Pharmacology & Pharmacy
Vol.3 No.2(2012), Article ID:18728,6 pages DOI:10.4236/pp.2012.32027
Possible Factors Involved in Oral Inactivity of Meropenem, a Carbapenem Antibiotic
![]()
Department of Pharmaceutics, School of Pharmaceutical Sciences, Health Sciences University of Hokkaido, Ishikari-Tobetsu, Hokkaido, Japan.
Email: *saitoh@hoku-iryo-u.ac.jp
Received January 15th, 2012; revised February 12th, 2012; accepted March 5th, 2012
Keywords: Meropenem; Oral Inactivity; Secretory Transport; Degradation; Gastric pH
ABSTRACT
Meropenem, a carbapenem antibiotic, is inactive after oral administration and administered exclusively by injection. In this study, in order to address the factors involved in the oral inactivity of meropenem, in vitro permeation characteristics across rat ileal segments was investigated using diffusion cells. Moreover, stability of meropenem was evaluated in the Japanese Pharmacopoeia (JP) 1st and 2nd fluid for disintegration test. Cefotaxime, ceftibuten, and faropenem were used for comparison. The permeation of meropenem across rat ileal segments was approximately 5-fold greater in secretory direction than in absorptive direction. The secretory-oriented transport of meropenem markedly diminished by replacement of D-glucose in the experimental medium with unmetabolizing 3-O-methyl-D-glucose, suggesting that the secretory transport of meropenem was an energy-dependent process. Cefotaxime exhibited extensively secretory-oriented permeation. On the other hand, much weaker directionalities were observed in ceftibuten and faropenem. While meropenem as well as other three β-lactam antibiotics was stable in JP 2nd fluid (pH 6.8), it declined rapidly in JP 1st fluid (pH 1.2). These results suggest that, in addition to the hydrophilic property of meropenem, its instability at gastric pH and secretory transport in the small intestine are possible factors involved in the inactivity of meropenem after oral administration.
1. Introduction
Recently, various carbapenem antibiotics have been developed to overcome microbial resistance to cephems and penicillins [1-3]. However, currently available carbapenems such as imipenem, meropenem, panipenem, biapenem and doripenem are orally inactive and, thus, administered exclusively by injection. The lack of oral activity of carbapenems is attributed mainly to their hydrophilic properties, which disadvantage their passive diffusion across the intestinal epithelium. Development of oral dosage forms of carbapenems is considered to be beneficial to the treatment of outpatients suffering from infectious diseases. Therefore, prodrug approaches have been undertaken to overcome the poor oral availability of carbapenems [4]. Some prodrugs have been reported to afford increases in the oral availability of parent carbapenems [5,6].
There are many reports demonstrating that various efflux transporters, which locate on the apical membrane of enterocytes, behave as potent absorption barriers against clinically relevant drugs [7]. Previously, it was shown that a secretory transporter different from P-glycoprotein (MDR1, ABCB1) was involved in limiting absorption of b-lactam antibiotics such as cefazolin, cefoperazone, and cefaloridine [8,9]. It is, therefore, feasible that similar secretory transport system potentially interferes with the translocation of carbapenems from intestinal lumen into portal vein. Until now, however, little information has been available about involvement of these factors in the oral inactivity of carbapenems. Also, some factors besides low lipophilicity may be responsible for the poor availability of carbapenems following oral administration. For example, degradation in the gastrointestinal lumen or intestinal epithelium often becomes a critical reason for poor oral availability. It has long since been shown that some b-lactam antibiotics are unstable in aqueous solution [10].
Meropenem was introduced into clinical use worldwide in the 1990s as a parenteral carbapenem [11]. This carbapenem exerts an excellent intrinsic stability to human renal dehydropeptidase I (DHP-I), which is known to extensively inactivate imipenem and panipenem [12]. Although some carbapenemases have been reported to compromise the efficacy of meropenem [13], the relatively low toxicity of meropenem has led to its increasing use in the treatment of serious infections in adult and pediatric patients [14]. In this study, using meropenem as a model carbapenem, we addressed factors restricting oral availability of carbapenems, with special interest in its permeation manner across the rat intestinal segments and its stability at the gastrointestinal pH.
