Pharmacology & Pharmacy
Vol.3 No.1(2012), Article ID:16952,10 pages DOI:10.4236/pp.2012.31010
Indolin-2-Ones in Clinical Trials as Potential Kinase Inhibitors: A Review
![]()
1Department of Medicinal Chemistry, DCRM Pharmacy College, Jawaharlal Nehru Technological University, Hyderabad, India; 2Department of Pharmaceutical Chemistry, PES’s Rajaram and Tarabai Bandekar College of Pharmacy, Faramagudi, India; 3Department of Pharmaceutical Chemistry, GITAM Institute of Pharmacy, GITAM University, Visakhapatnam, India.
Email: *crp2020@gmail.com
Received October 18th, 2011; revised November 13th, 2011; accepted December 8th, 2011
Keywords: Isatin; Enzyme Inhibition; Anticancer
ABSTRACT
The kinases have been intensely studied because of their involvement in regulating essential cellular activation of signaling cascades in response to extracellular and intracellular stimuli to control cell growth, proliferation, and survival. Recent cancer genomic sequencing studies have revealed that many more kinases contribute to tumor genesis and are potential targets for inhibitor drug development intervention. Herein we review recent results that have helped to unravel the indolin-2-ones underlying the conflicting roles of the kinase inhibition regulation. This review focuses on the potential of kinases as a chemotherapeutic target in cancer treatment and highlights important recent advances in the development of indolin-2-ones as kinase inhibitors.
1. Introduction
The kinases signify a large family of membrane-bound enzymes that play key roles in tumor growth, survival, and metastasis. Many kinases have been found to be closely involved in the processes leading to tumor cell proliferation and survival. Deregulation of kinase function has been implicated in other disorders, including neurological, immunological, metabolic and infectious disease. This has generated a great interest in the development of small molecule kinase inhibitors for the treatment of these disorders [1]. A number of small molecule kinase inhibitor like Sorafenib and Imatinib has been approved for the treatment of cancer. 3-Substituted indolin-2-ones have been reported in several literatures as potent and selective inhibitors of different kinases. Pyrrole-indoline-2-ones were among the first structures identified as kinase inhibitors and have been intensively studied for the inhibition of VEGFR, PDGFR α/β, c-kit, FLT3, and CSF1R [2]. Moreover the importance of indolin-2-one as kinase inhibitor is also reviewed by Underiner et al. [3]. The first compound of this type reaching the clinic was Sunitinib (Sutent; Pfizer) (Figure 1), has been approved and marketed for the treatment of renal cell carcinoma and Imatinib resistant GIST [4]. This review provides an overview of the indolin-2-one based molecules approved by USFDA and compounds under clinical trials as RTKIs.
2. Indolin-2-Ones Drugs in Market
The search for synthetic inhibitors of protein kinases as anti-tumor drugs has been invigorated by the successful approval of a number of molecules that target tyrosine kinases. The indolin-2-one based Sunitinib is a smallmolecule inhibitor of multiple RTK involved in cancer, including VEGFR, PDGFR. It was approved by the USFDA for the treatment of GIST and advanced renalcell carcinoma in January 2006 and European Union approval in January 2007 [5]. Sunitinib is the first anticancer drug which received simultaneous FDA approval for two different indications. It has potent anti-angiogenic effects and direct anti-tumor activities due to the selective inhibition of VEGFR-1 (also known as FLT1), VEGFR-2 (also known as FLK1/KDR), VEGFR3 (also known as FLT4), PDGFRα, PDGFRβ, c-kit, FLT3, RET, and CSF1R.
Toceranib Phosphate [6] (Palladia) an orally bioavailable compound is similar to Sunitinib, exhibiting potent inhibitory activity against members of the split-kinase receptor family, including VEGFR, PDGFR, and Kit, and was therefore predicted to have both anti-angiogenic and direct anti-tumor activity. Toceranib phosphate (Figure 2) is a small molecule with an indolinone chemical structure and approved by USFDA for the treatment of canine cu-
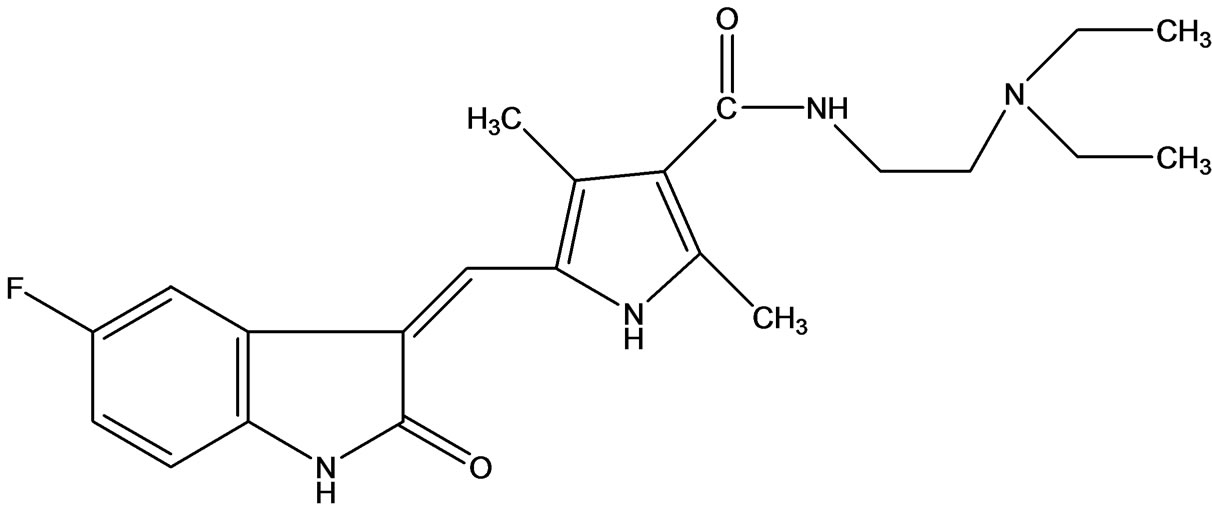
Figure 1. Sunitinib the first anticancer drug received FDA approval for dual indication.
