Advances in Chemical Engineering and Science
Vol.3 No.1(2013), Article ID:26973,11 pages DOI:10.4236/aces.2013.31010
Dehydrocyclization of n-Hexane over Heteropolyoxometalates Catalysts
1Laboratoire de Chimie Appliquée et de Genie Chimique, Universite Mouloud Mammeri, Tizi-Ouzou, Algeria
2Laboratory of Semiconductor and Solar Energy Research, Chemistry Department, An-Najah N. University, Nablus, Palestine
3Laboratoire de Chimie du Gaz Naturel, Institut de Chimie USTHB, Alger, Algeria
Email: abed_eed@yahoo.com
Received December 7, 2012; revised January 9, 2013; accepted January 15, 2013
Keywords: Dehydrocyclization; Heteropolyacid; Catalyst; n-Hexane; Keggin Ion
ABSTRACT
The catalytic dehydrocyclization of n-hexane was studied here for the first time using a number of compounds based on H3PMo12O40. The described catalysts were prepared by either replacing the acidic proton with counter-ions such as ammonium or transition metal cations (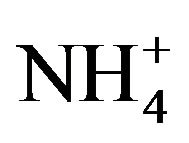 , Fe3+, K+), or by replacing Mo6+ with (Ni3+, Co3+, Mn3+) in the polyoxometalate framework, as reported earlier. For comparison purposes, the known (TBA)7PW11O39 catalyst system was used. All reactions were conducted at different temperatures in the range 200˚C - 450˚C. The Keggin structure of these heteropolycompounds was ascertained by XRD, UV and IR measurements. 31P NMR measurements and thermal behaviour of the prepared catalysts were also studied. These modified polyoxometalates exhibited heterogeneous superacidic catalytic activities in dehydrocyclization of n-hexane into benzene, cyclohexane, cyclohexene and cyclohexadiene. The catalysts obtained by substituting the acidic proton or coordination atom exhibited higher selectivity and stability than the parent compound H3PMo12O40. Catalytic activity and selectivity were heavily dependent on the composition of the catalyst and on the reaction conditions. At higher temperatures, the catalyst exhibited higher conversion efficiency at the expense of selectivity. Using higher temperatures (>400˚C) in the presence of hydrogen carrier gas, selectivity towards dehydrocyclization ceased and methane dominated. To explain the results, a plausible mechanism is presented, based on super-acidic nature of the catalyst systems.
, Fe3+, K+), or by replacing Mo6+ with (Ni3+, Co3+, Mn3+) in the polyoxometalate framework, as reported earlier. For comparison purposes, the known (TBA)7PW11O39 catalyst system was used. All reactions were conducted at different temperatures in the range 200˚C - 450˚C. The Keggin structure of these heteropolycompounds was ascertained by XRD, UV and IR measurements. 31P NMR measurements and thermal behaviour of the prepared catalysts were also studied. These modified polyoxometalates exhibited heterogeneous superacidic catalytic activities in dehydrocyclization of n-hexane into benzene, cyclohexane, cyclohexene and cyclohexadiene. The catalysts obtained by substituting the acidic proton or coordination atom exhibited higher selectivity and stability than the parent compound H3PMo12O40. Catalytic activity and selectivity were heavily dependent on the composition of the catalyst and on the reaction conditions. At higher temperatures, the catalyst exhibited higher conversion efficiency at the expense of selectivity. Using higher temperatures (>400˚C) in the presence of hydrogen carrier gas, selectivity towards dehydrocyclization ceased and methane dominated. To explain the results, a plausible mechanism is presented, based on super-acidic nature of the catalyst systems.
1. Introduction
Catalytic reforming of n-hexane is one of the most widely used processes in oil refining industry. Fuel octane number can be enhanced by increasing concentrations of aromatic compounds [1-3].
Catalytic reforming proceeds over bifunctional catalysts having both metallic and acidic functions are currently being considered [4,5]. Heteropoly acids (HPAs) are complex proton acids that incorporate polyoxometalate anions (heteropoly anions) having metal-oxygen octahedra as the basic structural units. HPAs have several advantages such as very strong Brönsted acidities approaching the super acid region [6] and high oxidant activities in multielectron redox reactions under mild conditions. Their acid-base and redox properties can be varied widely by changing their chemical compositions.
HPAs have a discrete ionic structure, containing the fairly mobile heteropoly anions and counter-cations (H+, H3O+, 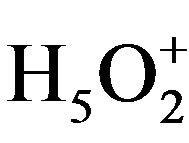 , etc.). This unique structure results in significantly high proton mobility and pseudo liquid phase behaviour. In addition, HPAs are highly soluble in polar solvents and their thermal stabilities are quite high. These properties make HPAs potentially promising acid, redox, and bifunctional catalysts in both homogeneous and heterogeneous systems [7,8].
, etc.). This unique structure results in significantly high proton mobility and pseudo liquid phase behaviour. In addition, HPAs are highly soluble in polar solvents and their thermal stabilities are quite high. These properties make HPAs potentially promising acid, redox, and bifunctional catalysts in both homogeneous and heterogeneous systems [7,8].
Heteropolyacids (HPA) usually act as strong acidic catalysts, with the ability to activate alkanes and produce carbonium ion intermediates [9,10]. Such intermediates are necessary for alkane cracking, alkylation and isomerization processes [11]. The large-scale industrial applications of these reactions drew attention to such catalyst systems for long time [12].
The transformation of n-hexane depends on the nature of the catalyst and on the operation conditions. The reactant may undergo three different competing reactions, namely: Hydrogenolysis to short linear chain alkanes such as methane, ethane, propane, butane and pentane in presence of H-[Al]-ZMS-5 leading [13]. Isomerization over Pt/zeolithe giving iso-alkanes, such as isobutane, isopentane, 2,2-dimethylbutane, 2,3-dimethylbutane, 2- methylpentane and 3-methylpentane [14]. Dehydrocyclization and aromatization giving cyclohexane, cyclohexene, and benzene on catalysts such as WO3/ZrO2 [15].
In our search for highly efficient, stable and selective catalyst systems for dehydrocyclization of hexane, synthesised and characterised a number of compounds based on modified polyoxomolybdates and polyoxytengestates. The investigated compounds are the heteropolyacid H3PMo12O40·xH2O (noted PMo12), the ammonium salt of the (Co, Ni, Mn) substituted species (NH4)4PMo11O39M (H2O)·xH2O (M: Co, Ni, Mn) (noted PMo11Co, PMo11Ni, PMo11Mn). (TBA)7PW11O39·xH2O (noted PW11) and KFePMo12O40·xH2O (noted FePMo12). Such complexes have been intensively studied for different organic reactions [16-20], and can activate alkanes due to their superacidic nature [21-24].
In this work, the compounds have been used in hexane dehydrocyclization reaction for the first time. The reaction may involve other products such as cyclohexane, cyclohexene and cyclohexadiene, in addition to the unwanted methane product. The main goal is to maximize benzene production and minimize methane in the product mixture, by investigating different catalysts, varying temperature, and carrier gas and retention time.
2. Experimental Part
2.1. Preparation of Catalysts
Pure heteropolyacid H3PMo12O40·xH2O was prepared as described earlier [25]. The mixed potassium-iron salt K0.5Fe0.1H0.2PMo12O40·xH2O was prepared as follows: to an aqueous solution of H3PMo12O40 (50 ml, 0.08 M) were added successively 9.0 ml of 0.10 M Fe(NO3)3 and 7.5 ml of 0.80 M KNO3. The obtained precipitate was filtered over glass frit and dried at 50˚C under vacuum for 5 h.
