Advances in Bioscience and Biotechnology
Vol.3 No.4A(2012), Article ID:21886,8 pages DOI:10.4236/abb.2012.324068
High concentration but low biological activity of hepatocyte growth factor in patients with chronic renal failure
![]()
1PEAS Institute, Linköping, Sweden
2Division of Clinical Medicine, School of Health and Medical Sciences, Örebro University, Örebro, Sweden
3Department of Nephrology UHL, County Council of Östergötland, Department of Medicine and Health Science, Faculty of Health Science, Linköping University, Linköping, Sweden
4Department of Molecular and Clinical Medicine, Division of Infectious Diseases, University Hospital, Linköping, Sweden
Email: *johanna.lonn@oru.se, *johanna@peasinstitut.se
Received 30 June 2012; revised 30 July 2012; accepted 16 August 2012
Keywords: Chronic Renal Failure; Hepatocyte Growth Factor; Biological Activity; Neutrophils
ABSTRACT
Hepatocyte growth factor (HGF) is a renotropic, antifibrotic and regenerative factor with cytoprotective effects that is produced by mesenchymal cells and shows high affinity to components of extra cellular matrix, such as heparan sulphate proteoglycan (HSPG), in healthy. Patients with chronic renal failure (CRF) suffer from a chronic inflammatory disorder. In order to assess the underlying mechanisms for development of CRF we aimed to assess the amounts and affinity of HGF in this patient group. ELISA, western blot and surface plasmon resonance (SPR) were used to study HGF in blood samples, as well as in isolated neutrophils, in CRF patients compared to healthy controls. Patients with CRF showed higher HGF levels in serum (P < 0.0001), but decreased affinity to HSPG (P < 0.0001). Addition of protease inhibitors decreased the difference between patients with CRF compared to healthy individuals. HGF with potent regenerative function during injury lacks affinity to HSPG in patients with CRF that may depend on production of proteases from activated immune cells. This information might be used to highlight underlying mechanisms for chronicity and leading to new strategies for treatment of chronic injuries.
1. INTRODUCTION
Chronic renal failure (CRF) is a global public health concern, and the underlying mechanisms and factors involved in the disease development are intensively studied but still not clarified. Non reversible damage of parenchymal tissue and renal fibrosis that ultimately leads to end-stage renal failure is the common pathogennetic mechanism in CRF. Simplified, renal fibrosis constitutes a failed wound-healing process of the kidney tissue after a chronic, sustained injury. Based on this fact we aimed to study the role of one of the most potent regenerative and anti-fibrotic factors, hepatocyte growth factor (HGF), in CRF in order to highlight the underlying mechanisms and eventually assess the therapy options.
In the genesis of chronic inflammation there is a shift in the balance between antiand pro-inflammatory mediators, such as growth factors, their receptors, extracellular matrix molecules and proteases [1]. HGF is a renotropic and regenerative factor with cytoprotective effects that has garnered substantial attention for its protective involvement in kidney disease [2-4]. This factor is multipotent and acts on various cell types [5], and in the kidney HGF plays an important role in renal development and maintenance of normal adult kidney structure and functions, e.g. by mediating motogenic, mitogenic, morphogenic and antiapoptotic actions in renal tubular epithelial cells [2,3,6].
Prior to the initiation of renal regeneration, HGF mRNA and protein levels increase in kidneys and intact organs (such as lungs) [7,8] and HGF is released after mechanical or chemical damage [9] mainly from fibroblasts. Once tissue injury occurs, active mature HGF is produced by proteolytic processing to a heterodimeric molecule consisting of a α-chain (62 kD) and a β-chain (32 - 36 kD). As the heterodimeric form is required for HGF bioactivity, the regulation of the proteolytic cleavage of the 92 kDa single-chain precursor from, pro-HGFis an important event to modulate HGF biological activity [10]. Additionally, polymorphonuclear neutrophils (PMNs), which are key components of the inflammatory response, are a potent source of HGF [11]. During inflammation PMNs are the first cells recruited from the circulation and they release HGF at the site of injury. Activated neutrophils are an important supply of both antiand pro-inflammatory mediators, as well as antimicrobial peptides [12,13]. HGF is stored as intracellular stock of pro-HGF in secretory vesicles, gelatinase/specific granules and in the plasma membrane of mature PMNs [11]. The infiltration of PMNs and their release of HGF in the inflammatory tissue may play an important role in wound healing and organ regeneration and is enhanced in patients with systemic inflammatory response [14]. During chronic inflammation, where a persistent infection or auto-immune processes may serve as inflammatory stimulus, recruited hyper-responsive neutrophils also contribute to host mediated tissue destruction and organ dysfunction. Protease secretion from neutrophils has been linked to pathological processes of a variety of inflammatory diseases, such as different types of kidney disorders [15]. Enhanced activation of neutrophils with an associated protease release into the circulation has been noticed in patients with CRF [16]. Proteolytic cleavage and inactivation of the HGF protein leading to inhibited signaling through the HGF receptor, c-Met, has been found in patients with chronic non-healing wounds [17].
C-Met is a transmembrane tyrosine kinase receptor expressed by almost all epithelial, endothelial, and erythroid progenitor cells [18]. In addition, a low-affinity binding site has been identified on heparan sulphate proteoglycan (HSPG) [19], a sulphated glycoprotein present on the cell surface in essentially all tissues and in the extracellular matrix [20]. Binding to HSPG is essential for the interaction of HGF with c-Met and induction of cellular responses. HSPG binds and facilitates the cytokine-receptor interaction [21-23], as well as the conversion of promitogen HGF to the two-chain mature form [22]. In addition, binding to HSPG is important for excretion of excess HGF from blood [24]. The binding to HSPG is thus important for the biological activity of HGF. A number of studies have demonstrated elevated HGF levels in patients with various chronic diseases, such as coronary artery disease, periodontitis, chronic ulcers and CRF [18,25-27]. However, despite high concentration, HGF may appear in a form with reduced biological activity, characterized by reduced binding affinity to HSPG and decreased regeneration in a model of cell injury in mouse skin epithelial cells (CCL-53.1) [25,28, 29].