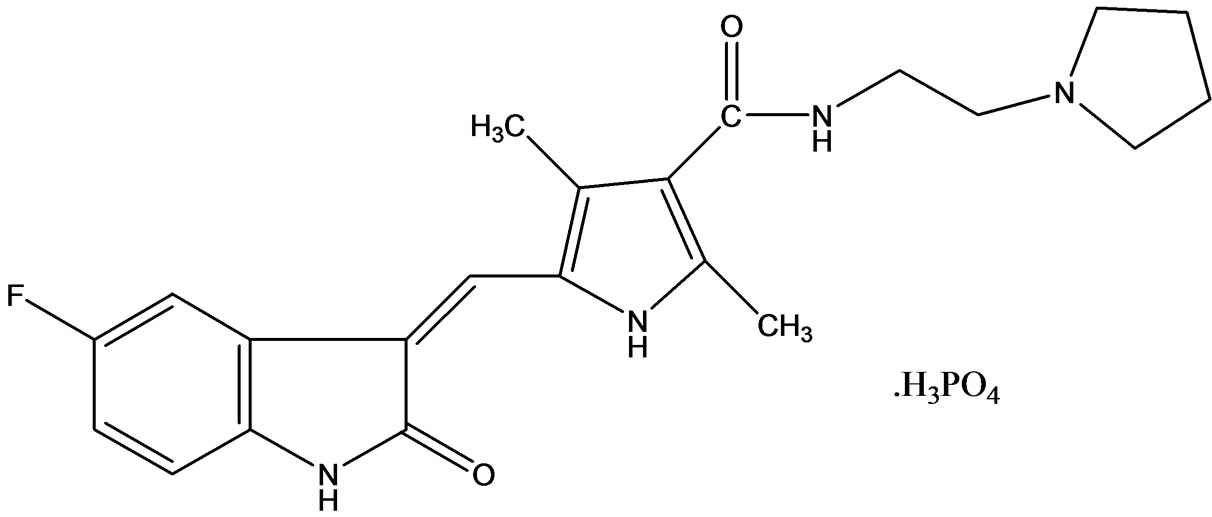
Figure 2. Toceranib phosphate.
taneous (skin-based) mast cell tumors, a type of cancer responsible for about 1 out of 5 cases of canine skin tumors. The drug is approved to treat the tumors with or without regional lymph node involvement in June 2009. Toceranib an TKI and works in two ways: by killing tumor cells and by cutting off the blood supply to the tumor. In a random clinical trial study, Toceranib showed a statistically significant variation in tumor shrinkage when compared to placebo.
The field of anti-viral drug development has made dramatic, impressive strides over the past half century, since the discovery of the first-ever medicinal antiviral, Methisazone [7] (for use in the treatment of smallpox and other related viruses), in the 1950s. Methisazone (Figure 3), an N-methyl derivative of thiosemicarbazone, is active against poxviruses and appears to be effective in the prophylaxis of smallpox and treatment of the complications of vaccination.
3. Indolin-2-Ones That Have Entered Clinical Studies
3.1. SU5416
SU5416 (Semaxanib) (Figure 4) is a synthetic small molecule inhibitor of the tyrosine kinase domain of VEGFR-2. SU5416 is also a potent competitive inhibitor of KIT and less potently the PDGFR (indirectly involved in angiogenesis), but is not directly cytotoxic by itself. Recent work has also suggested that SU5416 may medi-
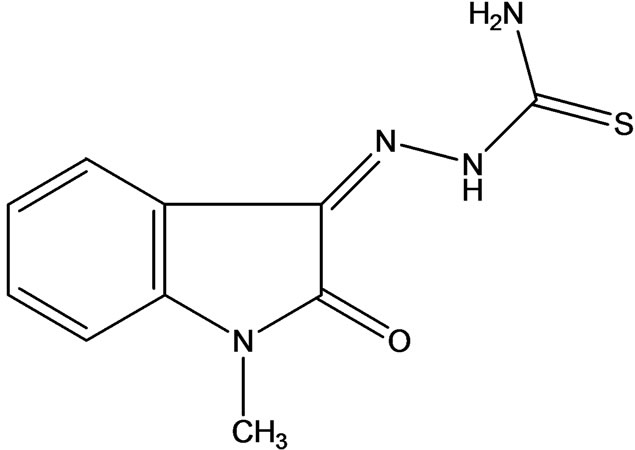
Figure 3. Anti-viral Methisazone.