The ammonium salt (NH4)4PMo11MO39 (H2O)·xH2O (M = Ni, Co, Mn) was prepared as described earlier [26]. A mixture of H3PO4 (5.0 ml; 1.00 M), H2SO4 (10.0 ml; 0.50 M) and MSO4 (5.0 ml; 1.00 M) was slowly added to 250 ml of an aqueous solution of ammonium paramolybdate [(NH4)6Mo7O24·4H2O] (48.8 g, 40 mmol) at 0˚C. The ammonium salt completely precipitated after addition of solid ammonium nitrate (NH4NO3).
(TBA)7PW11O39 was prepared by the method described in the literature [27]. A 15.00 g quantity of H3PW12O40·14H2O was dissolved in 20.0 mL of water; a 5.0 g quantity of [(n-C4H9)4N]Br (TBABr) was dissolved in 50.0 mL of CH2Cl2. The mixture was poured into the molybdophosphate solution with vigorous stirring. Three phases were formed: a yellow solid one, a lower yellow liquid one (CH2Cl2), and a poorly yellow coloured aqueous upper one. The solid was washed with Et2O, providing 8.50 g of product. A further yield of 2.50 g was obtained by addition of Et2O to the CH2Cl2 phase. Recrystalization was performed in CH2Cl2.
2.2. Equipment
The UV analysis of the catalyst samples (dissolved in acetonitrile/water 1:1 ratio, as dilute samples solutions 10-5-10-3 M) were performed on a UV-1601PC SHIMADZU spectrophotometer. IR spectra of the samples were obtained at room temperature with a BIO-RAD FTS 165 FTIR spectrometer using standard KBr pellet technique. Measurements were taken in the wave-number range of 4000 - 400 cm−1, 256 scans, and resolution 2 cm−1.
The X-ray powder diffraction (XRD) diagrams were recorded on a SIEMENS D5000 equipment in the 5 - 45˚ 2θ range, at a scanning speed of 1˚C per minute, using CuKα radiation. The 31P NMR measurements were performed on a Bruker CXP10 spectrophotometer at 10 MHz. Chemical shifts were referenced to Al(PO3)3.
Thermal behaviour of the prepared catalysts was studied using a tgA-dta Setaram apparatus. For each experiment 20 mg of powdered sample were heated at a temperature ramp rate of 5˚C/min under air flow. Differential scanning calorimetry experiments (DSC) were run on a DSC 111 SETARAM. Apparatus under similar conditions, with a nitrogen flow.
2.3. Catalytic Experiments
The conversion of n-hexane was carried out in a quartz fixed-bed flow reactor at different temperatures from 200˚C to 400˚C, using 0.5 g of catalyst. Catalysts were heated to the desired reaction temperature at 5˚C/min under nitrogen flow and then maintained at this temperature for one hour under nitrogen. A mixture of n-hexane (50 torr) and hydrogen was flowed over the catalyst (0.1 l/h). The reaction mixture was analysed by gas chromatography on a Hewlett Packard 5830A apparatus equipped with two successive separation columns, filled respectively with ®Porapak QS for a TCD detector and 5Å-molecular sieve for FIDCG detector.
Reaction rates (r) were calculated from the following equation:
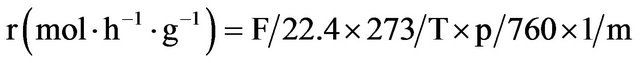 (1)
(1)
where F is the flow rate of the vector gas (l/h), T the reaction temperature (K), p the partial pressure of the products (torr) and m the mass of the catalyst (g).
The activation energy (Ea) was obtained from the Arrhenius plot according to:
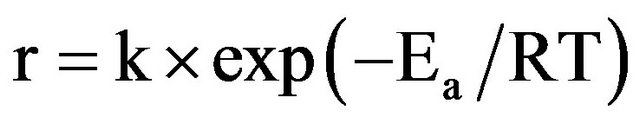 (2)
(2)
where R is the perfect gas constant and k the pre-exponential factor.
3. Results and Discussion
3.1. Characterization of Catalysts
In the UV, the Keggin heteropolyanions show two absorption bands, one around 200 nm and another around 280 nm. According to literature [28], when catalyst concentration inside the suspension decreases, the two bands disappear gradually indicating decomposition of the polyanion with dilution [29]. Therefore, intermediate concentrations of different catalyst systems were used here for characterization, as shown in Figure 1.
Values of absorption bands are summarized in Table 1. The Table shows main band observed for each prepared catalyst. The bands are assigned respectively to the vibrations of terminal Mo = Ot and bridging bonds. The bridging bonds are two types: the inter-bridges (M-Ob-M) that occur between two adjacent octahedra, and the intrabridges (M-Oc-M) that occur within same octahedron.
Solid-state FT-IR spectra measured for different prepared catalysts are given in Figure 2. The spectra display similar patterns characteristic for the Keggin structure [30,31]. Values of IF bands are summarized in Table 2.
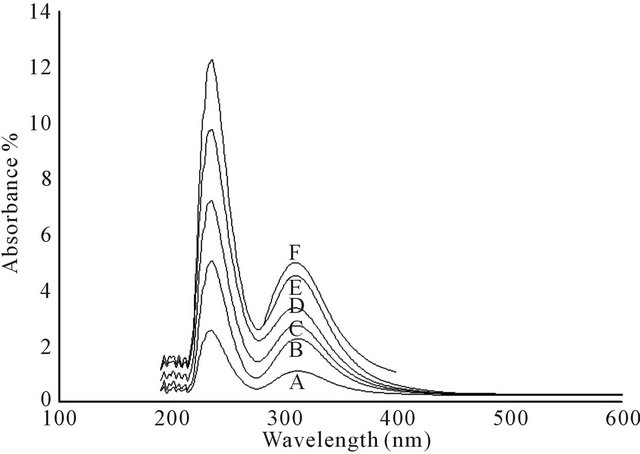
Figure 1. UV spectra for different catalyst systems (A: PMo12; B: FePMo12; C: PMo11Ni; D: PMo11Co; E: PMo11Mn; F: PW11). A suspension of each catalyst system in acetonitrile/water mixture was used for UV absorption spectra.
Table 1. Main UV bands observed for different HPA catalyst systems.
Figure 2. Solid state FT-IR spectra measured as KBr discs for a: PW11; b: PMo11Mn; c: PMo11Ni; d: PMo11Co; e: KFePMo12; f: PMo12.
Table 2. FT-IR absorption band values for different catalyst systems, together with their assignments.
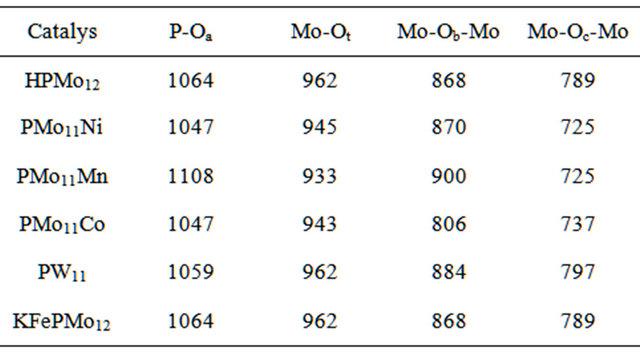
FT-IR spectra show that main peaks around 1064, 961, 861 and 786 cm−1 are attributed to P-Oa, Mo = Ot, MoOb-Mo, and Mo-Oc-Mo bonds, respectively.