2. Experimental
2.1. Materials
Meropenem trihydrate (Meropen® for intravenous drip infusion) was purchased from Dainippon Sumitomo Pharma Co. (Osaka, Japan). Cefotaxime sodium and glycyltryptophan were obtained from Wako Pure Chem. Ind. (Osaka, Japan) and Sigma (St. Louis, MO, USA), respectively. Faropenem sodium and ceftibuten were kindly supplied by Daiichi Suntory Pharma Co. (Tokyo, Japan) and Shionogi & Co. (Osaka, Japan). Other reagents were of the highest grade available.
2.2. In Vitro Permeation Experiments Using Rat Ileal Segments
In the present study, principles of good laboratory animal care were followed and animal experimentation was performed in compliance with the Guidelines for the Care and Use of Laboratory Animals in the Health Sciences University of Hokkaido.
Unless otherwise mentioned, Tyrode’s solution comprising of 137 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 12 mM NaHCO3, 0.4 mM NaH2PO4 and 6 mM D-glucose was used as experimental medium. In vitro permeation experiments using rat ileal segments were performed using diffusion cells (Corning Corstar, Acton, MA, USA) as described previously [9]. The surface area available for permeation was 1.78 cm2. After overnight fasting, male Wistar rats (Hokudo, Sapporo, Japan) weighing 300 - 400 g were anesthetized with ether and the entire small intestine was quickly removed and rinsed in ice-cold saline. The 10 cm portion immediately distal to the pylorus was discarded and the remaining small intestine was cut in two. The contents of the lower part of the small intestine were flushed out, and it was kept in ice-cold saline as ileal segment. The ileal segment was trimmed to approximately 4 cm in length without including Peyer’s patches and replaced in ice-cold Tyrode’s solution, which had been fully gassed with 95% O2 - 5% CO2, until use. Each piece was cut open longitudinally and its mucosal surface was gently washed with ice-cold saline. The piece was then mounted onto the pins of the diffusion cells, and the cell halves were clamped together. To donor compartment was added 7 mL of drug solution (1 mM), and to receiving compartment was added 7 mL of drug-free Tyrode’s solution. Both solutions had been pre-warmed to approximately 37˚C. The diffusion cells were fixed on exclusive heating blocks and their temperature was maintained at around 37˚C during the experiment. The fluid in each compartment was circulated by gas lift with 95% O2 - 5% CO2 and 0.5 mL receiver solution was taken at designated time points until 120 min for the measurement of drug that permeated either in the mucosal-to-serosal (M-to-S, absorptive) or serosal-tomucosal (S-to-M, secretory) directions. After each sampling, drug-free Tyrode’s solution (0.5 mL) was added to receiver compartment. The pH of mucosal and serosal solution was set at 6.5 and 7.4, respectively.
The cumulative permeation in either direction was plotted vs time for each permeation experiment. The permeation rate was calculated from the slope of the linear portion of the cumulative permeation vs time plot. The tissue mounting process was completed within 30 min after resection of the small intestine.
2.3. Stability Experiments Using the Japanese Pharmacopoeia (JP) 1st and 2nd Fluid for Disintegration Test
The JP 1st and 2nd fluid for disintegration test were prepared according to JP XV. To prepare the 1st fluids, 2 g of sodium chloride and 7 mL of hydrochloric acid were first dissolved in 100 mL of purified water and then diluted to 1000 mL with purified water. To prepare the 2nd fluid, 118 mL of 0.2 M sodium hydroxide solution was added to 250 mL of 0.2 M potassium dihydrogen phosphate (KH2PO4) solution and then diluted to 1000 mL with purified water. The pH of the 1st and 2nd fluid was 1.2 and 6.8, respectively. Drugs were dissolved in the 1st and 2nd fluid at a concentration of 10 µM and incubated at 37˚C for 6 h. At designed time points, the drug concentration in each solution was determined.