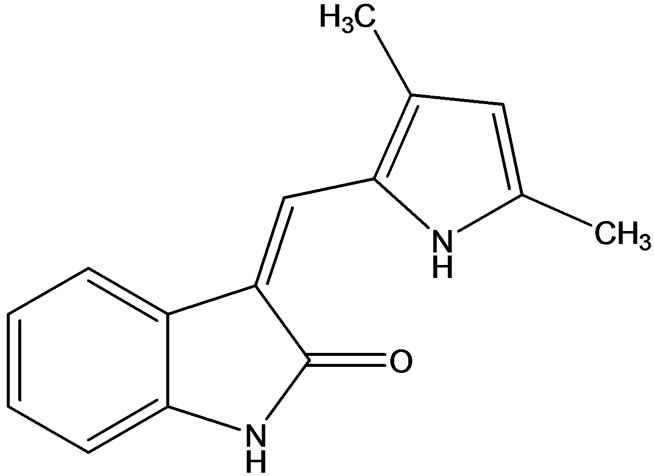
Figure 4. Semaxanib a potent inhibitor of VEGFR-2 and KIT.
ate some of its effects through inhibition of a second VEGFR [8]. Preclinical xenograft models have confirmed inhibition of tumor growth and a reduction in the number of metastases following treatment with SU5416, as well as a decrease in vascular density in treated tumors as measured by intravital video-microscopy. SU5416 recently underwent phase II clinical trials for its safety and efficacy in patients with recurrent or metastatic head and neck cancer [9].
3.2. SU5614
Karsten et al. [10] disclosed the expression of VEGFR-1/2 and its ligand VEGF in AML cell lines and characterized the inhibitory action of the protein TKI SU5614 (Figure 5) (5-Chloro-3-[(3,5-dimethylpyrrol-2-yl)methylene]-2indolinone) on human endothelial and leukemic cells. The results of this study showed intracellular VEGF expression was detected in 9 of 10 leukemic cell lines. In contrast, VEGFR-1 and VEGFR-2 expression was restricted to 6 and 2 out of 10 cell lines, respectively. Although SU5614 was a effective inhibitor of the VEGFinduced endothelial cell sprouting in vitro, the sensitivity of leukemic cells toward the growth inhibitory activity of the compound was determined by the c-kit, but not by the VEGFR-1/2 expression. SU5614 induced growth arrest and apoptosis in c-kit expressing Kasumi-1, UT-7, and M-07e cells and inhibited the stem cell factor induced tyrosine phosphorylation of c-kit.
3.3. SU6668
SU6668 (Figure 6) is a selective inhibitor of the recaptors for VEGFR, PDGFRβ and FGFR1 [11]. SU6668 inhibits the growth of tumor cell lines in vitro and has significant anti-tumor activity in xenografts after daily oral treatment. Laird and co-workers [12] determined the kinase inhibitory profile of SU6668, which is a more water soluble 2-oxindole derivative than SU5416. Using steady state enzyme kinetics with purified recombinant VEGFR-2, they reported that this compound is a competitive inhibitor with respect to ATP.
3.4. SU14813
SU14813 (Figure 7), an orally active indolinone multitargeted TKIs, blocks VEGFR-1, -2, and -3, PDGFR-α, -β, c-kit, and FLT3 at nanomolar concentrations [13]. In vitro, SU14813 inhibited VEGFR-2, PDGFRβ, and FLT3 internal tandem duplication phosphorylation, with cellular IC50 values of 0.04, 0.02, and 0.05 µmol/l, respecttively, and showed anti-tumor efficacy in tumor xenograft models. Preclinical studies in mice demonstrated a lack of major active metabolites, linear pharmacokinetics and oral bioavailability of 40%.
Shem et al. [14] reported a encouraging preclinical data for SU14813 and the clinical implications for its use based on a similar potency to Sunitinib but distinct pharmacokinetic profile, which may favorably affect tolerability and allow greater dosing flexibility, SU14813
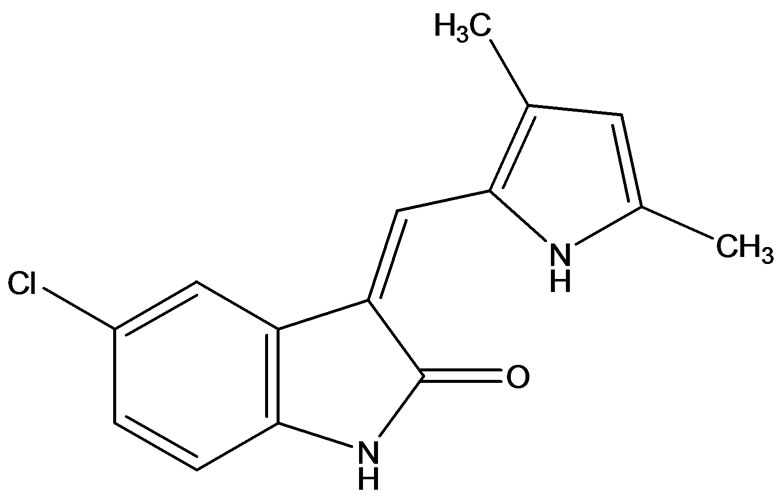
Figure 5. SU5614 inducer of growth arrest and apoptosis.
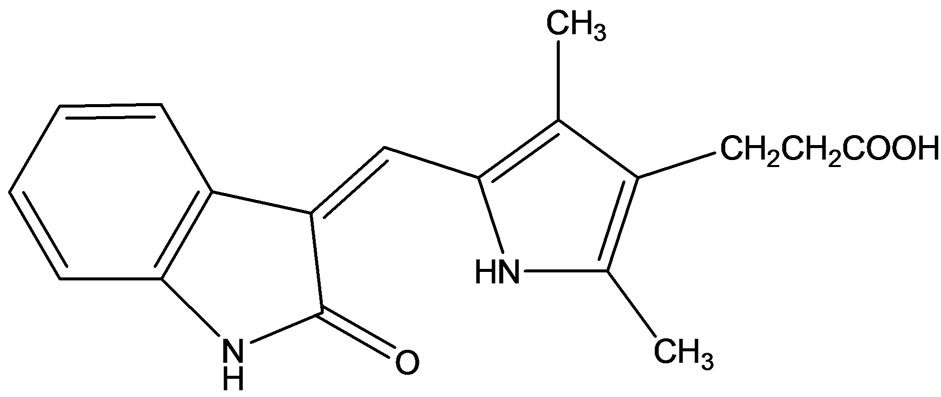
Figure 6. SU6668 Selective inhibitor of VEGFR, PDGFRβ, and FGFR1.
warrants further clinical evaluation in human malignancies, both alone and in combination with cytotoxic chemotherapy.