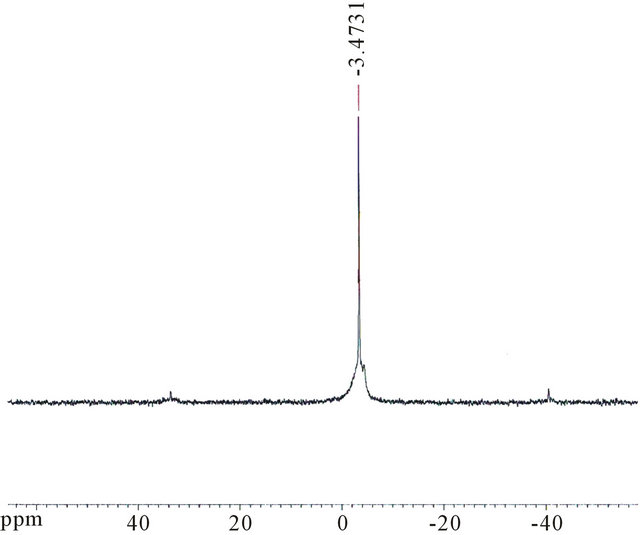
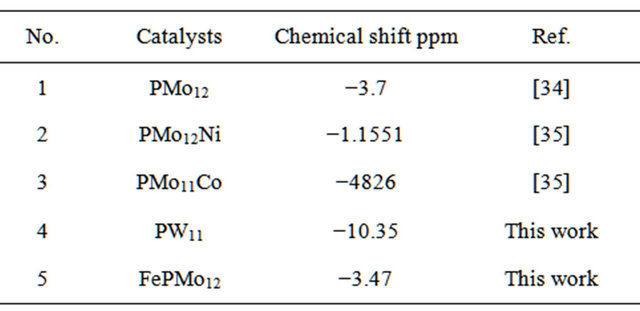
31P NMR spectra were measured for only two catalyst types, as shown in Figures 3(a) and (b). Figure 3(a) shows a single peak at −3.47 ppm for salt Fe0.1PMo12O40, which indicates purity of the sample. The KPW11 heteropolyanion showed a signal at 10.35 ppm, Figure 3(b).
The signal is characteristic for the lacunary species K7[α-PW11O39] [32,33]. 31P NMR measured and literature data are summarized in Table 3.
XRD patterns were measured for all solid catalysts systems in the powder form. Figure 4 shows the diffractograms of HPMo12, FePMo12 and PMo11Ni. The diffractogram of H3PMo12O40·13H2O was consistent with literature [36] and corresponded to a triclinic structure.
Other diffractograms show that the salt FePMo12 crystallises in a cubic structure with intense (222) peak [16, 37]. This corresponds to same diffraction plane for ammonium salts of (PMo11Ni, PMo11Co, PMo11Mn) with a monoclinic structure.
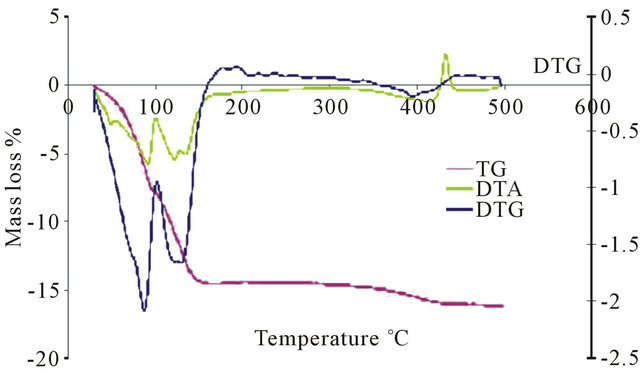
Thermogravimetric (TGA) analysis of the H3PMo12O40 acid shows two mass loss signals between 40˚C and
 (a)
(a)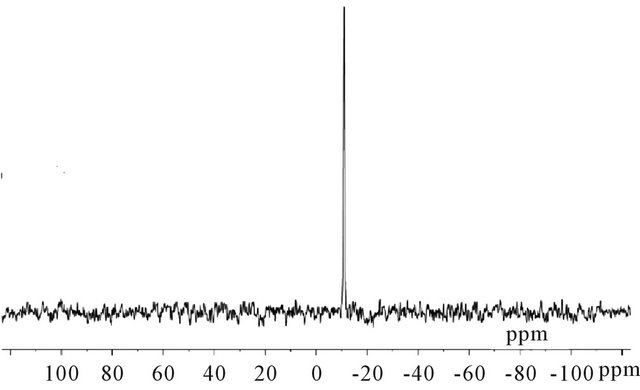 (b)
(b)
Figure 3. 31P NMR spectra for: (a) FePMo12; (b) PW11.
Table 3. 31P NMR measured and literature data for different catalyst systems.
140˚C corresponding to the departure of water of crystallization or hydration. A second mass loss at temperatures between 250˚C and 350˚C, was attributed to water content resulting from combination of H+ ions and network oxygen. This mass loss leads to a reversible modification of the polyanion.
In differential thermal (DT) analysis, the loss of water corresponds to endothermic peaks. An exothermic peak was observed above 350˚C and was attributed to acid decomposition into P2O5 and MoO3 oxides.
 (a)
(a)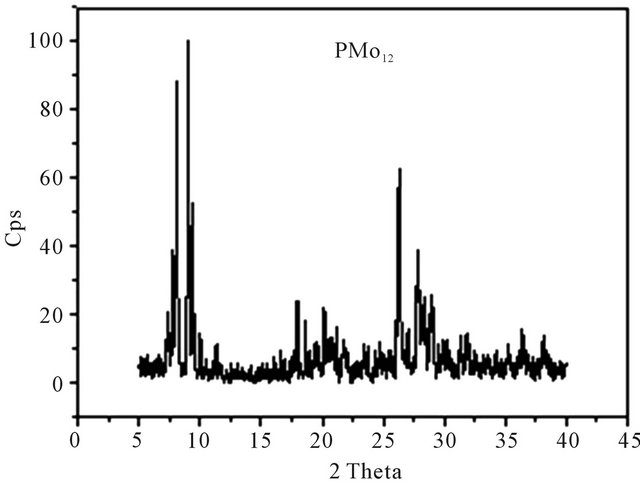 (b)
(b)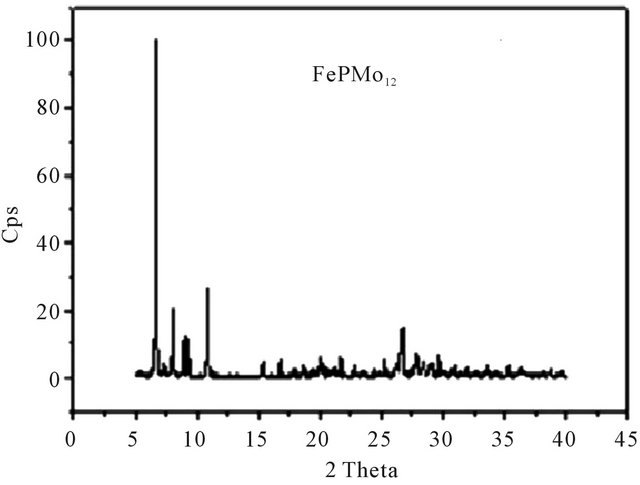 (c)
(c)
Figure 4. XRD spectrum of: (a) PMo11Ni; (b) PMo12; (c) FePMo12.
TGA analysis for the salts PMo11Ni, PMo11Co, PMo11Mn, FePMo12, PW11 shows three mass losses. A first mass loss occurred around 100˚C, which is interpreted as the start of physisorbed water loss. A second departure is at around 240˚C to 300˚C, which is due to loss of constitution water molecule. Water molecules coordinated to nickel and cobalt ions are involved here. Finally, a loss around 300˚C to 400˚C, corresponding to departure of molecular ammonia or molecular nitrogen, was observed. Salt catalysts exhibit higher thermal stability than H3PMo12O40 (Figure 5). The temperature at which their degradation starts depends on the position of the ion M3+. When the counter ion involved metal ionic species, the decomposition temperature exceeded 500˚C. On the other hand, the heteropolyacid H3PMo12O40 is less thermally stable than the salt, and its decomposition started at 350˚C.