2.4. Degradation Experiments Using Supernatant Fluid Obtained by Centrifugation of Mucosal Homogenates
Under ether anesthesia, the entire small intestine was quickly isolated. The mucosa was scraped off with a spatula and homogenized in 2 mL of Tyrode’s solution (pH 7.4). The homogenates obtained were diluted to 10 mL with Tyrode’s solution (pH 7.4) and centrifuged at 27,000× g for 30 min at 2˚C. An aliquot (0.5 mL) of the resultant supernatant fluid was taken and mixed with an equal volume of meropenem solution (10 µM) and the mixture was then stood at 37˚C. At designated time points, the mixture was mingled with 1 mL of methanol, stood for 5 min in ice-cold water, and then centrifuged at 27,000× g for 30 min at 2˚C. The resultant supernatant fluid was used for meropenem assay. For time 0 sample, methanol was added immediately after mixing the supernatant fluid and meropenem solution.
2.5. Determination of Drugs by HPLC
All drugs were assayed using a Shimadzu LC-10A HPLC system (Shimadzu, Kyoto, Japan) equipped with a Shimadzu SPD-10A spectrophotometer. Chromtographic conditions were as follows: column, Inertsil ODS-3 (4.6 mm i.d. × 250 mm, GL Sciences Inc., Tokyo, Japan) for meropenem, glycyltryptophan and faropenem, and Cosmosil 5C18-AR (4.6 mm i.d. × 150 mm, Nakarai Tesque, Kyoto, Japan) for cefotaxime and ceftibuten; mobile phase, 0.05 M KH2PO4/acetonitrile (9:1 for meropenem and 8:2 for faropenem) and 0.05 M KH2PO4/methanol (8:2 for cefotaxime and glycyltryptophan); wave length, 310 nm for meropenem, 305 nm for faropenem, 260 nm for cefotaxime and ceftibuten and 220 nm for glycyltryptophan; column temperature, 50˚C; flow rate, 0.8 - 1 mL/min; and injection volume, 20 µL. All drugs were reproducibly assayed with a coefficient of variance of less than 3%.
2.6. Data Analysis
Data have been expressed as the mean with standard errors (S.E.) of 4 experiments. Statistical analysis was performed using unpaired Student’s t-test, and p < 0.05 was considered to be significant.
3. Results
3.1. In Vitro Permeation of Meropenem across Rat Ileal Segments
The cumulative permeation of meropenem (1 mM) across rat ileal segments almost linearly increased with time both in M-to-S and S-to-M directions (Figure 1). The Sto-M permeation of meropenem across rat ileal segments was much greater than the M-to-S permeation, and the S-to-M/M-to-S ratio of permeation rate was approximately 4.9 (Table 1). When D-glucose in Tyrode’s solution was replaced with 3-O-methyl-D-glucose (3-O-MG) that is not metabolized in cells, the permeation rate of meropenem in M-to-S direction became slightly greater compared with that in the presence of D-glucose. On the other hand, the permeation rate in S-to-M direction markedly decreased and became approximately 40% of that in the presence of D-glucose (Table 1). As a result, the secretory orientation of meropenem permeation across rat ileal segments almost completely disappeared (Table 1).