Several studies reveal evidence for the protective effects of HGF in renal disease and demonstrate the importance of endogenous HGF in maintaining renal homeostatis, either by showing that neutralization of endogenous HGF supporting fibrosis and worsening regeneration, or that administration of exogenous HGF prevents fibrosis formation and enhance renal regeneration [30,31]. According to previous data we wanted in this study to analyse the concentration and evaluate the biological activity of HGF, in patients with chronic kidney disease compared to a healthy control group.
2. MATERIALS AND METHODS
2.1. Study Subjects and Protocol
Thirty six patients (mean age 71.4 years; range 48 - 88 years; 30 men and 6 women) with chronic renal disease that had developed to total renal insufficiency and were on on-going haemodialysis at Linköping University Hospital between 2007 and 2012, were enrolled in this study. A control group of thirty seven subjects (mean age 39.4 years; range 19 - 65; 23 men and 14 women) were recruited from healthy blood donors. Subjects with an active infection and/or treatment with antibiotics were excluded. Peripheral venous blood was drawn, and collected in serum vacutainer tubes (Venosafe, Terumo Europe). The blood samples were centrifuged at 3000 × g for 10 minutes, the serum was collected and frozen at –70˚C and later stored in –20˚C. The study protocol was approved by the Regional Ethical Review Board in Linköping, Sweden, and all participants provided written, informed consent.
2.2. Isolation of Neutrophils
Blood was drawn prior to haemodialysis from patients suffering from chronic renal failure (n = 11), as well as from healthy volunteers (n = 8) into EDTA-containing (2 mg/ml) vacutainer tubes (Venosafe) at Linköping university hospital in Sweden. Polymorphonuclear cells were isolated by density centrifugation as previously described [32]. Briefly, whole blood was layered upon PolymorphprepTM (Axis-Shield PoC AS, Oslo, Norway), and PMNs were isolated through density centrifugation (480 × g, 40 min, RT). Contaminating erythrocytes were lysed using hypotonic water, after which the osmotic pressure was restored. The cells were washed (400 × g, 5 min, 4˚C), counted (by using light microscope and a Bürker chamber) to get a final concentration of 1 × 107 cells/ml, and re-suspended in Krebs-Ringer glucose buffer (KRG; 120 mM NaCl, 4.9 mM KCl, 1.2 mM MgSO4, 1.7 mM KH2PO4, 8.3 mM Na2HPO4, and 10 mM glucose, pH 7.3) supplemented with 1.1 mmol/L CaCl2, and kept on ice until further usage.
2.3. Neutrophil Degranulation Experiment
PMNs (1 × 107 cells/ml) were pre-incubated 5 min with 5 μg/ml cytochalasin B (Sigma-Aldrich, St Louis, MO, USA) at 37˚C, thereafter additionally 5 min in the presence or absence of protease inhibitors (p-8340; AEBSF 40 μM, aprotinin 0.32 μM, bestatin 16 μM, E-64 5.6 μM, leupeptin 8 μM, pepstatin A 6 μM; Sigma-Aldrich), and subsequently stimulated with 10 μM N-formylmethionylleucyl-phenylalanine (fMLP;) for 30 min. As negative control, samples without fMLP-stimulation were incubated at 37˚C for 30 min. The PMN cell suspension was kept on ice and then centrifuged at 400 × g at 4˚C for 10 min to get the cell-free supernatant. The supernatants were stored immediately at –20˚C until further analysis.
2.4. ELISA Measurement
The HGF concentration in serum and in neutrophil suspensions was determined using an ELISA kit (Quantikine Human HGF immunoassay, minimum detectable limit: 0.04 ng/mL; R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. The HGF in the neutrophil suspension was concentrated by using both a 100 kDa and a 30 kDa filter (Millipore, Solna, Sweden). All measurements were performed in duplicate at 450 nm using an ELISA reader (Expert 96; Asys Hitech GmbH, Eugendorf, Austria), and calibrated using the recombinant human HGF reference samples and standards that were provided in the ELISA kit.
2.5. SPR Measurements and Ligand Immobilisation Procedures
Biologically relevant ligands of HGF; monoclonal antiHGF antibody (unknown epitope), recombinant HGF receptors, and D-19 goat polyclonal IgG (Table 1), were obtained commercially and immobilised on a Biacore CM5 chip (GE-Healthcare GmbH, Uppsala, Sweden). The affinity of HGF to the different ligands was analyzed by SPR, and the binding to HSPG has previously been proved to be important and correlate with the biological activity and growth in an in vitro model of cell injury and was used in this study to evaluate the biological activity of HGF [28,29]. Briefly, SPR measurements and ligand immobilization procedures were conducted at 760 nm in a fully automatic Biacore 2000 instrument (GE-Healthcare GmbH) equipped with four flow cells, with a temperature of 25˚C. HBS-EP buffer (0.01 M HEPES [pH 7.4], 0.15 M NaCl, 3 mM EDTA, 0.005 % surfactant P20) (GE-Healthcare GmbH) was used as running buffer. Ligands were coupled to carboxymethylated dextran CM5 chips by conventional carbodiimide chemistry using 200 mM N-ethyl-N0-(3-diethylaminopropyl) carbodiimide and 50 mM N-hydroxysuccinimide. The activation time was 7 min, followed by a 7 min ligand injection. Deactivation of the remaining active esters was performed by a 7 min injection of ethanolamine/hydrochloride [pH 8.5].