3.2. Catalytic Study
3.2.1. Effect of Catalyst Type
Steady-state activity for catalytic n-hexane dehydrocyclization was reached within about 4 h. The observed products were benzene, (one most preferred product), cyclohexane, cyclohexene, cycloheadiene and methane (least preferred product). As expected, the catalytic activity and selectivity were dependent on the nature of the catalyst.
Figure 6 shows the conversion of n-hexane using different catalysts. The conversion of the solid catalyst was sensitive to the nature of the coordination ion (Mo6+, W6+, Ni3+, Co3+, Mn3+) and counter ion H3O+, 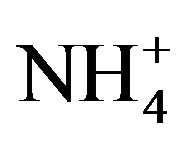 , Fe3+). The replacement of mobile M (Ni3+, Co3+, Mn3+) ions in the HPA framework increased the activity of the catalyst. Replacing H+ ion with Fe3+ lowered the activity. PW11 exhibited relatively high activity as well.
, Fe3+). The replacement of mobile M (Ni3+, Co3+, Mn3+) ions in the HPA framework increased the activity of the catalyst. Replacing H+ ion with Fe3+ lowered the activity. PW11 exhibited relatively high activity as well.
Detailed studies on n-hexane isomerization catalysis with HPC are summarized in (Tables 5 and 6). The results show that benzene was the major product at low conversion level while using different catalysts. Cyclohexane, cyclohexene, cyclohexadiene, benzene and methane were also observed. As the conversion level increased benzene product ratio was lowered in ratio of methane product.
The 30% activity for H3PMo12O40 (or even 100% for other catalysts excluding FePMo) shows that the protons in anhydrous HPA are accessible to the reactant molecules. The high catalytic activity of HPA may thus be related to high acid strength and high mobility of protons, as reported earlier [38]. The resulting protons are responsible of the aromatization of n-hexane. Cyclohexane, cyclohexene cyclohexadiene were also obtained, with variable ratios depending on type of catalyst. An important selectivity obtained by PMoNi catalyst involved cyclohexane 14%, cyclohexene 2%, cyclohexadiene 40%. A lower selectivity with PMoCo and PMoMn catalysts was observed. The results indicate that both relatively strong acidity and metallic propriety are responsible for
 (a)
(a)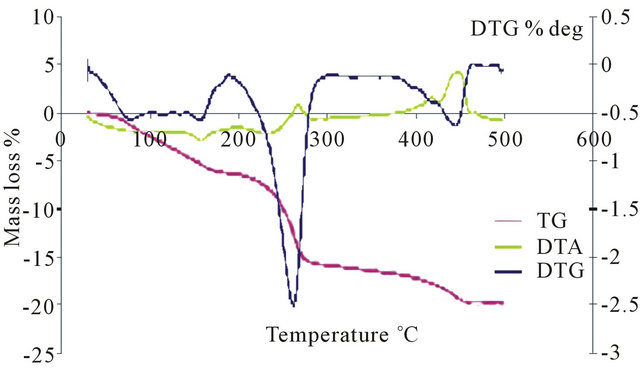 (b)
(b)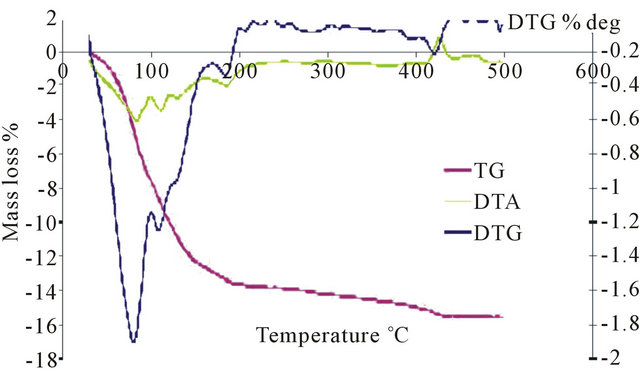 (c)
(c)
Figure 5. Thermal analysis of: (a) PMo12; (b) PMo11Ni; (c) FePMo12.
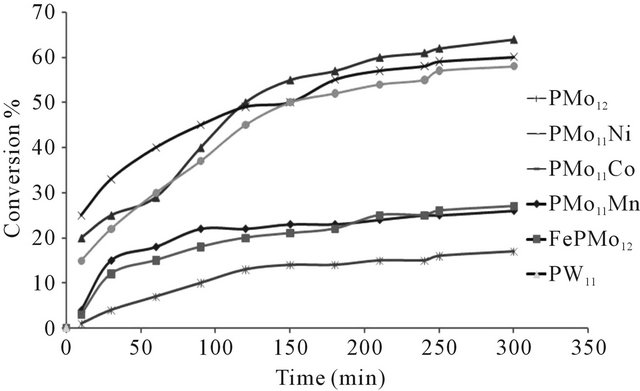
Figure 6. Conversion of n-hexane as function of time at 350˚C, (using N2 as carrier gas).
Table 4. Lattice parameters (A˚) measured for different catalyst systems.
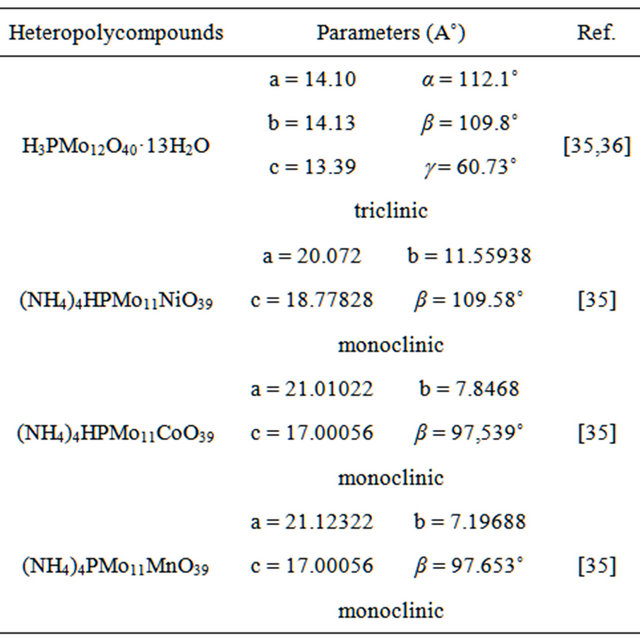
Table 5. Catalytic performances of catalysts (Carrier gas H2, flow rate: 0.1 I\h).