For comparison, the permeation manners of cefotaxime, ceftibuten, and faropenem were investigated under the experimental conditions containing D-glucose. Similarly to meropenem, the permeation of cefotaxime, an orally inactive cephem antibiotic, across rat ileal seg-
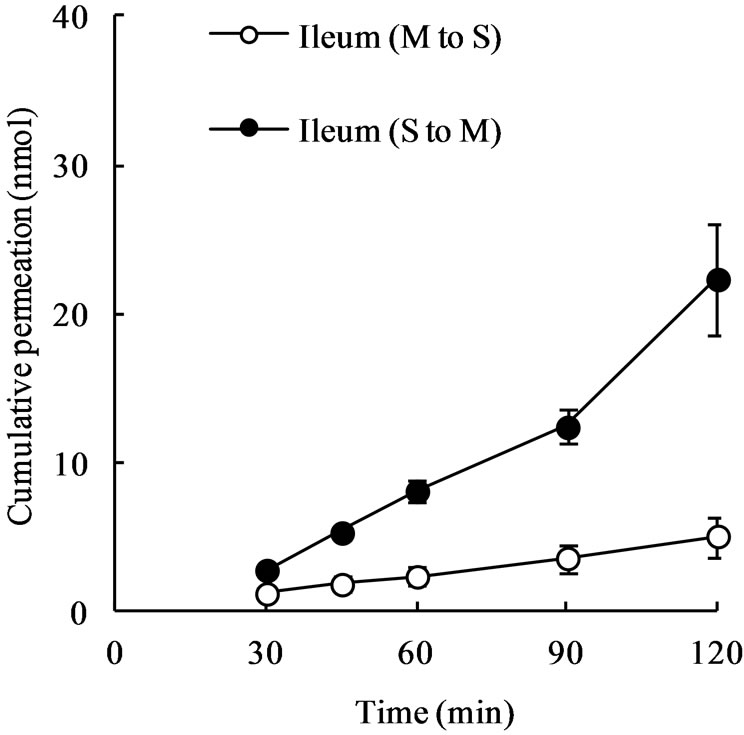
Figure 1. Mucosal-to-serosal (M-to-S) and serosal-to-mucosal (S-to-M) permeation of meropenem across rat ileal segments. Ileal segments (approximately 4 cm in length) were mounted onto diffusion cells and 7 mL of meropenem solution (1 mM) was introduced into either the mucosal or serosal compartment. At designated time points, 0.5 mL of sample solution was taken from the receiver compartment for drug assay. Each point represents the mean ± S.E. of 4 experiments. Some error bars are hidden behind symbols.
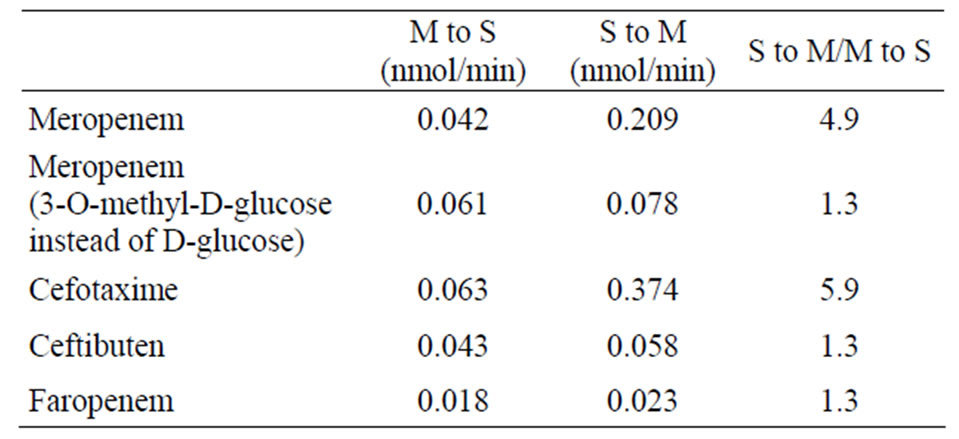
Table 1. Permeation rates of meropenem, cefotaxime, ceftibuten, and faropenem across rat ileum.
ments was greatly secretory-oriented (Figure 2) and the S-to-M/M-to-S ratio of permeation rate was approximately 5.9 (Table 1), being greater than that in meropenem. On the other hand, the permeation of ceftibuten and faropenem was slightly greater in S-to-M direction than in M-to-S direction (Figure 2) and the S-to-M/M-to-S ratio of permeation rates in these two drugs was 1.3 (Table 1).