3.5. SU11274
SU11274 (Figure 8) is an ATP competitive small molecule inhibitor of the catalytic activity of Met which exhibited selectivity for Met enzyme versus a panel of other tyrosine kinases [15]. Chemically SU11274 is a [(3Z)-N- (3-chlorophenyl)-3-({3,5-dimethyl-4-[(4-methylpiperazin-1-yl)carbonyl]-1H-pyrrol-2-yl} methylene)-N-methyl-2- oxoindoline-5-sulfonamide]. Recent studies demonstrate that SU11274, a prototypic anti-MET small molecule, is an effective inhibitor of both HGF dependent and independent signaling, which results also from the presence of point mutations. Inhibition of the Met kinase activity by SU11274 led to time and dose dependent reduced cell growth and induced G1 cell cycle arrest and apoptosis. The bulk of clinical and experimental evidence has suggested that Met is a potential therapeutic target for development of inhibitors for treatment of cancers. Xueyan et al. [16] reported the identification and characterization of small molecule inhibitors SU11274 that potently and selectively inhibit the enzymatic activity of Met kinase. SU11274 displayed much lower IC50 values for inhibittion of Met, then towards other tyrosine kinases includes EGFR, FGFR1 or Src, thus, suggesting its high speci- ficity. SU11274 belongs to a class of small molecule RTK inhibitors that exert their activity by competing for the Mg-ATP complex binding pocket, which results in inhibition of kinase activity and subsequent downstream signaling.
3.6. SU12662
SU12662 (Figure 9) is an active N-desethyl metabolite of Sunitinib. SU12662 has receptor tyrosine kinase inhibitory profile similar to that of Sunitinib in vitro and has similar plasma protein binding [17].
3.7. SU5402
SU5402 (Figure 10) chemically 3-[(3-(2-carboxyethyl)- 4-methylpyrrol-2-yl) methylene]-2-indolinone shown a selective inhibitory effect of tyrosine kinase with known anti-FGFR activity [18] against control Ba/F3 cells expressing the constitutively active ZNF198-FGFR1 fusion protein, and also against KMS-11 and OPM-2, t(4;14) positive MM cell lines that express activated FGFR3.
3.8. SU10944
Neela et al. [19] reported a novel small molecule inhibitor of VEGFR-2, SU10944 (Figure 11). This indolinone 2- one derivative is an effective, ATP-competitive inhibitor of VEGFR-2 biochemical activity and is active in the nanomolar range in cellular assays. SU10944 can be administered in vivo by the oral route and achieves sufficient exposure to inhibit nearly all VEGF stimulated ne-
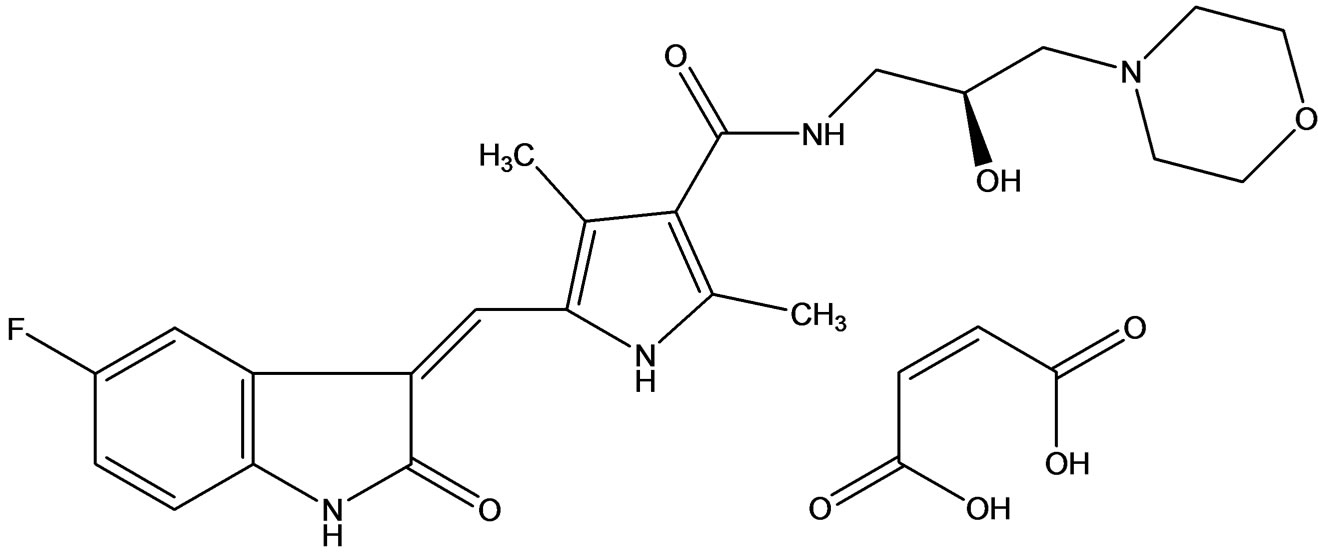
Figure 7. SU14813 an inhibitor of c-kit and FLT-3.

Figure 8. SU11274 Met kinase inhibitor.