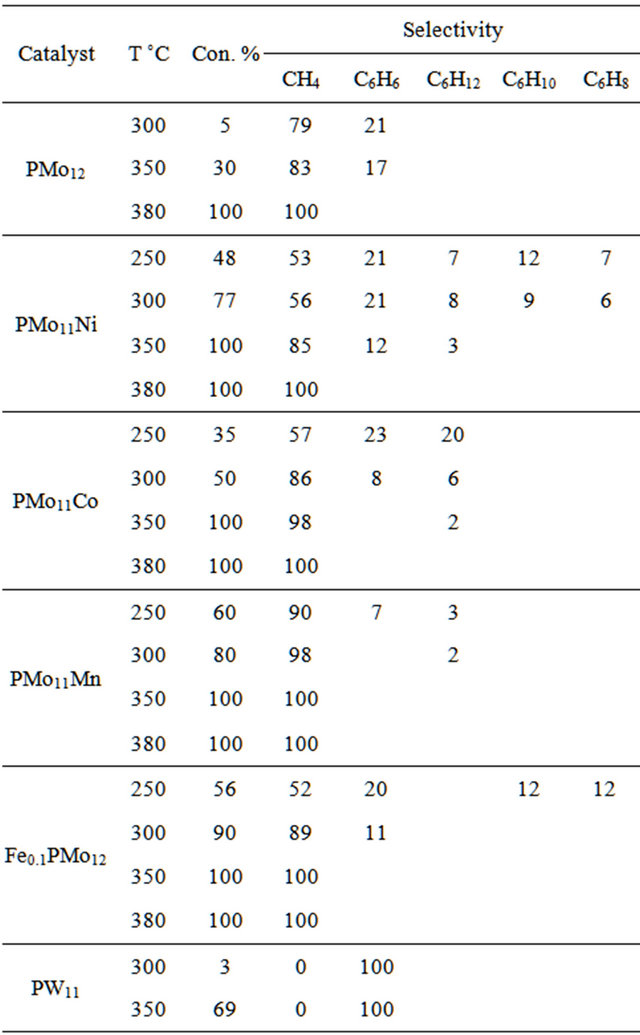
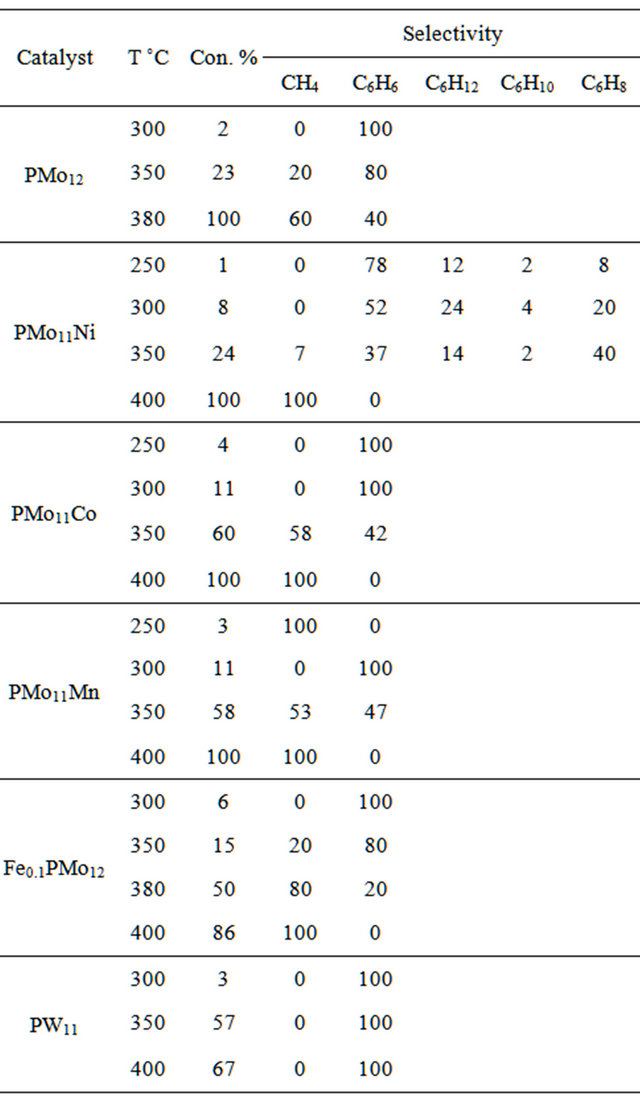
Table 6. Catalytic performances of catalysts (carrier gas N2, flow rate: 0.1 L\h).
catalyst efficiency at lower temperature (lower than 350˚C).
The PW11 catalyst showed specially high efficiency, selectivity and stability towards benzene formation. For example, PW11 showed high activity (69%) conversion and high selectivity (100%) at 350˚C compared to PMo12 (30% conversion and 17% selectivity), PMoNi (100% and 11.5%), and PMoCo and PMoMn (100% and 0%). Such behaviour should be ascribed to the Lewis acid properties of the TBA cations, Also, we may conclude that Lewis acid species should be important components of the active site in the isomerization of n-hexane reaction over HPAs, in agreement with other studies [39,40].
3.2.2. Effect of Temperature
The effect of temperature on efficiency and benzene selectivity has been studied for different catalysts. Figure 7 shows how conversion varies with reaction temperature. The temperature curves exhibit linear character. For dif-
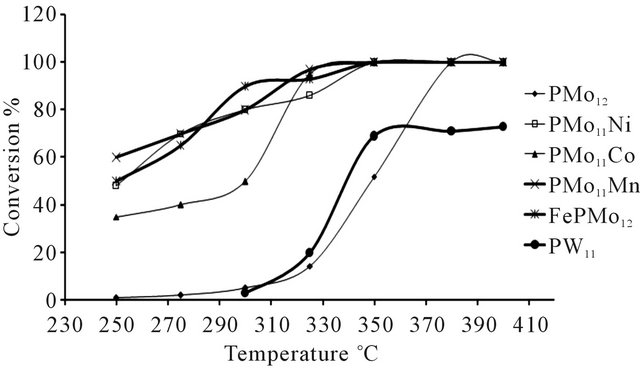
Figure 7. Conversion of n-hexane as function of temperature, (using N2 as carrier gas).
ferent catalysts, overall hexane conversion increased with temperature. The transformation of n-hexane on the tested catalysts started only above 250˚C and the conversion reached 100% at 350˚C - 400˚C.
Until 300˚C, benzene was the unique or major product. The selectivity to benzene decreased with increasing temperature (Tables 4 and 5). The conversion increased from 2 up to 30%, whereas benzene selectivity decreased from 100% to 80% when the reaction temperature was increased from 300˚C to 350˚C when using HPMo12O40 heteropolyacid catalyst.
Methane was obtained with all catalysts at temperature above 300˚C. Methane production selectivity increased with temperature at the expense of benzene. At 400˚C n-hexane was converted completely to methane with all catalysts. The high methane selectivity was with PMoMn catalysts.
At 400˚C the H3PMo12 heteropolyacid was decomposed into Mo3 and P2O5. This result agrees with literature [41] which showed that the isomerization of n-hexane was due to surface Bronsted acid sites.
3.2.3. Activation Energy
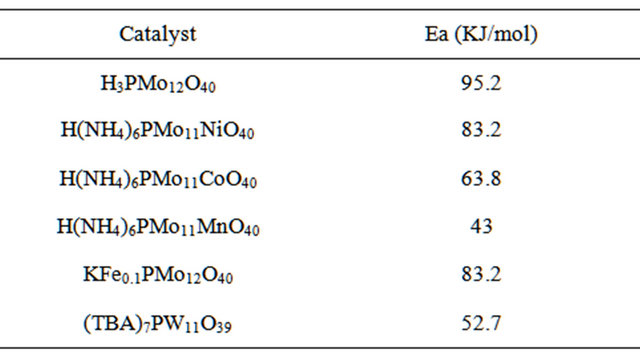
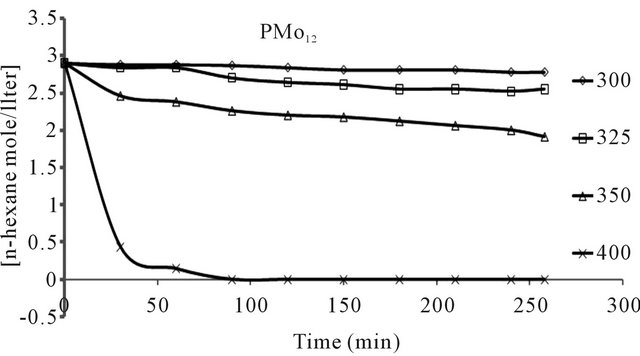
n-hexane conversion rate were determined over the temperature range 200˚C - 450˚C. From (Figures 8(a)-(f) n-hexane dehydrocyclization rate constants were determined over the temperature range 200˚C - 450˚C. From Arrhenius plots (Figure 9), Activation energies for the transformation of n-hexane over acidic catalysts were calculated as shown in Table 7.