3.2. Stability of Meropenem in JP 1st and 2nd Fluids
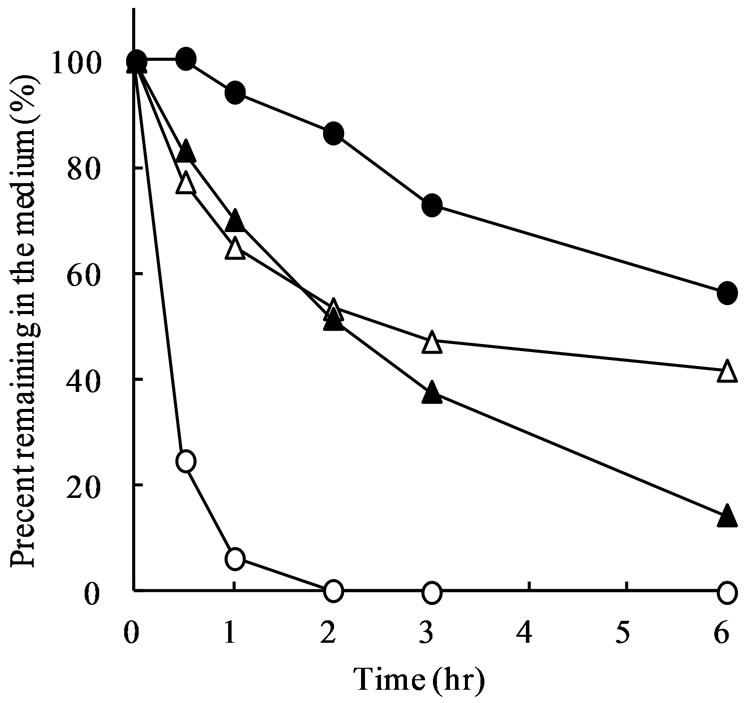
Figure 3 shows the percentage of meropenem, cefotaxime, ceftibuten, and faropenem remaining in JP 1st and 2nd fluid during incubation at 37˚C. In the JP 2nd fluid with pH of 6.8, meropenem was stable and only 10% was disappeared after 6 h (Figure 3(b)). However, meropenem was very unstable in the JP 1st fluid with pH of 1.2 and approximately 80% of meropenem disappeared by 30 min after the start of incubation. After 1 h, mero-
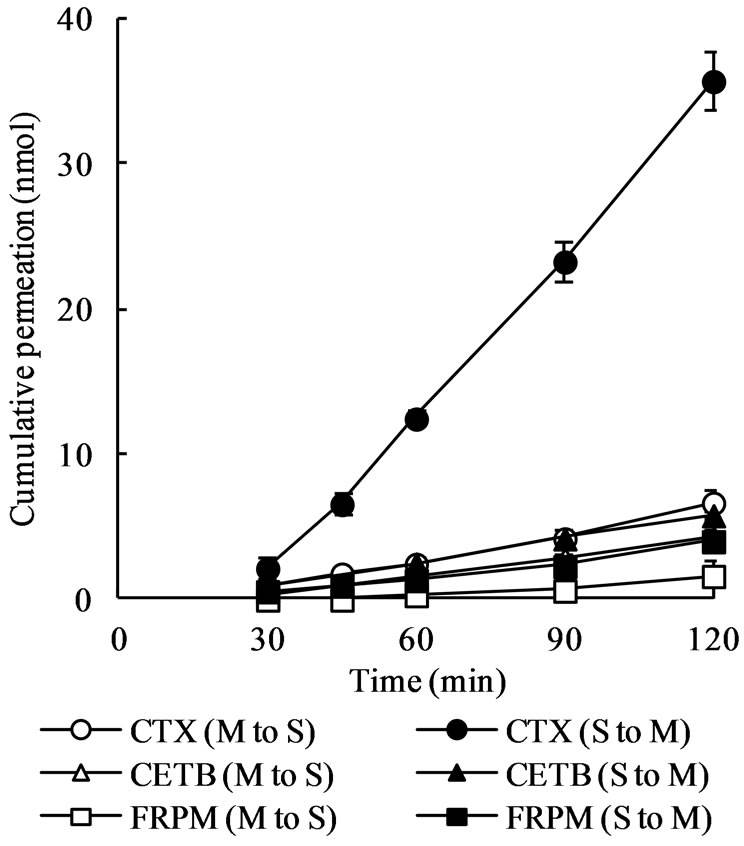
Figure 2. Mucosal-to-serosal (M-to-S) and serosal-to-mucosal (S-to-M) permeation of cefotaxime (CTX), ceftibuten (CETB), and faropenem (FRPM) across rat ileal segments. Ileal segments (approximately 4 cm in length) were mounted onto diffusion cells and 7 mL of drug solution (1 mM) was introduced into either mucosal or serosal compartment. At designated time points, 0.5 mL of sample solution was taken from the receiver compartment for drug assay. Each point represents the mean ± S.E. of 4 experiments. Some error bars are hidden behind symbols.