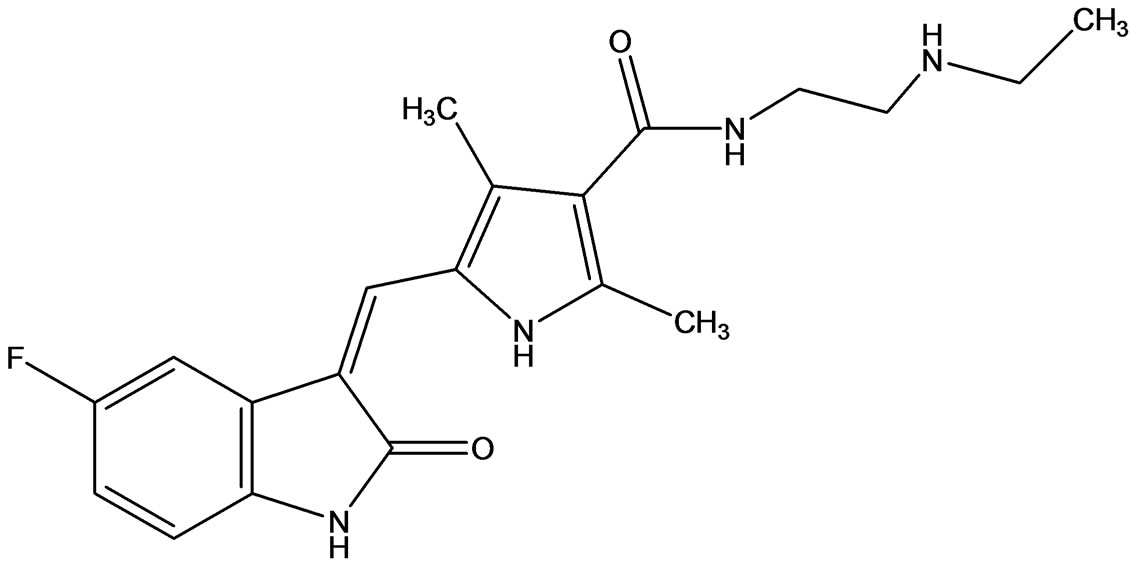
Figure 9. SU12662 active metabolite of Sunitinib.
ovascularization and vascular permeability. Robert disclosed steady state kinetic analysis of SU10944, he found that this compound is a potent ATP competitive inhibittor of VEGFR-2 in vitro with an IC50 value of 96 nM and a Ki value (a dissociation constant) of 21 nM [20]. They found that SU10944 is a more potent inhibitor of VEGFR- 1 when compared with VEGFR-2. However, the compound is not an effective inhibitor of PDGFRβ, KIT or FGFR1.
3.9. SU6656
SU6656 (Figure 12) is an inhibitor of the Src family of tyrosine kinases [21], a group of enzymes that regulate many important signaling pathways, including RAS/ MAPK, PI3K/AKT and STAT, and which represent molecular targets for the treatment of various cancers. SU6656 showed cross reactivity with certain other kinases such as the Aurora B kinases, proteins that control cell ploidy and centrosome number. SU6656 can also significantly augment the anti-angiogenic effect of irradiation and may be a useful agent in the treatment of drug resistant cancers.
3.10. SU9516
3-substituted indolin-2-ones represent a potent and selec-
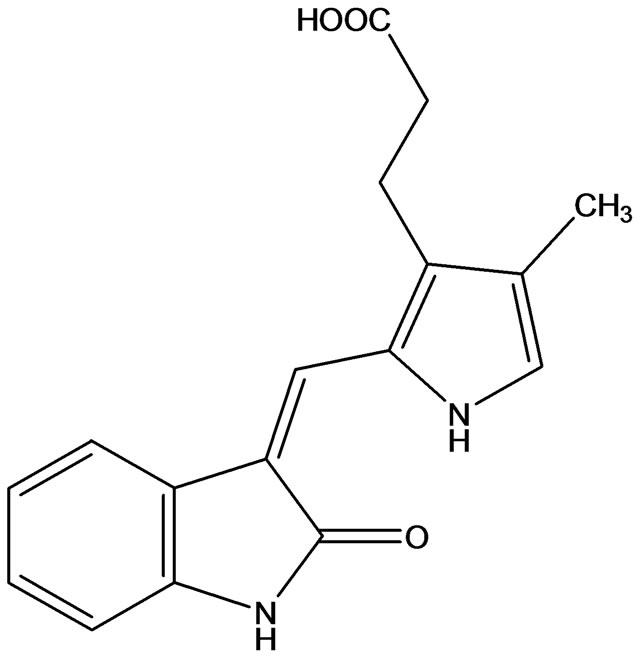
Figure 10. SU5402 anti-FGFR.
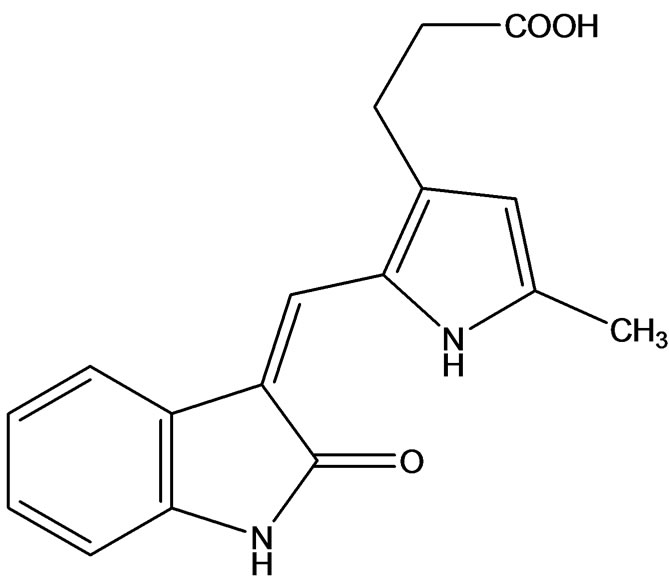
Figure 11. SU10944 potent inhibitor of VEGFR-2.