Values of activation energies for the overall process of dehydrocyclization and aromatization of n-hexane using the acidic catalyst systems (43 to 95.2 kJ∙mol−1) are much lower than those known for metal type catalysts (typically 230 to 293 kJ∙mol−1) [42]. Acidic catalytic activity is expected to occur by the highly acidic heteropoly compounds. Actually the activation energies are comparable to those found for the transformation of the n-hexane in benzene using acidic catalysts. [43].
Furthermore, the selectivity of the benzene decreases, Table 7. Reaction activation energy for different catalyst systems measured based on amount of n-hexane consumption.
while the conversion increases, with higher temperatures or longer retention times. In case of all catalysts, the benzene is the secondary product of the reaction.
3.2.4. Effect of Time of the Reaction
The conversion of n-hexane was increase with time for all of the catalysts at 350˚C as it is clear from Figure 10. The selectivity of benzene only slightly decreased with time on all catalysts Figure 8, when keeping temperature constant at 350˚C. This due to high thermodynamic stability of benzene.
3.2.5. Effect of Type of Carrier Gas
The type of carrier gas affected both conversion and selectivity of the reaction. The overall conversion increased by using H2 as a carrier gas, while benzene selectivity increased when using N2 as a carrier gas. This applied to all catalytic systems, as shown in Tables 5 and 6. Comparison between the two Tables shows that for each catalyst system, using H2 gas gives higher conversion than using N2. On the other hand, the Tables show using N2 gave higher benzene selectivity than using H2, for each catalyst.
3.2.6. Mechanism
A plausible mechanism has been suggested to explain the observations discussed above. As shown in Scheme I, two possible products can be expected by the HPA catalyst systems. The Scheme shows that with excess H2 (carrier gas, as discussed above) the carbonium ion formed by super-acid activation of hexane will be cracked into CH4 [44]. Such a process involves complete saturation of all carbon atoms with hydrogen, via complete cracking of the hydrocarbon chain. On the other hand, in case of nitrogen carrier gas (with no hydrogen) dehydrocylization process is favored to dominate, with no significant carbon chain cracking, as shown in the Scheme. The process is multi-stage involving β-H elimination in each step. This explains the production of cyclohexane, cyclohexene and cyclohexadiene as ac-
 (a)
(a)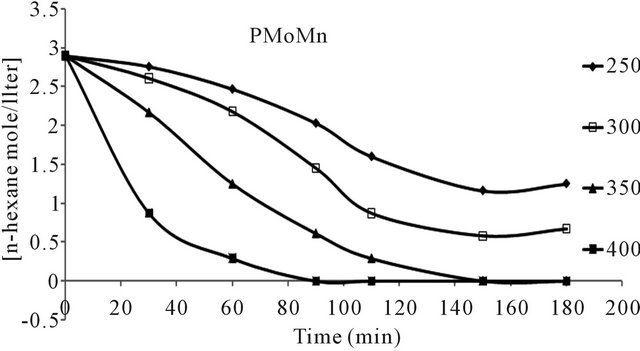 (b)
(b)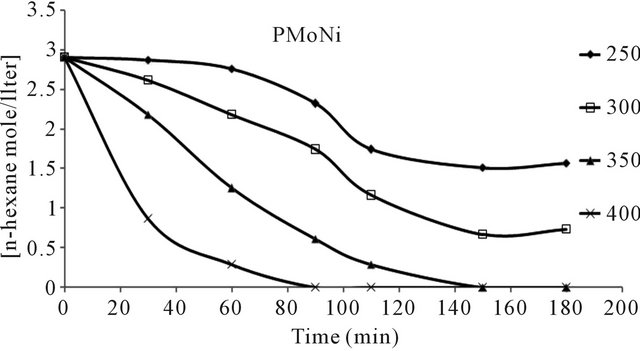 (c)
(c)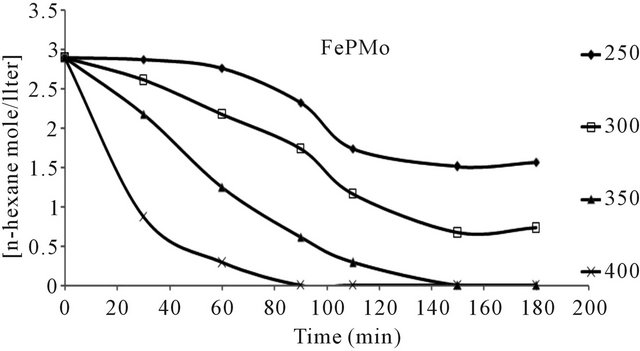 (d)
(d)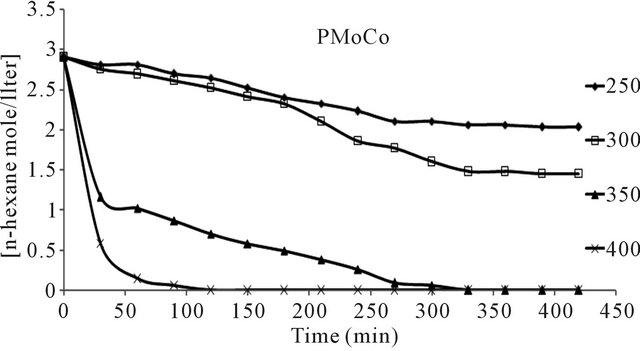 (e)
(e)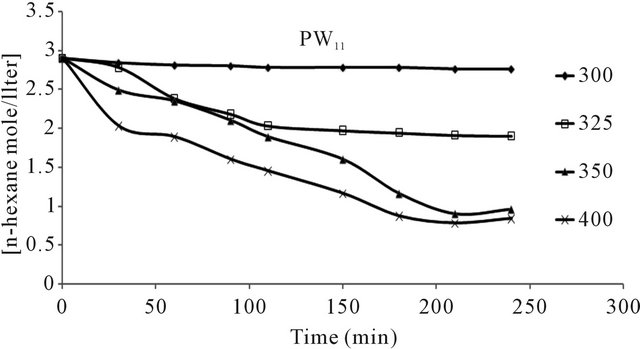 (f)
(f)
Figure 8. n-Hexane conversion as a function of time over: (a) PMo12 catalyst; (b) PMo11Mn catalyst; (c) PMo11Ni catalyst; (d) FePMo12 catalyst; (e) PMo11Co catalyst; (f) PW11 catalyst.
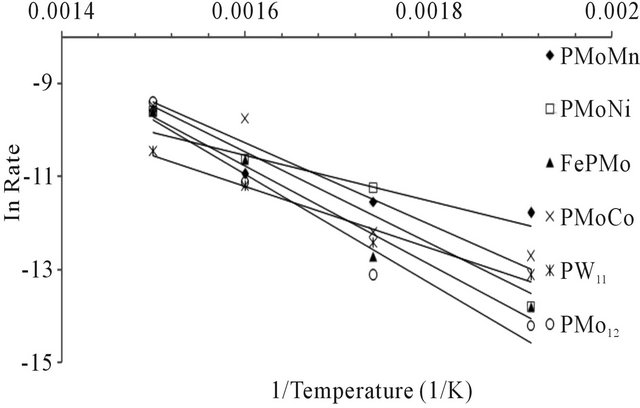
Figure 9. Arrhenuis plots for different catalyst systems (using N2 as carrier gas).
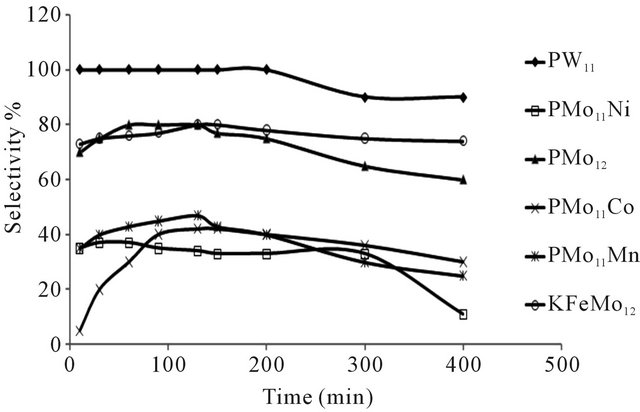
Figure 10. Selectivity of benzene as function of time at 350˚C, (using N2 as carrier gas).
