. Annals of the New York Academy of Sciences, 1207, E75-E85. doi:10.1111/j.1749-6632.2010.05757.x
- [135] Lai, W.K. and Adams, D.H. (2005) Angiogenesis and chronic inflammation; the potencial for novel therapeutic approaches in chronic liver disease. Journal of Hepatology, 42, 7-11. doi:10.1016/j.jhep.2004.11.008
- [136] Shimamura, K., Nakatani, T., Ueda, A., Sugama, J. and Okuwa, M. (2009) Relationship between lymphangiogenesis and exudates during the wound-healing process of mouse skin full-thickness wound. Wound Repair and Regeneration, 17, 598-605. doi:10.1111/j.1524-475X.2009.00512.x
- [137] Thaunat, O., Kerjaschki, D. and Nicoletti, A. (2006) Is defective lymphatic drainage a trigger for lymphoid neogenesis? Trends in Immunology, 27, 441-445. doi:10.1016/j.it.2006.08.003
- [138] Kuiper, J.J., Boomsma, F., Van Buren, H., De Man, R., Danser, A.H. and Van den Meiracker, A.H. (2008) Components of the renin-angiotensin-aldosterone system in plasma and ascites in hepatic cirrhosis. European Journal of Clinical Investigation, 38, 939-944. doi:10.1111/j.1365-2362.2008.02044.x
- [139] Bansal, S., Lindenfeld, J. and Schrier, R.W. (2009) Sodium retention in heart failure and cirrhosis: Potential role of natriuretic doses of mineralocorticoid antagonist? Circulation. Heart Failure, 2, 370-376. doi:10.1161/CIRCHEARTFAILURE.108.821199
- [140] Mayberry, J.C., Welker, K.J., Goldman, R.K. and Mullins, R.J. (2003) Mechanism of acute ascites formation after trauma resuscitation. Archives of Surgery, 138, 773-776. doi:10.1001/archsurg.138.7.773
- [141] Dong, M.H. and Saab, S. (2008) Complications of cirrhosis. Disease-a-Month, 54, 445-446. doi:10.1016/j.disamonth.2008.03.006
- [142] Beavis, J., Harwood, J.L., Coles, G.A. and Williams, J.D. (1994) Synthesis of phospholipids by human peritoneal mesothelial cells. Peritoneal Dialysis International, 14, 348-355.
- [143] Bourbon, J.R. and Chailley-Heu, B. (2001) Surfactant protein in the digestive tract, mesentery, and other organs: evolutionary significance. Comparative Biochemistry and Physiology, 129, 151-161. doi:10.1016/S1095-6433(01)00312-9
- [144] Obradovic, M.M., Stojimirovic, B.B., Trpinac, D.P., Milutinovic, D.D., Obradovic, D.I. and Nesic, V.B. (2001) Ultrastructure of peritoneal mesothelial cells. Serbian archives of entire medicine, 129, 175-179.
- [145] Schilte, M.N., Celie, J.W.A.M., Ter-Wee, P.M., Beelen, R.H.J. and Van der Born, J. (2009) Factors contributing to peritoneal tissue remodeling in peritoneal dialysis. Peritoneal Dialysis International, 29, 605-617.
- [146] Li, M.K. and Crawford, J.M. (2004) The pathology of cholestasis. Seminars in Liver Disease, 24, 21-24. doi:10.1055/s-2004-823099
- [147] Aller, M.A., Mendez, M., Nava, M.P., Lopez, L., Arias, J.L. and Arias, J. (2009) The value of microsurgery in liver research. Liver International, 29, 1132-1140. doi:10.1111/j.1478-3231.2009.02078.x
- [148] Aller, M.A., Prieto, I., Cruz, M., Losada, M., Arias, J.I., García-Domínguez, J., Argudo, S., Arias, J.L. and Arias, J. (2009) Extrahepatic cholestasis. In: Aller, M.A. and Arias, J., Eds, Microsurgery in Liver Research, Bentham Ebook, 137-156.