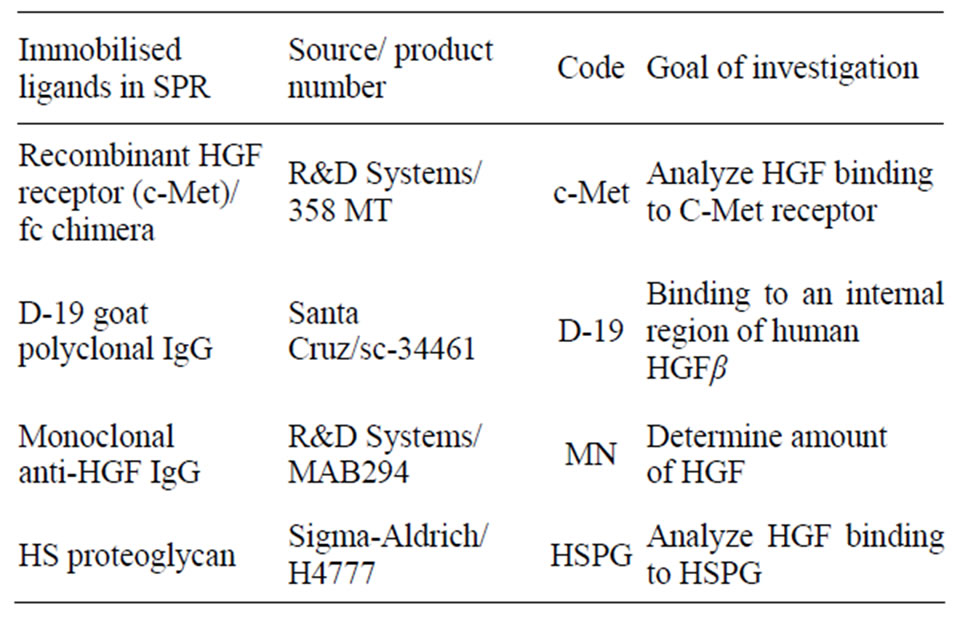
Table 1. Ligands used to investigate the binding response of HGF by SPR.
A flow rate of 5 μL/min was used during immobilization and measurement procedures. The ligands were diluted in a ratio of 1:10 or 1:5 in 10 mM acetate buffer [pH 4.5] below the isoelectric point of the protein, thus enhancing the electrostatic interactions between the dextran matrix and the ligands. The contact time was 7 min, which resulted in immobilization levels between 1000 and 10,000 response units (RU). Serum samples were diluted in a ratio of 1:20 and the PMN supernatants were diluted in a ratio of 1:5 with PBS [pH 7.4] (Apoteket AB, Umeå, Sweden). An equal mixture of 1 M NaCl and 10 mM glycine [pH 2], followed by one injection of borate [pH 8.5], was used as a regeneration buffer after each run of sample. A positive and a negative control were included at the beginning and at the end of each sensogram to confirm the reliability of the surfaces. The SPR data of the affinity to the ligands on the sensor chip surface was obtained as response unit (RU).
2.6. Western Blotting
The supernatant of the neutrophil suspensions was filtered through a 10 kDA filter (Millipore) and the concentrate was mixed with Laemmli sample buffer (5×) containing 2-Mercaptoethanol (98˚C for 5 min) to denature and reduce the proteins. The proteins were separated using standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to a polyvinylidene difluoride membrane. Unspecific binding was blocked by incubating the membranes in 5% milk-Tris-buffer saline (TBS; consisting of 25 mM Tris base, 150 mM NaCl, 2 mM KCl pH 7.4, and 0.1% Tween-20) for 1 h at RT prior to incubation with a goat polyclonal anti-HGF antibody (1:1000; AF-294-NA; R& D systems) for 1 h at RT. The membranes were thereafter incubated with a polyclonal HRP-conjugated donkey anti-goat antibody (1:1000; HAF109; R&D systems). Recombinant HGF (294-HA; R&D systems) was used as a positive control.
2.7. Statistical Analysis
The serum data were not normally distributed and therefore statistically analyzed by Mann-Whitney U-test and the medians are presented. The data of the neutrophil experiments were normally distributed and analysed by un-paired or paired student t-test and expressed as mean ± SEM. Software used was Graph Pad Prism version 5, and P < 0.05 was considered to be statistically significant.
3. RESULTS
3.1. The Serum Concentration of HGF
The concentration of HGF, measured by ELISA, was analyzed in serum collected prior to haemodialysis of patients with CRF that had developed to total renal insufficiency, and from healthy controls. The median concentration of HGF was elevated in the patient group (2.25 ng/ml), and differed significantly from the healthy control group (0.98 ng/ml; P < 0.0001) (Figure 1).
3.2. Binding Affinity to Biological Relevant Ligands of HGF
To analyse the ability of HGF to interact with its receptors or other biological relevant ligands important for the biological activity of HGF, the binding affinity to different ligands of HGF (c-Met, D-19, MN and HSPG) was analysed by SPR. The ligands were immobilised on a CM5 chip and the samples were run over the chip surface and binding response was measured in response units (RU). The binding affinity to all ligands tested (Table 1) was significantly reduced in serum from the CRF
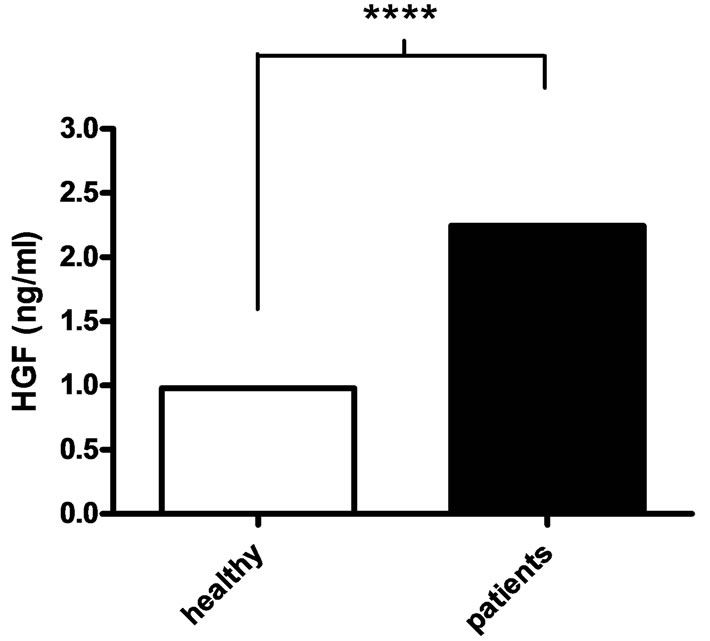
Figure 1. Serum concentration of HGF. The serum concentration (ng/ml) of HGF, measured by ELISA, was higher in patients with chronic renal failure (black bars) compared to healthy controls (white bars). Mann-Whitney U-test was used for statistical analysis and the medians are presented (n = 36 for patients and n = 37 for healthy controls). ****P < 0.0001.