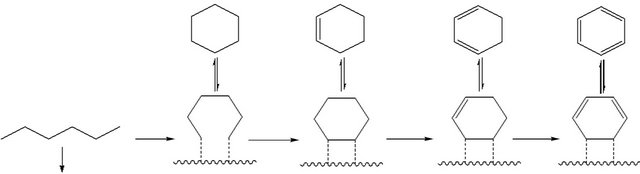
CH4
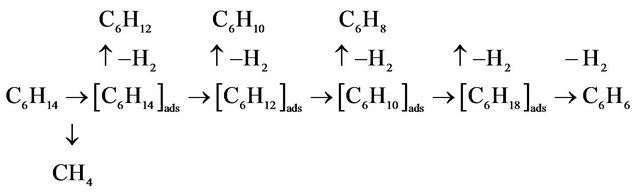
Figure 11. Reaction scheme of n-hexane dehydrocylization over heteropolycompounds catalysts.
companying products to benzene.
It should also be noted that this logic does not fully work under higher temperatures, where CH4 production dominates even in the absence of hydrogen gas. In such a case, high temperature experiments without hydrogen yielded high molecular weight aromatics and tars, in parallel to CH4 formation, which caused some technical difficulties such as reactor blockage during experiments. Therefore, If benzene is the desired product from nhexane, hydrogen gas should not be used, and the reaction must be conducted under mild temperatures (350˚C or lower).
4. Conclusion
A number of molybdenum-based compounds H3PMo12O40, KFePMo12O40, (TBA)7PW12O40·xH2O, (NH4)4PMo11CoO40, (NH4)4PMo11NiO40, (NH4)4PMo11MnO40 have been prepared and characterized by UV, IR, NMR Spectra, TGA and XRD. The compounds were of Keggin type structure. Due to their super-acidic nature, the compounds showed catalytic efficiency in the dehydrocyclization of n-hexane. The product distribution (benzene, cyclohexane, cyclohexene, cyclohexadiene and methane) strongly depends on the nature of the catalyst, the type of carrier gas and the reaction temperature. The observed differences in the behaviour of the studied catalysts should be firstly ascribed to the superacidic nature of the catalysts, which can activate saturated alkanes.
REFERENCES
- B. C. Gates, “Catalytic Chemistry,” John Wiley & Sons, Inc., New York, 1992.
- A. Corma, J. M. Serra and A. Chica, “Discovery of New Paraffin Isomerization Catalysts Based on SO42−/ZrO2 and WOx/ZrO2 Applying Combinatorial Techniques,” Catalysis Today, Vol. 81, No. 3, 2003, pp. 495-506. doi:10.1016/S0920-5861(03)00148-2
- Y. Ono, “A Survey of the Mechanism in Catalytic Isomerization of Alkanes,” Catalysis Today, Vol. 81, No. 1, 2003, pp. 3-16. doi:10.1016/S0920-5861(03)00097-X
- B. Bachiller-Baeza, J. Alvarez-Rodríguez, A. GuerreroRuiz and I. Rodríguez-Ramos, “Support Effects on RuHPA Bifunctional Catalysts: Surface Characterization and Catalytic Performance,” Applied Catalysis A: General, Vol. 333, No. 2, 2007, pp. 281-289. doi:10.1016/j.apcata.2007.09.027
- M. H. Jordao, V. Simoes and D. Cardoso, “Zeolite Supported Pt-Ni Catalysts in n-Hexane Isornerization,” Applied Catalysis A: General, Vol. 319, 2007, pp. 1-6. doi:10.1016/j.apcata.2006.09.039
- Y. Gucbilmez, A. S. Yargic and I. Calis, “A Comparative Characterization of the HPA-MCM-48 Type Catalysts Produced by the Direct Hydrothermal and Room Temperature Synthesis Methods,” Journal of Nanomaterials, Vol. 2012, 2012, Article ID: 210437. doi:10.1155/2012/210437
- I. V. Kozhevnikov, “Heteropoly Acids and Related Compounds as Catalysts for Fine Chemical Synthesis,” Catalysis Reviews, Vol. 37, No. 2, 1995, pp. 311-352. doi:10.1080/01614949508007097
- I. V. Kozhevnikov, K. R. Kloetstra, A. Sinnema, H. W. Zandbergen and H. Van Bekkum, “Study of Catalysis Comprising Keteropoly Acid H3PW12O40 Supported on MCM-41 Molecular Sieve and Amorphous Silica,” Journal of Molecular Catalysis A: Chemical, Vol. 114, No. 1-3, 1996, pp. 287-298. doi:10.1016/S1381-1169(96)00328-7
- H. Hayashi and J. B. Moffat, “Methanol Conversion over Metal Salts of 12-Tungstophosphoric Acid,” Journal of Catalysis, Vol. 81, No. 1, 1983, pp. 61-66. doi:10.1016/0021-9517(83)90146-X
- T. Baba, H. Watanabe and Y. Ono, “Generation of Acidic Sites in Metal Salts of Heteropoly Acids,” Journal of Physical Chemistry, Vol. 87, No. 13, 1983, pp. 2406-2411. doi:10.1021/j100236a033
- K. Nowińska, “Catalytic Activity of Supported Heteropoly Acids for Reactions Requiring Strong Acid\Centres,” Journal of Chemical Society, Faraday Transactions, Vol. 87, No. 5, 1991, pp. 749-753. doi:10.1039/ft9918700749
- V. Haensel, “Process of Reforming a Gasoline with an Alumina-Platinum-Halogen Catalyst,” US Patent No. 24- 79109 and 2479110, 1949.
- H. E. Kluksdahl, “Reforming a Sulfur-Free Naphtha with a Platinum-Rhenium Catalyst,” US Patent No. 3415737, 1968.
- M. H. Jordão, V. Simões and D. Cardoso, “Zeolite Supported Pt-Ni Catalysts in n-Hexane Isomerization,” Applied Catalysis A: General, Vol. 319, 2007, pp. 1-6. doi:10.1016/j.apcata.2006.09.039
- C. Rocchioccioli-Deltcheff, M. Fournier, R. Franck and R. Thouvenot, “Vibration Investigatios of Polyoxometalates. 2. Evidence for Anion-Anion Interaction in Molybdenum (VI) and Tungsten(VI) Compounds Related to the Keggin Structure,” Inorganic Chemistry, Vol. 22, No. 2, 1983, pp. 207-216. doi:10.1021/ic00144a006
- T. Mazari, S. Hocine, N. Salhi and C. Rabia, “Oxidation of Propane over Ammonium-Transition Metal Mixed Keggin Phosphomolybdate Salts,” Journal of Natural Gas Chemistry, Vol. 19, No. 1, 2010, pp. 54-60.
- O. Benlounes, S. Cheknoun, S. Mansouri, C. Rabia and S. Hocine, “Catalytic Activation of C-H Bonds of Hydrocarbons by Heteropolycompounds,” Journal of the Taiwan Institute of Chemical Engineers, Vol. 42, No. 1, 2011, pp. 132-137.