- [149] Aller, M.A., Duran, M., Ortega, L., Arias, J.L., Nava, M.P., Prieto, I. and Arias, J. (2004) Comparative study of macroand microsurgical extrahepatic cholestasis in the rat. Microsurgery, 24, 442-447. doi:10.1002/micr.10153
- [150] Aller, M.A., Lorente, L., Alonso, S. and Arias, J. (1993) A model of cholestasis in the rat, using a microsurgical technique. Scandinavian Journal of Gastroenterology, 28, 10-14. doi:10.3109/00365529309096038
- [151] Sanchez-Patan, F., Anchuelo, R., Corcuera, M.T., Casado, I., Gómez-Aguado, F., Aller, M.A., Cruz, A., Alonso, M.J. and Arias J. (2008) Biliary fibrosis in microsurgical extrahepatic cholestasis in the rat. Microsurgery, 28, 361- 366. doi:10.1002/micr.20495
- [152] Ramadori, G. and Saile, B. (2004) Portal tract fibrogenesis in the liver. Laboratory Investigation, 84, 153-159. doi:10.1038/labinvest.3700030
- [153] Aller, M.A., Arias, J.L., Garcia-Dominguez, J., Arias, J.I., Duran, M. and Arias, J. (2008) Experimental obstructive cholestasis: The wound-like inflammatory liver response. Fibrogenesis and Tissue Repair, 1, 6. doi:10.1186/1755-1536-1-6
- [154] Aller, M.A., Arias, J.L., Prieto, I., Losada, M. and Arias, J. (2010) Bile duct ligation: Step-by-step to cholangiocyte inflammatory tumorigenesis. European Journal of Gastroenterology and Hepatology, 22, 651-661.
- [155] Garcia-Moreno, L.M., Aller, M.A., Conejo, M., Gómez, M.A., Martin, F.R., Arias, J. and Arias, J.L. (2002) Brain Ag-NOR activity in cholestatic rats with hepatic encephalopathy. Hepatology Research, 24, 275-281. doi:10.1016/S1386-6346(02)00132-8
- [156] Aller, M.A., Nava, M.P., Arias, J.L., Duran, M., Prieto, I., Llamas, M.A. and Arias J. (2004) Microsurgical extrahepatic cholestasis in the rat: A long-term study. Journal of Investigative Surgery, 17, 99-104. doi:10.1080/08941930490422537
- [157] Pereira, R.M., Dos Santos, R.A.S., Oliveira, E.A., Leite, V.H., Dias, F.L., Rezende, A.S., Costa, L.P., Barcelos, L.S., Teixeira, M.M. and Simoes e Silva, A.C. (2008) Development of hepatorenal síndrome in bile duct ligated rats. World Journal of Gastroenterology, 28, 4505-4511. doi:10.3748/wjg.14.4505
- [158] Thabut, D., Tazi, K.A., Bonnefont-Rousselot, D., Aller, M.A., Farges, O., Guimont, M.C., Tellier, Z., Guichard, C., Ogier-Denis, E., Poynard, T., Moreau, R. and Lebrec, D. (2007) High-density lipoprotein administration attenuates liver proinflammatory response, restores liver endothelial nitric oxide synthase activity, and lowers portal pressure in cirrhotic rats. Hepatology, 46, 1893-1906. doi:10.1002/hep.21875
- [159] Assimakopoulos, S.F. and Vagianos, C.E. (2009) Bile duct ligation in rats: Areliable model of hepatorenal syndrome? World Journal of Gastroenterology, 15, 121-123. doi:10.3748/wjg.15.121
- [160] Wang, G., Shen, H., Rajaraman, G., Roberts, M.S., Gong, Y., Jiang, P. and Burczynski, F. (2007) Expression and antioxidant function of liver fatty acid binding protein in normal and bile-duct ligated rats. European Journal of Pharmacology, 560, 61-68. doi:10.1016/j.ejphar.2007.01.015
- [161] Portincasa, P., Grattagliano, I., Testini, M., Caruso, M.G., Wang, D.Q., Moschetta, A., Calamita, G., Vacca, M., Valentini, A.M., Renna, G., Lissidini, G. and Palasciano, G. (2007) Parallel intestinal and liver injury during early cholestasis in the rat: Modulation by bile salts and antioxidants. Free radical biology and medicine, 42, 1381- 1391. doi:10.1016/j.freeradbiomed.2007.01.039
- [162] Assimakopoulos, S.F., Vagianos, C.E., Patsoukis, N., Georgiou, C., Nikolopoulou, V. and Scopa, C.D. (2004) Evidence for intestinal oxidative stress in obstructive jaundice-induced gut barrier dysfunction in rats. Acta Physiologica Scandinavica, 180, 177-185. doi:10.1046/j.0001-6772.2003.01229.x
- [163] Soylu, A.R., Aydogtu, N., Basaran, U.N., Altaner, S., Tarcin, O., Gedik, N., Umit, H., Tezel, A., Dokmeci, G., Baloglu, H., Ture, M., Kutlu, K. and Kaymak, K. (2006) Antioxidants vitamins E and C attenuate hepatic fibrosis in biliary obstructed rats. World Journal of Gastroenterology, 12, 6835-6841.