patients (P ≤ 0.0001 for all ligands) (Figure 2).
3.3. The Release of HGF from Activated Neutrophils
To study the concentration of HGF released by neutronphils (1 × 107 cells/ml) isolated from patients with CRF and healthy controls, the cells were stimulated with cytochalasin B (5 μg/ml) and fMLP (10 μM) and the concentration of HGF in the supernatant was measured by ELISA. The fMLP-stimulated neutrophils from CRF patients released a higher amount of HGF (2.16 ± 0.65 ng/ml), compared to controls (1.06 ± 0.386 ng/ml), however, the difference was not statistically significant. Protease inhibitors reduced the amount of HGF in supernatant from stimulated CRF neutrophils (P = 0.039) (Figure 3). There were no differences comparing non-stimulated neutrophils (with or without protease inhibitors) of CRF patients and healthy controls (data not shown).
3.4. The Affinity to HSPG in Supernatant from fMLP-Stimulated Neutrophils
The affinity of HGF released from fMLP-stimulated (10 μM) CRF and healthy control neutrophils (1 × 107 cells/ml), respectively, was evaluated by measuring binding affinity to HSPG using SPR. The supernatant of the CRF neutrophil suspension showed a reduced binding affinity to HSPG (12.37 ± 4.2 RU) compared to control neutrophils (45.19 ± 12.03; P < 0.01). The presence of protease inhibitors (pi) increased the binding affinity for HSPG in the suspension of fMLP-stimulated neutrophils in the patient group (P = 0.032), however, the protease inhibitors did not significantly affect the healthy control
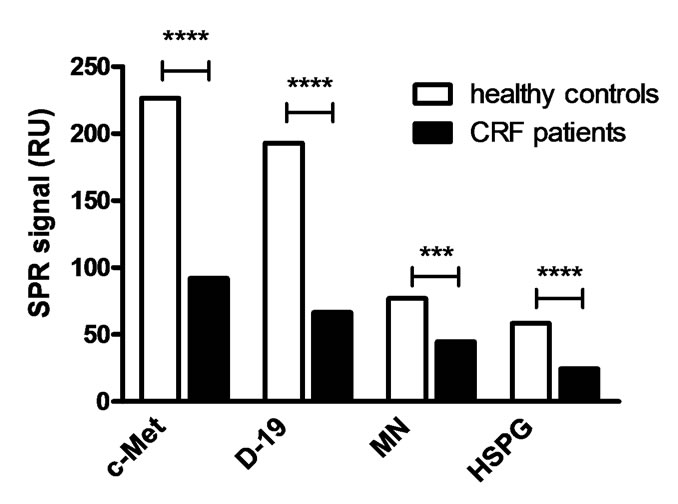
Figure 2. Binding affinity to biological relevant ligands of HGF. The binding affinity to different ligands of HGF (c-Met, polyclonal ab D-19, monoclonal anti-HGF ab (MN) and heparan sulphate proteoglycan (HSPG)) was analysed by surface plasmon resonance (SPR) in a Biacore 2000. For statistical analysis Mann-Whitney U-test was used and the medians are presented (n = 36 for patients and n = 30 for healthy controls). ****P < 0.0001, ***P = 0.0001.
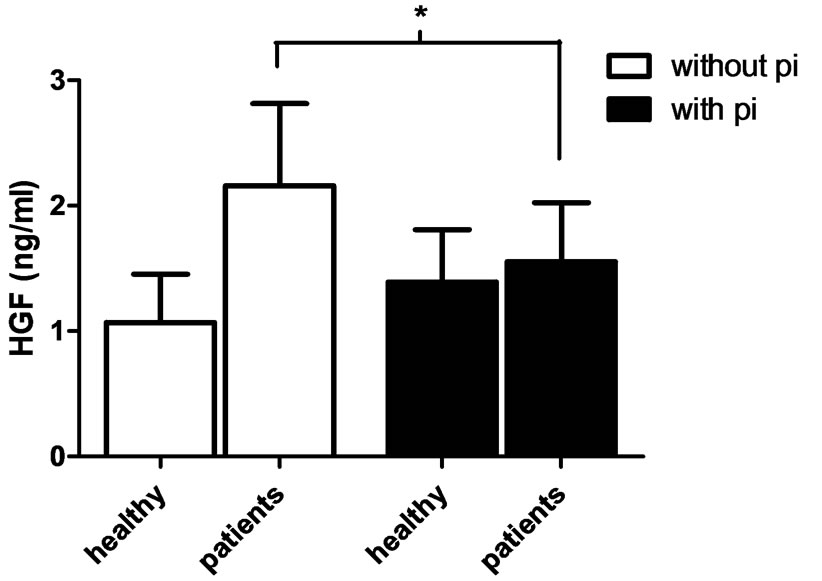
Figure 3. Concentration of HGF in supernatant from CRF neutrophils. Neutrophils (1 × 107 cells/ml) isolated from patients with CRF and from healthy controls were pre-incubated with 5 μg/ml cytochalasin B for 5 min at 37˚C and additionally 5 min in the absence (white bars) or presence (black bars) of protease inhibitors (pi), thereafter stimulated with 10 μM fMLP for 30 min. The HGF concentration (ng/ml) was measured by ELISA. Unpaired and paired student t-test was used for statistical analysis and data are presented as mean ± SEM (healthy controls n = 7, CRF patients n = 10). *P < 0.05.
samples (Figure 4). When comparing non-stimulated neutrophils of CRF patients and controls there were no differences found, neither did the protease inhibitor cocktail by its own bind to HSPG (data not shown).