penem remaining in the JP 1st fluid was less than 5% (Figure 3(a)). Although cefotaxime, ceftibuten, and faropenem time-dependently disappeared in the JP 1st fluid, they are rather stable compared with meropenem (Figure 3(a)). The percentage of cefotaxime, ceftibuten, and faropenem remaining in the JP 1st fluid after incubation for 1 h were 65.0%, 94.1%, and 70.2%, respectively.
3.3. Stability of Meropenem in Supernatant Fluid Obtained from Mucosal Homogenate of Rat Small Intestine
In order to assess whether meropenem is susceptible to degradation in the enterocytes, its stability was investigated in Tyrode’s solution (pH 7.4) containing supernatant fluid obtained by high-speed centrifugation of mucosal homogenates from the entire small intestine. However, only slight decrease (approximately 5%) in meropenem concentration was observed in the absence or presence of the supernatant fluids for 60 min. For comparison, the disappearance of glycyltryptophan was investigated under the same experimental conditions. The dipeptide was disappeared very rapidly in the medium containing the supernatant fluid and not detected even at the first sampling time point of 5 min, implying that the supernatant fluid from the mucosal homogenates possessed potent enzymatic activity. It was, therefore, considered that meropenem was resistant to the enzymatic
 (a)
(a)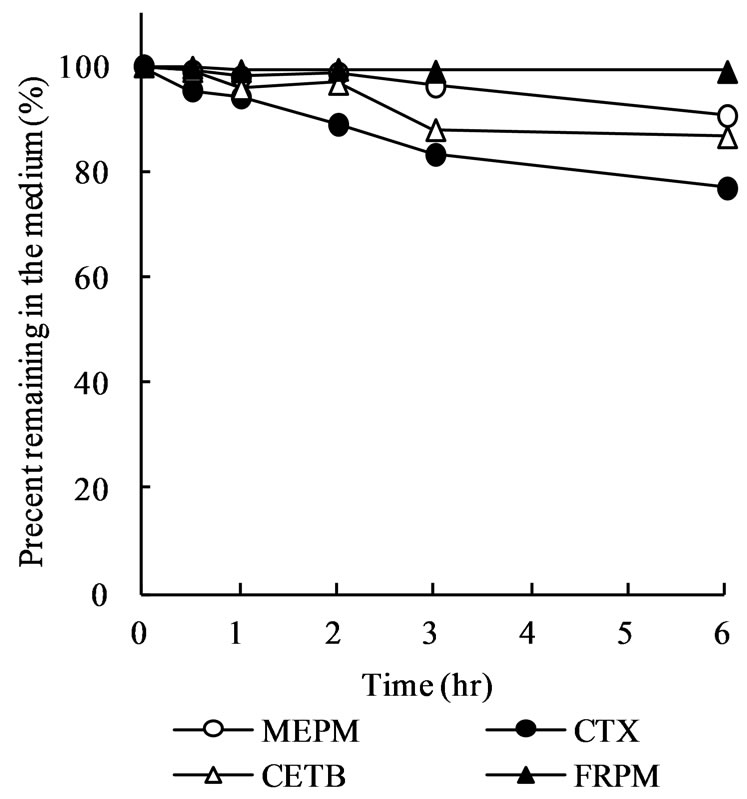 (b)
(b)
Figure 3. Stability of meropenem, cefotaxime, ceftibuten, and foropenem in JP 1st (a) and 2nd (b) fluid. Drugs were dissolved at a concentration of 10 μM and incubated at 37˚C for 6 h. At designated time points, the drug concentration in the solution was determined. Each point represents the mean of 4 experiments.