- F. M. Zhang, M. P. Guo, H. Q. Ge and J. Wang, “Hydroxylation of Benzene with Hydrogen Peroxide over Highly Efficient Molybdovanadophos Phoric Heteropoly Acid Catalysts,” Chinese Journal of Chemical Engineering, Vol. 15, No. 6, 2007, pp. 895-898. doi:10.1016/S1004-9541(08)60021-X
- O. Benlounesa, S. Mansouria, C. Rabiab and S. Hocine, “Direct Oxidation of Methane to Oxygenates over Heteropolyanions,” Journal of Natural Gas Chemistry, Vol. 17, No. 3, 2008, pp. 309-312. doi:10.1016/S1003-9953(08)60070-5
- H. Kima, P. Kima, K. Leeb, S. H. Yeomc, J. Yia and I. K. Song, “Preparation and Characterization of Heteropoly Acid/Mesoporous Carbon Catalyst for the Vapor-Phase 2- Propanol Conversion Reaction,” Catalysis Today, Vol. 111, No. 3-4, 2006, pp. 361-365. doi:10.1016/j.cattod.2005.10.048
- J. N. Beltramini, “Studies in Surface Science and Catalysis,” Proceedings of the 3rd International Mesostructured Materials Symposium, Nanotechnology in Mesostructured Materials, Jeju, 8-11 July 2002, pp. 653-656.
- R. S. Drago, J. A. Dias and T. O. Maier, “An Acidity Scale for Brönsted Acids Including H3PW12O40,” Journal of American Chemical Society, Vol. 119, No. 33, 1997, pp. 7702-7710. doi:10.1021/ja9639123
- K. Nowinska, R. Fiedorow and J. Adamiec, “Catalytic Activity of Supported Heteropoly Acids for Reactions Requiring Strong Acid Centres,” Journal of the Chemical Society, Faraday Transactions, Vol. 87, No. 5, 1991, pp. 749-753. doi:10.1039/ft9918700749
- K. J. Nowinska, “Evidence for Superacid Sites on the Ammonium Salt of 12-Tungstophosphoric Acid from a Catalytic Test Reaction,” Journal of the Chemical Society, Chemical Communications, 1990, pp. 44-45. doi:10.1039/c39900000044
- C. Marchal-Roch, J. M. Millet and C. R. Acad, “Phosphomolybdic Heteropolycompounds as Oxidation Catalysts. Effect of Transition Metals as Counter-Ions,” Science Chemistry, Vol. 4, No. 5, 2001, pp. 321-329.
- S. Hocine, C. Rabia, M. M. Bettahar and M. Fournier, “Oxidative Dehydrogenation of Cyclohexane over Heteropolymolybdates,” Studies in Surface Science and Catalysis, Vol. 130, 2000, pp. 1895-1900. doi:10.1016/S0167-2991(00)80478-4
- R. Tayebee, “Simple Heteropoly Acids as Water-Tolerant Catalysts in the Oxidation of Alcohols with 34% Hydrogen Peroxide,” Journal of the Korean Chemical Society, Vol. 52, No. 1, 2008, pp. 23-29. doi:10.5012/jkcs.2008.52.1.023
- T. Okuhara, “Catalytic Chemistry of Heteropoly Compounds,” Advances in Catalysis, Vol. 41, 1996, pp. 113- 252. doi:10.1016/S0360-0564(08)60041-3
- Y. He, W. Sang, J. Wang, R. Wu and J. Min, “Vertically Well-Aligned ZnO Nanowires Generated with Self-Assembling Polymers,” Materials Chemistry and Physics, Vol. 94, No. 1, 2005, pp. 29-33.
- C. L. Hill and C. M. Prosser-Mccartha, “Homogeneous Catalysis by Transition Metal Oxygen Anion Clusters,” Coordination Chemistry Reviews, Vol. 143 ,1995, pp. 407- 455.
- A. V. Churakov, E. A. Legurova, A. A. Dutov, P. V. Prikhodchenko and T. A. Tripol’skaya, “Peroxide Derivatives of Heteropoly Compounds with Keggin Anions [PW12O40]3− and [SiW12O40]4−: Synthesis and Structure,” Russian Journal of Inorganic Chemistry, Vol. 53, No. 8, 2008, pp. 1187-1192. doi:10.1134/S0036023608080068
- T. J. R. Weakley and S. A. Malik, “Triheteropolyanions Containing Copper(II), Manganese(II), or Manganese (III),” Journal of Inorganic and Nuclear Chemistry, Vol. 32, No. 12, 1970, pp. 3875-3890.
- O. Benlounesa, “Oxidation of Methane on Heteropoly Compounds,” Ph.D. Dissertation, University of Mouloud Mammeri-Tizi-Ouzou, Tizi-Ouzou, 2010.
- S. S. Lima, G. I. Park, I. K. Song and W. Y. Lee, “Heteropolyacid (HPA)-Polymer Composite Films as Catalytic Materials for Heterogeneous Reactions,” Journal of Molecular Catalysis A: Chemical, Vol. 182-183, 2002, pp. 175-183. doi:10.1016/S1381-1169(01)00464-2
- S. Hocine, “Heteropolyphosphomolybdates. Preparation, Charactarization, Catalytic Activity of Cyclohexane Oxydehydrogenation,” Ph.D. Dissertation, Houari Boume Diene University, Algiers, 2003.
- A. Popa, V. Sasca, M. Stefanescu, E. Kis and R. Marinkovic-Neducin, “The Influence of the Nature and Textural Properties of Different Supports on the Thermal Behavior of Keggin Type Heteropolyacids,” Journal of the Serbian Chemical Society, Vol. 71, 2006, pp. 235-249. doi:10.2298/JSC0603235P
- G. A. Tsigdinos and C. J. Hallada, “Molybdovanadophosphoric Acids and Their Salts Investigation of Methods of Preparation and Characterization,” Inorganic Chemistry, Vol. 7, No. 3, 1968, pp. 437-441. doi:10.1021/ic50061a009
- T. Okuhara, N. Mizuno and M. Misono, “Catalysis by HeteroPolycompounds—Recent Developments,” Applied Catalysis A: General, Vol. 222, No. 1-2, 2001, pp. 63-77. doi:10.1016/S0926-860X(01)00830-4
- R. Hubaut, B. Ouled Ben Tayeb, W. Kuang, A. Rives and M. Fournier, “Mechanical Mixtures of Me(Ni, Pd)Ce Oxides and Silica-Supported Heteropolyacids: Role and Optimal Content of Each Active Species for n-Hexane Isomerization,” Kinetics and Catalysis, Vol. 47, No. 1, 2006, pp. 20-24. doi:10.1134/S0023158406010046
- V. V. Brei, O. V. Melezhyk, S. V. Prudius, M. M. Levechuk and K. I. Patrylak, “Superacid WOx/ZrO2 Catalysts for Isomerization of n-Hexane and for Nitration of Benzene,” Studies in Surface Science and Catalysis, Vol. 143, 2002, pp. 387-395. doi:10.1016/S0167-2991(00)80679-5
- B. Demirel and E. N. Givens, “Transformation of Phosphor Molybdic Acid into an Active Catalyst with Potential Application in Coal Liquefaction,” Catalysis Today, Vol. 50, No. 1, 1999, pp. 149-158. doi:10.1016/S0920-5861(98)00472-6
- F. Garin and F. G. Gault, “Mechanisms of Hydrogenolysis and Isomerization of Hydrocarbons on Metals. VIII. Isomerization of Carbon-13 Labeled Pentanes on a 10% Platinum-Aluminum Oxide Catalyst,” Journal of the American Chemical Society, Vol. 97, No. 16, 1975, pp. 4466- 4476. doi:10.1021/ja00849a004
- F. R. Ribeiro, C. Marcilly and M. Guisnet, “Hydroisomerization of n-Hexane on Platinum Zeolites,” Journal of Catalysis, Vol. 78, No. 2, 1982, pp. 267-280. doi:10.1016/0021-9517(82)90311-6
- S. Kotrel, H. Knozinger and B. C. Gates, “The HaagDessau Mechanism of Protolytic Cracking of AlKanes,” Microporous and Mesoporous Materials, Vol. 35-36, 2000, pp. 11-20. doi:10.1016/S1387-1811(99)00204-8