3.5. Western Blotting
To visualize the HGF released from CRF and control neutrophils western blot was used. The isolated neutronphils (1 × 107 cells/ml) were stimulated with 5 μg/ml cytochalasin B and 10 μM fMLP for 30 min, with and without the addition of protease inhibitor cocktail, and the proteins were concentrated by ultrafiltration. Five blots (including five CRF patients and five healthy controls) were analyzed. The HGF released from the neutrophils of the controls showed bands of 90 kDa, which corresponds to the size of pro-HGF. Some controls also showed weaker bands at 60 kDa, which is the size of the α-chain of HGF. The patient neutrophils varied more in the forms of HGF, showing mostly bands of 60 kDa, one patient also showed bands of 34 kDa, which is the size of the β-chain. No differences were found between samples incubated with or without protease inhibitors (Figure 5).
4. DISCUSSION
In the present study, we have shown that systemic HGF, as well as HGF produced by immune cells, differs significantly between patients with CRF and healthy controls. Regarding to regenerative properties of HGF, this observation suggests a lack of healing of injuries and fibrosis in the course of development of total renal fail-
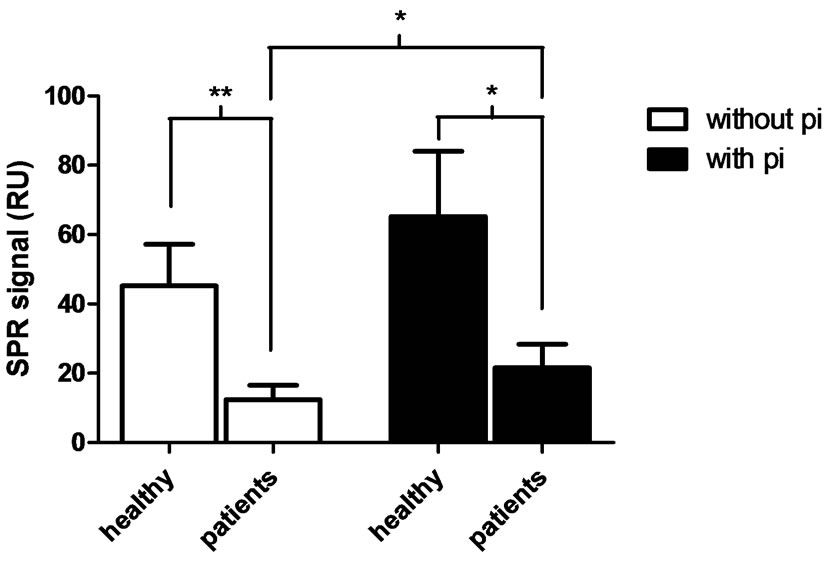
Figure 4. Binding affinity to HSPG of supernatants of activated neutrophils. Neutrophils (1 × 107 cells/ml) isolated from patients with CRF and from healthy controls were pre-incubated with 5 μg/ml cytochalasin B for 5 min at 37˚C and additionally 5 min in the absence (white bars) or presence (black bars) of protease inhibitor cocktail (pi), thereafter stimulated with 10 μM fMLP for 30 min. The binding affinity to heparan sulphate proteoglycan (HSPG) was measured by surface plasmon resonance (SPR) and presented in response units (RU). Unpaired and paired student t-test was used for statistical analysis and data are presented as mean ± SEM (healthy controls n = 8, CRF patients n = 11). **P < 0.01, *P < 0.05.