- [164] Garcia-Dominguez, J., Aller, M.A., Garcia, C., de Vicente, F., Corcuera, M.T., Gómez-Aguado, F., Alonso, M.J., Vara, E. and Arias, J. (2010) Splanchnic Th2 and Th1 cytokine redistribution in microsurgical cholestatic rats. Journal of Surgical Research, 162, 203-212. doi:10.1016/j.jss.2009.06.010
- [165] Waynforth, H.B. and Flecknell, P.A. (1992) Methods of obtaining body fluids. In: Waynforth, H.B. and Flecknell, P.A., Eds., Techniques in the Rat, Elsevier Academic Press, London, 68-99.
- [166] Hauss, D., Fogal, S. and Ficorilli, J. (1998) Chronic collection of mesenteric lymph from conscious, tethered rats. Contemp Topics in Laboratoy Animal Science, 37, 56-58.
- [167] Aller, M.A., Nava, M.P. and Arias, J. (2009) Techniques of blood, bile and lymph samples extraction. In: Aller, M.A. and Arias, J., Eds., Microsurgery in Liver Reserch, Bentham Ebook, 71-92.
- [168] Mittal, A., Middleditch, M., Ruggiero, K., Buchanan, C.M., Jullig, M., Loveday, B., Cooper, G.J., Windsor, J.A. and Phillips, A.R. (2008) The proteome of rodent mesenteric lymph. American Journal of Physiology Gastrointestinal Liver Physiology, 295, G895-G903. doi:10.1152/ajpgi.90378.2008
- [169] Fang, J.F., Shih, L.Y., Yuan, K.C., Fang, K.Y., Hwang, T.L. and Hsieh, S.Y. (2010) Proteomic analysis of posthemorrhagic shock mesenteric lymph. Shock, 34, 291-298. doi:10.1097/SHK.0b013e3181ceef5e
- [170] Ohtani, O. and Ohtani, Y. (2008) Structure and function of rat lymph nodes. Archives of Histology and Cytology, 71, 69-76. doi:10.1679/aohc.71.69
- [171] Sztrymf, B., Libert, J.M., Mougeot, C., Lebrec, D., Mazmanian, M., Humbert, M. and Herve, P. (2005) Cirrhotic rats with bacterial translocation have higher incidence and severity of hepatopulmonary syndrome. Journal of Gastroenterology and Hepatology, 20, 1538-1544. doi:10.1111/j.1440-1746.2005.03914.x
- [172] Frances, R., Chiva, M., Sanchez, E., González-Navajas, J.M., Llovet, T., Zapater, P., Soriano, G., Muñoz, C., Balanzó, J., Pérez-Mateo, M., Song, X.Y., Guarner, C. and Such, J. (2007) Bacterial translocation is downregulated by anti-TNFα monoclonal antibody administration in rats with cirrhosis and ascites. Journal of Hepatology, 46, 797- 803. doi:10.1016/j.jhep.2006.11.018
- [173] Cheatham, M.L. (2009) Abdominal compartment syndrome: Pathophysiology and definitions. Scandinavian Journal of Trauma and Resuscitation Emergency Medicine, 17, 10. doi:10.1186/1757-7241-17-10
- [174] Hou, W. and Sanyal, A.J. (2009) Ascites: Diagnosis and management. MedicineClinics of North America, 93, 801- 817. doi:10.1016/j.mcna.2009.03.007
- [175] Kim, S.U., Kim, Do.Y., Lee, C.K., Park, J.Y., Kim, S.H., Kim, H.M., Choi, E.H., Kim, S., Han, K.H., Chon, C.Y. and Ahn, S.H. (2010) Ascitic fluid infection in patients with hepatitis B virus-related liver cirrhosis: Culturenegative neutrocytic ascites versus spontaneous bacterial peritonitis. Journal of Gastroenterology and Hepatology, 25, 122-128. doi:10.1111/j.1440-1746.2009.05970.x
- [176] Chan-Kyung, J.C., Shan, S.J., Winsor, E.J. and Diamandis, E.P. (2007) Proteomics analysis of human amniotic fluid. Molecular & Cell Proteomics, 6, 1406-1415. doi:10.1074/mcp.M700090-MCP200