tive inhibitor for certain RTKs [22]. These small molecules typically act as adenine mimetics that occupy the ATP binding pocket in specific RTKs, with the specificity being governed by the substituents around the indolinone core. Compounds of this class have reached clinical trials for the treatment of several cancers. One such compound is SU9516 (Figure 13) (3-[1-(3H-Imidazol- 4-yl)-meth-(Z)-ylidene]-5-methoxy-1,3-dihydro-indol-2one). From crystal structure of the inhibitor bound to cdk2 and kinetically characterized studies about the inhibition of cdks by SU9516 suggest that this compound could have usefulness as a core molecule for the study of cdk inhibition. Furthermore, information gained from the structural analysis should aid in the development of more potent and specific inhibitors in the future. SU9516 is also reported to inhibit GSK-3 which is involved in normal cell death [23].
3.11. SU4984
SU4984 (Figure 14) chemically 3[4-(1-Formylpiperazin- 4-yl)-benzylidenyl]-2-indolinone derivative inhibits the tyrosine kinase activity of FGFR1 by interacting with the catalytic domain of FGFR1. SU4984 also inhibits FGF-
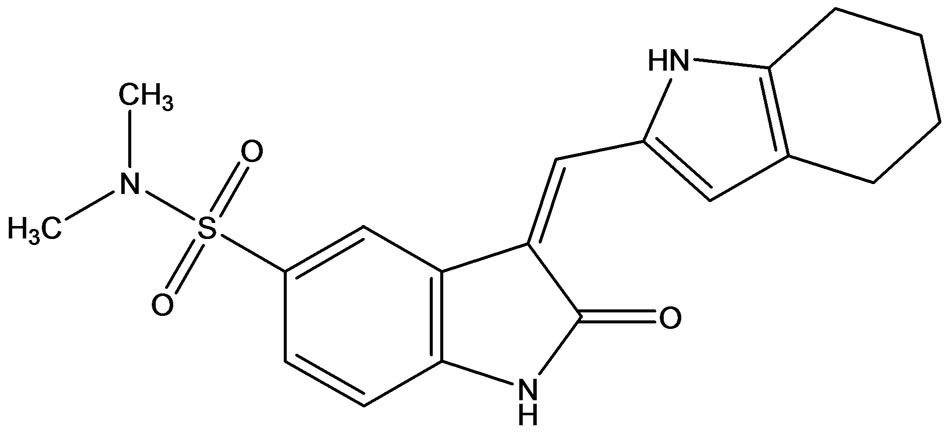
Figure 12. SU6656 inhibitors of Src family kinases.
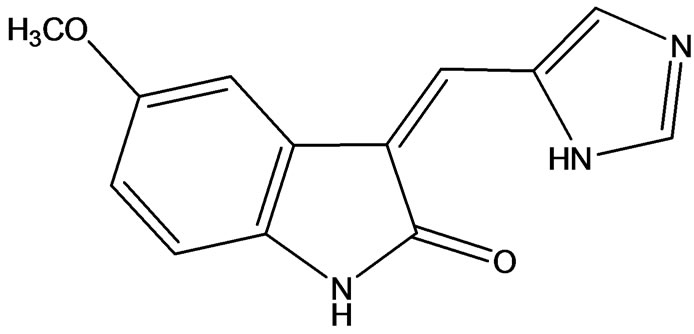
Figure 13. SU9516 cdk and GSK-3 inhibitor.
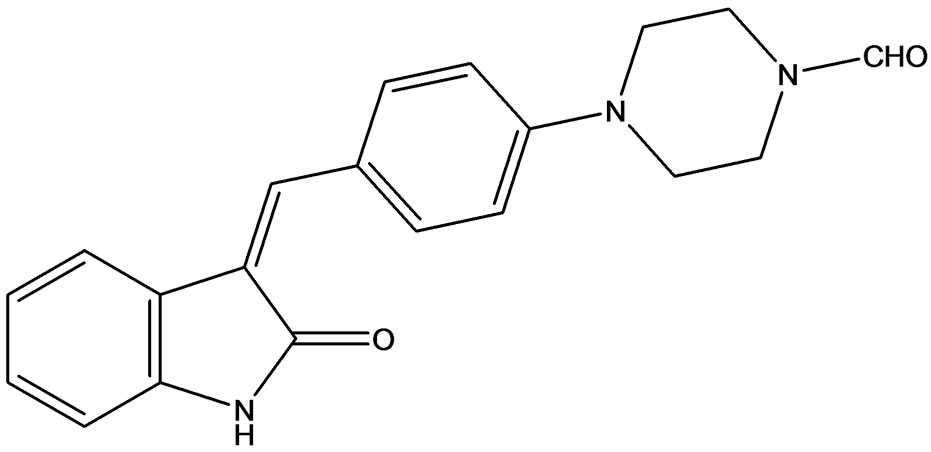
Figure 14. SU4984 inhibitors of ERK1 and 2.