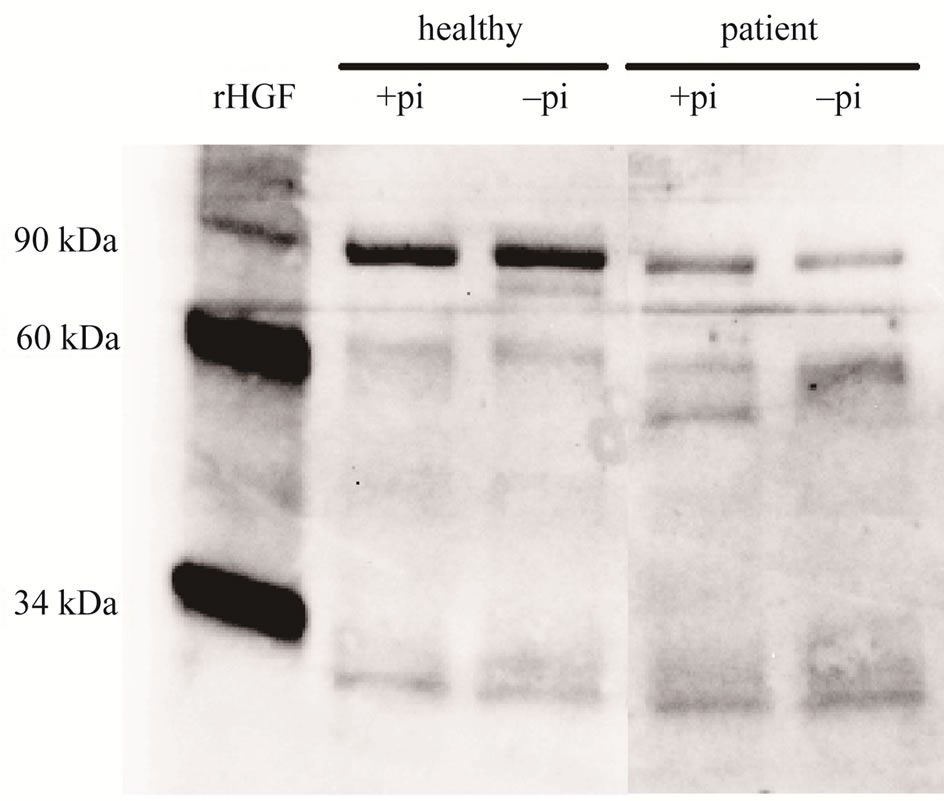
Figure 5. Western blot results. Neutrophils (1 × 107 cells/ml) isolated from patients with CRF and from healthy controls were pre-incubated with 5 μg/ml cytochalasin B for 5 min at 37˚C and additionally 5 min in the absence (−pi) or presence (+pi) of protease inhibitor cocktail (pi), thereafter stimulated with 10 μM fMLP for 30 min. The proteins in the suspensions were concentrated by using a 10 kDa filter and then reduced by heating with Laemmli sample buffer containing 2-Mercaptoethanol. An equivalent volume was loaded in each lane, and the electrophoretically separated proteins were detected by western blot using a goat polyclonal anti-HGF antibody (1:1000; AF- 294-NA) and a polyclonal HRP-conjugated donkey antigoat antibody (1:1000; HAF109). Recombinant HGF (294-HA; R&D systems, Minneapolis, MN, USA) was used as a positive control. One representative blot out of five is presented.
ure, which might have an impact in development of new therapeutic methods.
Previous studies have shown that administration of recombinant HGF prevents fibrosis and enhances renal regeneration, and that neutralization of endogenous HGF supports fibrosis and worsens tissue regeneration [4,30]. This indicates that despite increased endogenous levels of HGF in patients with CRF, it is insufficient for restoring the homeostatic balance and to heal the tissue and reverse the chronic inflammatory process. We have previously studied the concentration and biological activity of HGF in various infectious diseases, and observed a significant correlation between binding affinity of HGF to HSPG in an SPR-based system [29] and the biological activity of HGF in an in vitro cell injury model [28]. In the present study, we hypothesized that the inflammatory process in patients with CRF is associated with changes in the expression and activity of HGF. Interestingly, this study shows that the biological activity of HGF (measured by binding affinity to the co-receptor HSPG) is markedly reduced in the CRF patients, despite that the amount of HGF measured by ELISA was increased. Additionally, the binding of HGF to the other biological relevant ligands analysed (c-Met, D-19 and MN) was reduced in the patient group. In previous studies by our group, we have found reduced affinity to these ligands by biological inactive HGF, and that HSPG was the strongest indicator of biological activity of HGF in vitro [29]. It is possible that the HGF configuration is altered during the chronic inflammation and therefore not able to bind to its ligands/receptors. The western blot analysis showed that neutrophils of the healthy controls mostly secreted proHGF and also the α-chain, whereas the neutrophil suspension of the CRF patients varied more in the forms of HGF by showing already proteolytic processed HGF, however, the β-chain was only visible in one patient. The α-chain alone binds with high-affinity to c-Met, although the binding of the α-chain does not activate the receptor [33] . Several proteases in the serum or cell membranes are involved in the activation of single-chain pro-HGF. Perhaps pro-HGF binds to HSPG and is processed in the wounded tissue for further activation of c-Met signaling, however, this is a question for further studies.
Binding to HSPG was previously shown to be important for excretion of excess HGF from blood [24]. A lower binding affinity of HGF to HSPG during chronic inflammation, and a concurrent increased need for regenerative factors, may thus explain the higher systemic amounts of HGF determined by ELISA. In addition, binding to the co-receptor HSPG is essential for the interaction of HGF with its high-affinity receptor (c-Met) and induction of cellular responses [21-23]. Since HGF in CRF patients lacks these binding properties it may suggest reduced regenerative and protective effects in the tissue. The inability of HGF to induce c-Met signalling was previously shown in ulcer secretion of chronic nonhealing wounds, and that this HGF was modified and inactivated by neutrophil proteases [17]. The absent c-Met signalling may be explained by the reduced binding to HSPG seen in our study, together with the decreased binding to c-Met. Wilfred et al. [34] also found that neutrophil proteases could inactivate HGF, generating an N-terminal segment and four kringle domains (NK4)-like fragment, which antagonized the biological effect of HGF. Additionally, we found that protease inhibitors positively affected the biological activity of HGF (P = 0.03), but reduced the concentration of HGF (P < 0.04) in the supernatant of stimulated neutrophils from CRF patients. These data suggest that proteases released from neutrophils are involved in the modulation of the expression of HGF, leading to reduced biological activity in the patients. Possibly proteases stimulate HGF release and thereby counteract the effects of proteolytic cleavage of host-molecules and tissue destruction following increased levels of proteases, by activating protease-activating receptors (PARs) [35]. PAR-2 is expressed on neutrophils [36], and activation of PAR-2 by neutrophil proteases may through intracellular mechanisms, including caspase-mediated cleavage of pro-HGF, cause HGF secretion. However, it has not been shown whether neutrophil proteases exert such autocrine effects.
Several studies have highlighted the role of HGF in treatment of injuries of organ and tissues, e.g. kidney [2, 37] and cardiovascular systems [38] . We have previously reported positive effects of exogenous HGF, with high affinity towards HSPG, in the treatment of patients with chronic ulcers [39]. Since mechanisms during the chronic inflammation in CRF may down-regulate the biological activity of HGF, our study suggests that biologically active HGF with high affinity to HSPG may be useful as a therapeutic tool for treatment of CRF.