- [177] Tong, X.-L., Wang, L., Gao, T.-B., Qin, Y.-G., Qi, Y.-Q. and Xu, Y.-P. (2009) Potential function of amniotic fluid in fetal development-Novel insights by comparing the composition of human amniotic fluid with umbilical cord and maternal serum at mid and late gestation. Journal of Clinical Medical Association, 72, 368-373. doi:10.1016/S1726-4901(09)70389-2
- [178] Gray, T. and Huestis, M. (2007) Bioanalytical procedures for monitoring in utero drug exposure. Analytical and Bioanalytical Chemistry, 388, 1455-1465. doi:10.1007/s00216-007-1228-9
- [179] Schmidt, W. (1992) The amniotic fluid compartment: The fetal habitat. Advances in Anatomy, Embryology, and Cell Biology, 127, 1-100. doi:10.1007/978-3-642-77300-6_1
- [180] Bellini, C., Boccardo, F., Bonioli, E. and Campisi, C. (2006) Lymphodynamics in the fetus and newborn. Lymphology, 39, 110-117.
- [181] Brace, R. (1997) Physiology of amniotic fluid volume regulation. Clinical Obstetrics & Gynecology, 40, 280- 289. doi:10.1097/00003081-199706000-00005
- [182] Koski, K.G. and Fergusson, M.A. (1992) Amniotic fluid composition responds to changes in manternal dietary carbohydrate and is related to metabolic status in term fetal rats. Journal of Nutrition, 122, 385-392.
- [183] Wagner, C.L., Taylor, S.N. and Johnson, D. (2008) Host factors in amniotic fluid and breast milk that contribute to gut maturation. Clinical Reviews in Allergy and Immunology, 34, 191-204. doi:10.1007/s12016-007-8032-3
- [184] Moran, E.T. (2007) Nutrition of the developing embryo and hatchling. Poultry Science, 86, 1043-1049.
- [185] Lopez de Torre, B., Tovar, J.A., Uriarte, S. and Aldazabal, P. (1992) The nutrition of the fetus with intestinal atresia: Studies in the chick embryo model. Journal of Pediatric Surgery, 27, 1325-1328. doi:10.1016/0022-3468(92)90288-I
- [186] Delo, D.M., De Coppi, P., Bartsch, G. and Atala, A. (2006) Amniotic fluid and placental stem cells. Methods in Enzymology, 419, 426-438. doi:10.1016/S0076-6879(06)19017-5
- [187] Insausti, C.L., Blanquer, M., Bleda, P., Iniesta, P. Majado, M.J., Castellanos, G. and Moraleda, J.M. (2010) The amniotic membrane as a source of stem cells. Histology and Histopathology, 25, 91-98.
- [188] Schlievert, P., Johnson, W. and Galask, R.P. (1976) Isolation of a low-molecular-weight antibacterial system from human amniotic fluid. Infection and Immunology, 14, 1156-1166.
- [189] Elinson, R.P. and Beckham, Y. (2002) Development in frogs with large eggs and the origin of amniotes. Zoology, 105, 105-117.
- [190] Blackburn, D.G. and Flemming, A.F. (2009) Morphology, development and evolution of fetal membranes and placentation is squamate reptiles. Journal of Experimental Zoology. Part B, Molecular and Developmental Evolution, 312, 579-589. doi:10.1002/jez.b.21234
- [191] Aller, M.A., de las Heras, N., Blanco-Rivero, J., Arias, J.I., Lahera, V., Balfagon, G. and Arias, J. (2012) Portal hypertensive cardiovascular pathology: The rescue of ancestral survival mechanisms? Clinical Research in Hepatlogy andl Gastroenteroogy, 36, 35-46.
NOTES
*Corresponding author.