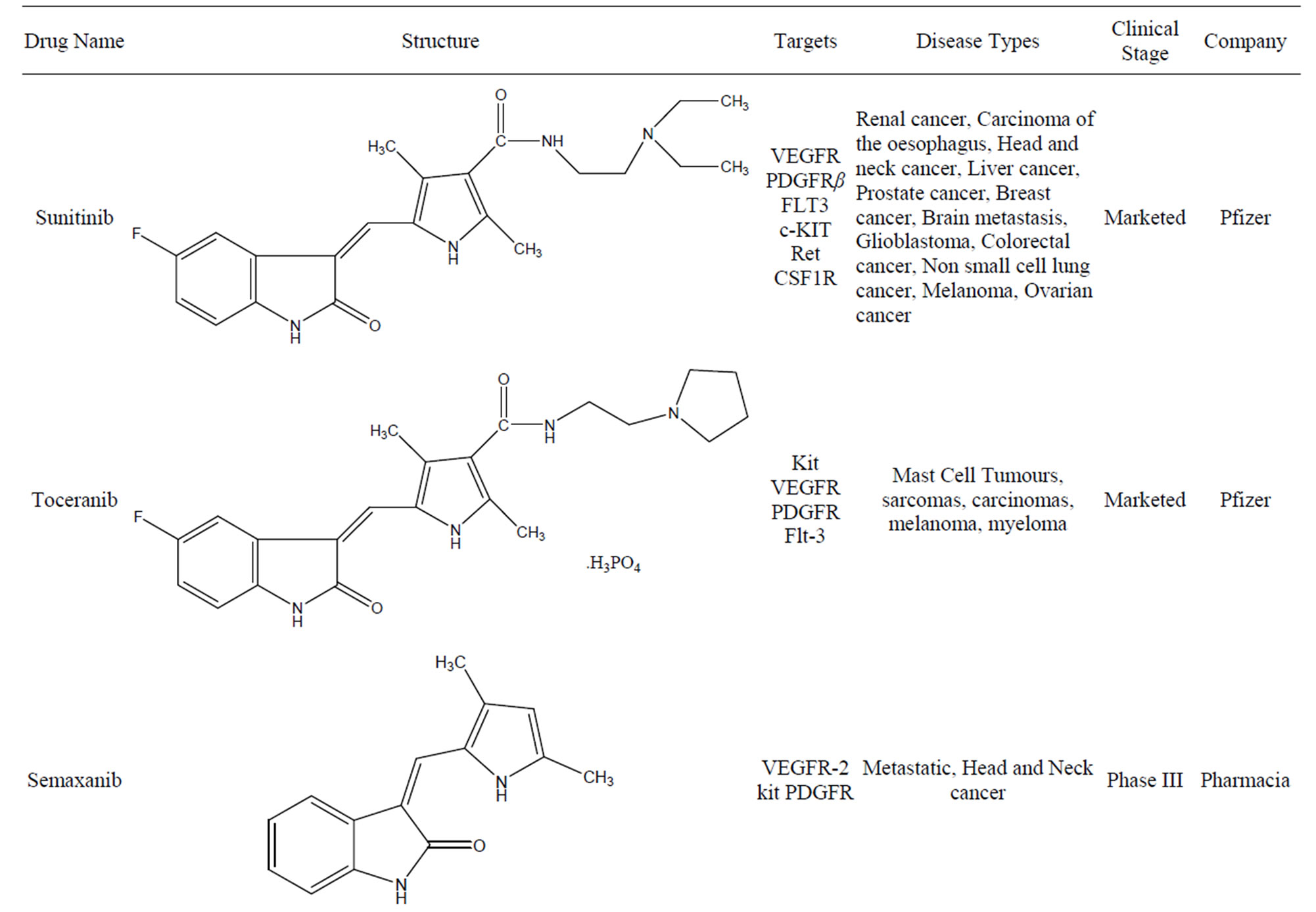
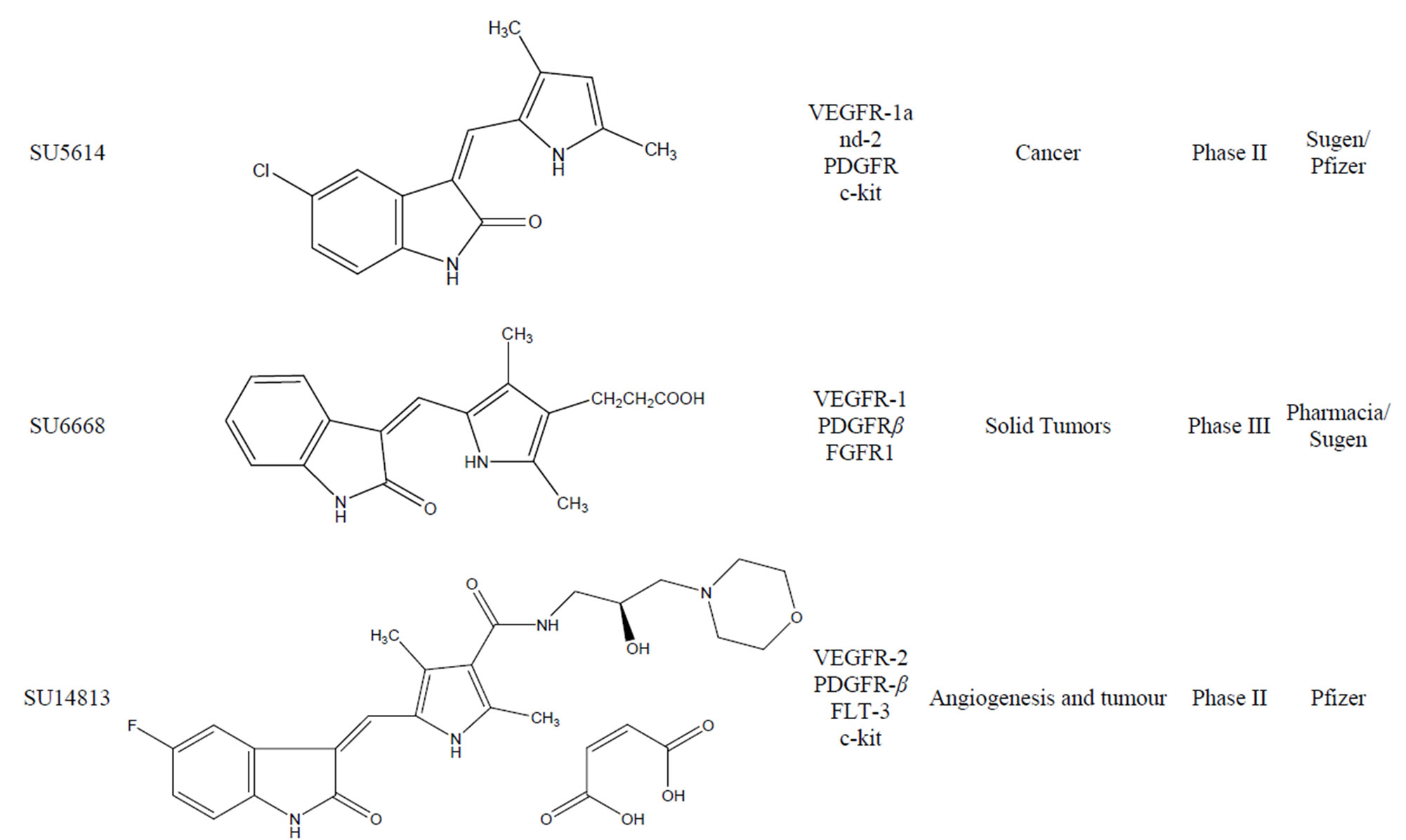