In summary, we conclude that patients with CRF have a systemic HGF profile reflecting a chronic inflammatory condition with high concentration, but low biological activity, of HGF. These observations might have an impact on future development of strategies for diagnosis, evaluation, and treatment of chronic renal failure.
REFERENCES
- Gurtner, G.C., Werner, S., Barrandon, Y. and Longaker, M.T. (2008) Wound repair and regeneration. Nature, 453, 314-321. doi:10.1038/nature07039
- Mizuno, S., Matsumoto, K. and Nakamura, T. (2008) HGF as a renotrophic and anti-fibrotic regulator in chronic renal disease. Front Bioscience, 13, 7072-7086. doi:10.2741/3211
- Matsumoto, K. and Nakamura, T. (2001) Hepatocyte growth factor: Renotropic role and potential therapeutics for renal diseases. Kidney International, 59, 2023-2038.
- Iekushi, K., Taniyama, Y., Azuma, J., Sanada, F., Kusunoki, H., Yokoi, T., et al. (2010) Hepatocyte growth factor attenuates renal fibrosis through TGF-β1 suppression by apoptosis of myofibroblasts. Journal of Hypertension, 28, 2454-2461.
- Nakamura, T. and Mizuno, S. (2010) The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proceedings of the Japan Academy—Series B: Physical & Biological Sciences, 86, 588-610. doi:10.2183/pjab.86.588
- Yo, Y., Morishita, R., Nakamura, S., Tomita, N., Yamamoto, K., Moriguchi, A., et al. (1998) Potential role of hepatocyte growth factor in the maintenance of renal structure: Anti-apoptotic action of HGF on epithelial cells. Kidney International, 54, 1128-1138. doi:10.1046/j.1523-1755.1998.00092.x
- Nagaike, M., Hirao, S., Tajima, H., Noji, S., Taniguchi, S., Matsumoto, K., et al. (1991) Renotropic functions of hepatocyte growth factor in renal regeneration after unilateral nephrectomy. Journal of Biological Chemistry, 266, 22781-22784.
- Igawa, T., Matsumoto, K., Kanda, S., Saito, Y. and Nakamura, T. (1993) Hepatocyte growth factor may function as a renotropic factor for regeneration in rats with acute renal injury. American Journal of Physiology, 265, F61- F69.
- Matsumoto, K. and Nakamura, T. (1997) Hepatocyte growth factor (HGF) as a tissue organizer for organogenesis and regeneration. Biochemical and Biophysical Research Communications, 239, 639-644. doi:10.1006/bbrc.1997.7517
- Naka, D., Ishii, T., Yoshiyama, Y., Miyazawa, K., Hara, H., Hishida, T., et al. (1992) Activation of hepatocyte growth factor by proteolytic conversion of a single chain form to a heterodimer. Journal of Biological Chemistry, 267, 20114-20119.
- Grenier, A., Chollet-Martin, S., Crestani, B., Delarche, C., El Benna, J., Boutten, A., et al. (2002) Presence of a mobilizable intracellular pool of hepatocyte growth factor in human polymorphonuclear neutrophils. Blood, 99, 2997- 3004. doi:10.1182/blood.V99.8.2997
- Luerman, G.C., Uriarte, S.M., Rane, M.J. and McLeish, K.R. (2010) Application of proteomics to neutrophil biology. Journal of Proteomics, 73, 552-561. doi:10.1016/j.jprot.2009.06.013
- Cowburn, A.S., Condliffe, A.M., Farahi, N., Summers, C. and Chilvers, E.R. (2008) Advances in neutrophil biology: Clinical implications. Chest, 134, 606-612. doi:10.1378/chest.08-0422
- Matsushima, A., Ogura, H., Koh, T., Fujita, K., Yoshiya, K., Sumi, Y., et al. (2004) Hepatocyte growth factor in polymorphonuclear leukocytes is increased in patients with systemic inflammatory response syndrome. Journal of Trauma, 56, 259-264. doi:10.1097/01.TA.0000111752.60500.DA
- Costa, E., Rocha, S., Rocha-Pereira, P., Nascimento, H., Castro, E., Miranda, V., et al. (2008) Neutrophil activation and resistance to recombinant human erythropoietin therapy in hemodialysis patients. American Journal of Nephrology, 28, 935-940. doi:10.1159/000142147
- Polańska, B., Augustyniak, D., Makulska, I., Niemczuk, M., Zwolińska, D. and Elastase, J.A. (2010) α1-proteinase inhibitor, and interleukin-8 in pre-dialyzed and hemodialyzed patients with chronic kidney disease. Pediatrics International, 52, 735-743.
- Buchstein, N., Hoffmann, D., Smola, H., Lang, S., Paulsson, M., Niemann, C., et al. (2009) Alternative proteolytic processing of hepatocyte growth factor during wound repair. American Journal of Pathology, 174, 2116- 2128. doi:10.2353/ajpath.2009.080597
- Nakamura, T., Sakai, K. and Matsumoto, K. (2011) Hepatocyte growth factor twenty years on: Much more than a growth factor. Journal of Gastroenterology and Hepatology, 26, 188-202. doi:10.1111/j.1440-1746.2010.06549.x
- Liu, K.X., Kato, Y., Narukawa, M., Kim, D.C., Hanano, M., Higuchi, O., et al. (1992) Importance of the liver in plasma clearance of hepatocyte growth factors in rats. American Journal of Physiology, 263, G642-G649.