attack in the medium.
4. Discussion
A variety of drugs are transported as substrates for efflux transporters such as MDR1, multidrug resistance-associated protein 2 (MRP 2, ABCC2) or breast cancer resistance protein (BCRP, ABCG2), located on the brushborder membranes of enterocytes. Such events often lead to poor absorption of substrate drugs. In order to address whether an efflux transport system is involved in oral inactivity of meropenem, we first investigated its in vitro permeation manner across rat intestinal segments using diffusion cells. This method is popular in evaluating transport mediated by a specialized system.
As shown in Figure 1, meropenem exhibited extensively secretory-oriented permeation across rat ileal segments, implying the existence of an efflux transport system for meropenem in the rat small intestine. For comparison, we investigated cefotaxime permeation across rat ileal segments and found that the orally inactive cephem antibiotic exerted much greater permeation in the S-to-M direction than in the M-to-S direction (Table 1). The results from cefotaxime seemed in line with previous ones reported for other orally inactive cephem antibiotics such as cefazolin, cefoperazone, and cefaloridine [8,9], confirming the existence of an efflux transport system for b-lactam antibiotics in the rat small intestine. It was previously shown that the secretory-oriented transport of cefazolin greatly weakened by replacement of D-glucose in the experimental medium with unmetabolizing 3-OMG [8]. In the present study, the secretory-oriented transport of meropenem was almost completely disappeared in the absence of D-glucose (Table 1). Although it remains to be addressed whether the efflux of meropenem and cefotaxime occurs through a common pathway, the involvement of a secretory transport system could be an obstacle to the absorption of hydrophilic meropenem. At present, it is unclear whether the emerging efflux transport system for meropenem commonly carries other carbapenems.
Enzymatic degradation of drugs in the absorption process is often responsible for the low availability following oral administration. However, the present observation that meropenem was rather stable in the medium containing the supernatant fluid obtained from mucosal homogenate negates this possibility. In contrast, meropenem was very unstable in the JP 1st fluid (Figure 3), strongly suggesting that the instability at the gastric pH would lead to extensive loss of meropenem in the stomach following oral administration. Compared to meropenem, orally active faropenem was more stable in the JP 1st fluid (Figure 3(a)). Thus, it is thought that faropenem is capable of escaping extensive degradation in the stomach before intestinal absorption, which is probably connected with the oral activity of this antibiotic.
In conclusion, the present study suggests that, in addition to poor membrane permeability due to the hydrophilic property, intestinal secretory transport and instability in the gastric fluid are possible factors underlying the lack of oral activity of meropenem.
5. Acknowledgements
The authors would like to thank to Dr. Michiya Kobayashi, Professor, Health Sciences University of Hokkaido for his support to this study.
REFERENCES
- A. Dalhoff, N. Janjic and R. Echols, “Redefining Penems,” Biochemical Pharmacology, Vol. 71, No. 7, 2006, pp. 1085-1095. doi:10.1016/j.bcp.2005.12.003
- G. G. Zhanel, R. Wiebe, L. Dilay, K. Thomson, E. Rubinstein, D. J. Hoban, A. M. Noreddin and J. A. Karlowsky, “Comparative REVIEW of the CARBAPENEMs,” Drugs, Vol. 67, No. 7, 2007, pp. 1027-1052. doi:10.2165/00003495-200767070-00006
- M. I. El-Gamal and C. H. Oh, “Current Status of Carbapenem Antibiotics,” Current Opinion of Medicinal Chemistry, Vol. 10, No. 18, 2010, pp. 1882-1887.
- G. Bonfiglio, G. Russo and G. Nicoletti, “Recent Developments in Carbapenems,” Expert Opinion on Investigational Drugs, Vol. 11, No. 4, 2002, pp. 529-544. doi:10.1517/13543784.11.4.529
- T. Isoda, H. Ushirogochi, K. Satoh, T. Takasaki, I. Yamamura, C. Sato, A. Mihira, T. Abe, S. Tamai, S. Yamamoto, T. Kumagai and Y. Nagao, “Syntheses and Pharmacokinetic Studies of Prodrug Esters for the Development of Oral Carbapenem, L-084,” Journal of Antibiotics (Tokyo), Vol. 59, No. 4, 2006, pp. 241-247. doi:10.1038/ja.2006.34
- N. Sato, K. Kijima, T. Koresawa, N. Mitomi, J. Morita, H. Suzuki, H. Hayashi, S. Shibasaki, T. Kurosawa and K. Totsuka, “Population Pharmacokinetics of Tebipenem Pivoxil (ME1211), a Novel Oral Carbapenem Antibiotic, in Pediatric Patients with Otolaryngological Infection or Pneumonia,” Drug Metabolism and Pharmacokinetics, Vol. 23, No. 5, 2008, pp. 434-446. doi:10.2133/dmpk.23.434
- E. M. Leslie, R. G. Deeley and S. P. Cole, “Multidrug Resistance Proteins: Role of P-Glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in Tissue Defense,” Toxicology and Applied Pharmacology, Vol. 204, No. 3, 2005, pp. 216-237. doi:10.1016/j.taap.2004.10.012
- H. Saitoh, C. Gerard and B. J. Aungst, “The Secretory Intestinal Transport of Some Beta-Lactam Antibiotics and Anionic Compounds: A Mechanism Contributing to Poor Oral Absorption,” Journal of Pharmacology and Experimental Therapeutics, Vol. 278, No. 1, 1996, pp. 205-211.
- H. Saitoh, H. Fujisaki, B. J. Aungst and K. Miyazaki, “Restricted Intestinal Absorption of Some Beta-Lactam Antibiotics by an Energy-Dependent Efflux System in Rat Intestine,” Pharmaceutical Research, Vol. 14, No. 5, 1997, pp. 645-649. doi:10.1023/A:1012113430539
- P. C. van Krimpen, W. P. van Bennekom and A. Bult, “Penicillins and Cephalosporins. Physicochemical Properties and Analysis in Pharmaceutical and Biological Matrices,” Pharmaceutisch Weekblad, Scientific Edition, Vol. 9, No. 1, 1987, pp. 1-23.
- J. W. Mouton and J. N. van den Anker, “Meropenem Clinical Pharmacokinetics,” Clinical Pharmacokinetics, Vol. 28, No. 4, 1995, pp. 275-286. doi:10.2165/00003088-199528040-00002
- M. Hikida, K. Kawashima, M. Yoshida and S. Mitsuhashi, “Inactivation of New Carbapenem Antibiotics by Dehydropeptidase-I, from Porcine and Human Renal Cortex,” Journal of Antimicrobial Chemotherapy, Vol. 30, No. 2, 1992, pp. 129-134. doi:10.1093/jac/30.2.129
- L. Poirel, J. D. Pitout and P. Nordmann, “Carbapenemases: Molecular Diversity and Clinical Consequences,” Future Microbiology, Vol. 2, No. 5, 2007, pp. 501-512. doi:10.2217/17460913.2.5.501
- P. Linden, “Safety Profile of Meropenem: An Updated Review of over 6000 Patients Treated with Meropenem,” Drug Safety, Vol. 30, No. 8, 2007, pp. 657-668. doi:10.2165/00002018-200730080-00002
NOTES
*Corresponding author.