- Nakamura, T., Teramoto, H. and Ichihara, A. (1986) Purification and characterization of a growth factor from rat platelets for mature parenchymal hepatocytes in primary cultures. Proceedings of the National Academy of Sciences USA, 83, 6489-6493. doi:10.1073/pnas.83.17.6489
- Karihaloo, A., Kale, S., Rosenblum, N.D. and Cantley, L.G. (2004) Hepatocyte growth factor-mediated renal epithelial branching morphogenesis is regulated by glypican-4 expression. Molecular and Cellular Biology, 24, 8745-8752. doi:10.1128/MCB.24.19.8745-8752.2004
- Rubin, J.S., Day, R.M., Breckenridge, D., Atabey, N., Taylor, W.G., Stahl, S.J., et al. (2001) Dissociation of heparan sulfate and receptor binding domains of hepatocyte growth factor reveals that heparan sulfate-c-met interaction facilitates signaling. Journal of Biological Chemistry, 276, 32977-32983. doi:10.1074/jbc.M105486200
- Sakata, H., Stahl, S.J., Taylor, W.G., Rosenberg, J.M., Sakaguchi, K., Wingfield, P.T., et al. (1997) Heparin binding and oligomerization of hepatocyte growth factor/scatter factor isoforms. Heparan sulfate glycosaminoglycan requirement for Met binding and signaling. Journal of Biological Chemistry, 272, 9457-94563. doi:10.1074/jbc.272.14.9457
- Liu, K.X., Kato, Y., Kato, M., Kaku, T.I., Nakamura, T. and Sugiyama, Y. (1997) Existence of two nonlinear elimination mechanisms for hepatocyte growth factor in rats. American Journal of Physiology, 273, E891-E897.
- Nayeri, F., Olsson, H., Peterson, C. and Sundqvist, T. (2005) Hepatocyte growth factor; expression, concentration and biological activity in chronic leg ulcers. Journal of Dermatological Science, 37, 75-85. doi:10.1016/j.jdermsci.2004.11.002
- Funakoshi, H. and Nakamura, T. (2003) Hepatocyte growth factor: From diagnosis to clinical applications. Clinica Chimica Acta, 327, 1-23. doi:10.1016/S0009-8981(02)00302-9
- Nagaraja, C. and Pradeep, A.R. (2007) Hepatocyte growth factor levels in gingival crevicular fluid in health, disease, and after treatment. Journal of Periodontology, 78, 742-747. doi:10.1902/jop.2007.060249
- Nayeri, F., Holmgren-Pettersson, K., Perskvist, N., Forsberg, P., Peterson, C. and Sundqvist, T. (2007) An in vitro model for assessment of the biological activity of hepatocyte growth factor. Growth Factors, 25, 33-40. doi:10.1080/08977190600997200
- Nayeri, F., Nayeri, T., Aili, D., Brudin, L. and Liedberg, B. (2008) Clinical impact of real-time evaluation of the biological activity and degradation of hepatocyte growth factor. Growth Factors, 26, 163-171. doi:10.1080/08977190802128083
- Ohnishi, H., Mizuno, S. and Nakamura, T. (2008) Inhibition of tubular cell proliferation by neutralizing endogenous HGF leads to renal hypoxia and bone marrowderived cell engraftment in acute renal failure. American Journal of Physiology—Renal Physiology, 294, F326- F335. doi:10.1152/ajprenal.00480.2007
- Liu, Y., Rajur, K., Tolbert, E. and Dworkin, L.D. (2000) Endogenous hepatocyte growth factor ameliorates chronic renal injury by activating matrix degradation pathways. Kidney International, 58, 2028-2043. doi:10.1111/j.1523-1755.2000.00375.x
- Gunnarsson, P., Fornander, L., Pahlsson, P. and Grenegard, M. (2010) Sialic acid residues play a pivotal role in alpha(1)-acid glycoprotein (AGP)-induced generation of reactive oxygen species in chemotactic peptide pre-activated neutrophil granulocytes. Inflammation Research, 59, 89-95. doi:10.1007/s00011-009-0071-1
- Matsumoto, K., Kataoka, H., Date, K. and Nakamura, T. (1998) Cooperative interaction between alphaand betachains of hepatocyte growth factor on c-Met receptor confers ligand-induced receptor tyrosine phosphorylation and multiple biological responses. Journal of Biological Chemistry, 273, 22913-22920. doi:10.1074/jbc.273.36.22913
- Raymond, W.W., Cruz, A.C. and Caughey, G.H. (2006) Mast cell and neutrophil peptidases attack an inactivation segment in hepatocyte growth factor to generate NK4- like antagonists. Journal of Biological Chemistry, 281, 1489-1494. doi:10.1074/jbc.M511154200
- Uehara, A., Muramoto, K., Imamura, T., Nakayama, K., Potempa, J., Travis, J., et al. (2005) Arginine-specific gingipains from Porphyromonas gingivalis stimulate production of hepatocyte growth factor (scatter factor) through protease-activated receptors in human gingival fibroblasts in culture. Journal of Immunology, 175, 6076- 6084.
- Howells, G.L., Macey, M.G., Chinni, C., Hou, L., Fox, M.T., Harriott, P., et al. (1997) Proteinase-activated receptor-2: Expression by human neutrophils. Journal of Cell Science, 110, 881-887.
- Dai, C., Saleem, M.A., Holzman, L.B., Mathieson, P. and Liu, Y. (2010) Hepatocyte growth factor signaling ameliorates podocyte injury and proteinuria. Kidney International, 77, 962-973. doi:10.1038/ki.2010.40
- Urbanek, K., Rota, M., Cascapera, S., Bearzi, C., Nascimbene, A., De Angelis, A., et al. (2005) Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circulation Research, 97, 663-673. doi:10.1161/01.RES.0000183733.53101.11
- Nayeri, F., Strömberg, T., Larsson, M., Brudin, L., Söderström, C. and Forsberg, P. (2002) Hepatocyte growth factor may accelerate healing in chronic leg ulcers: A pilot study. Journal of Dermatological Treatment, 13, 81-86. doi:10.1080/095466302317584449
NOTES
*Corresponding author.