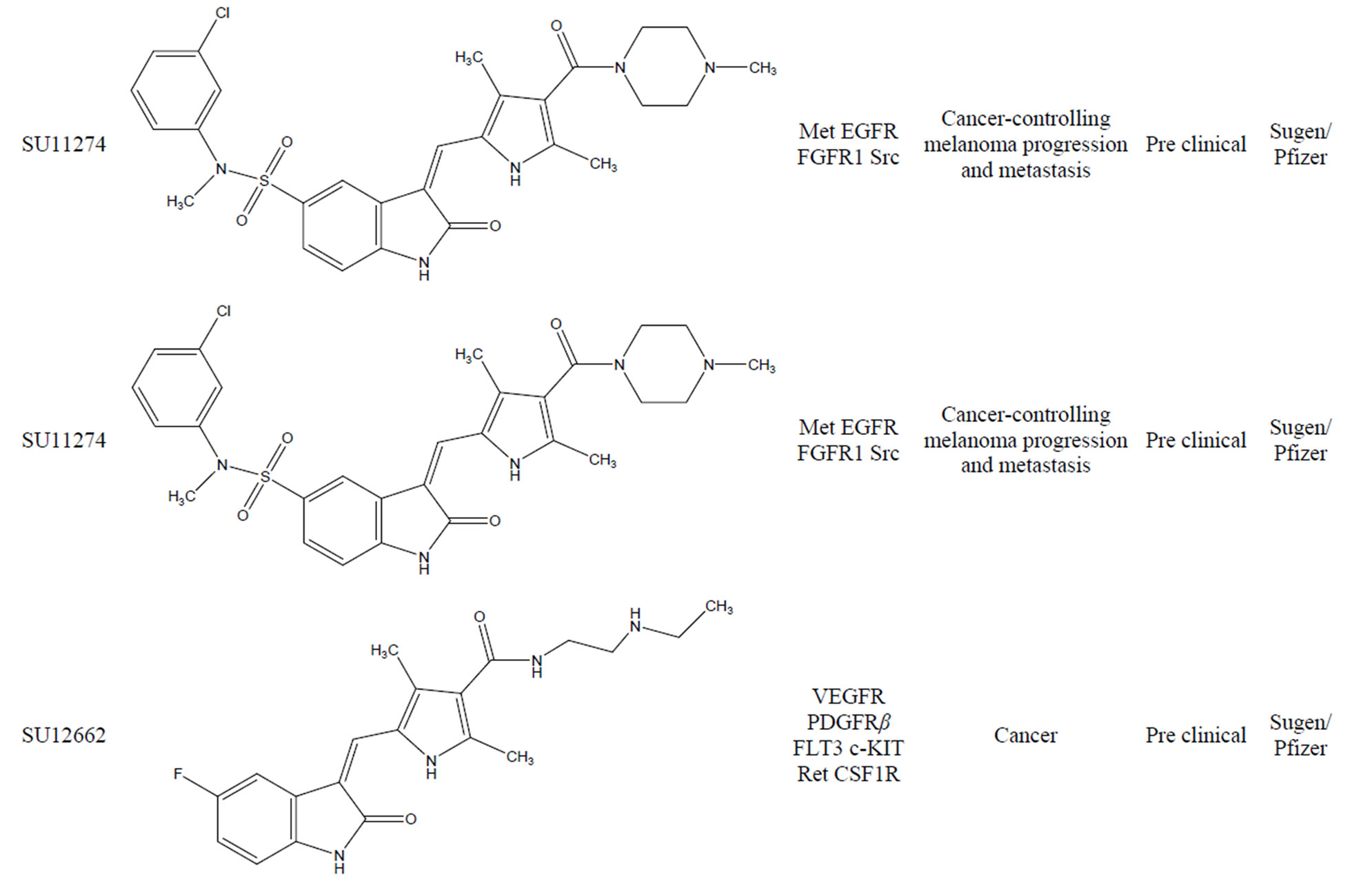
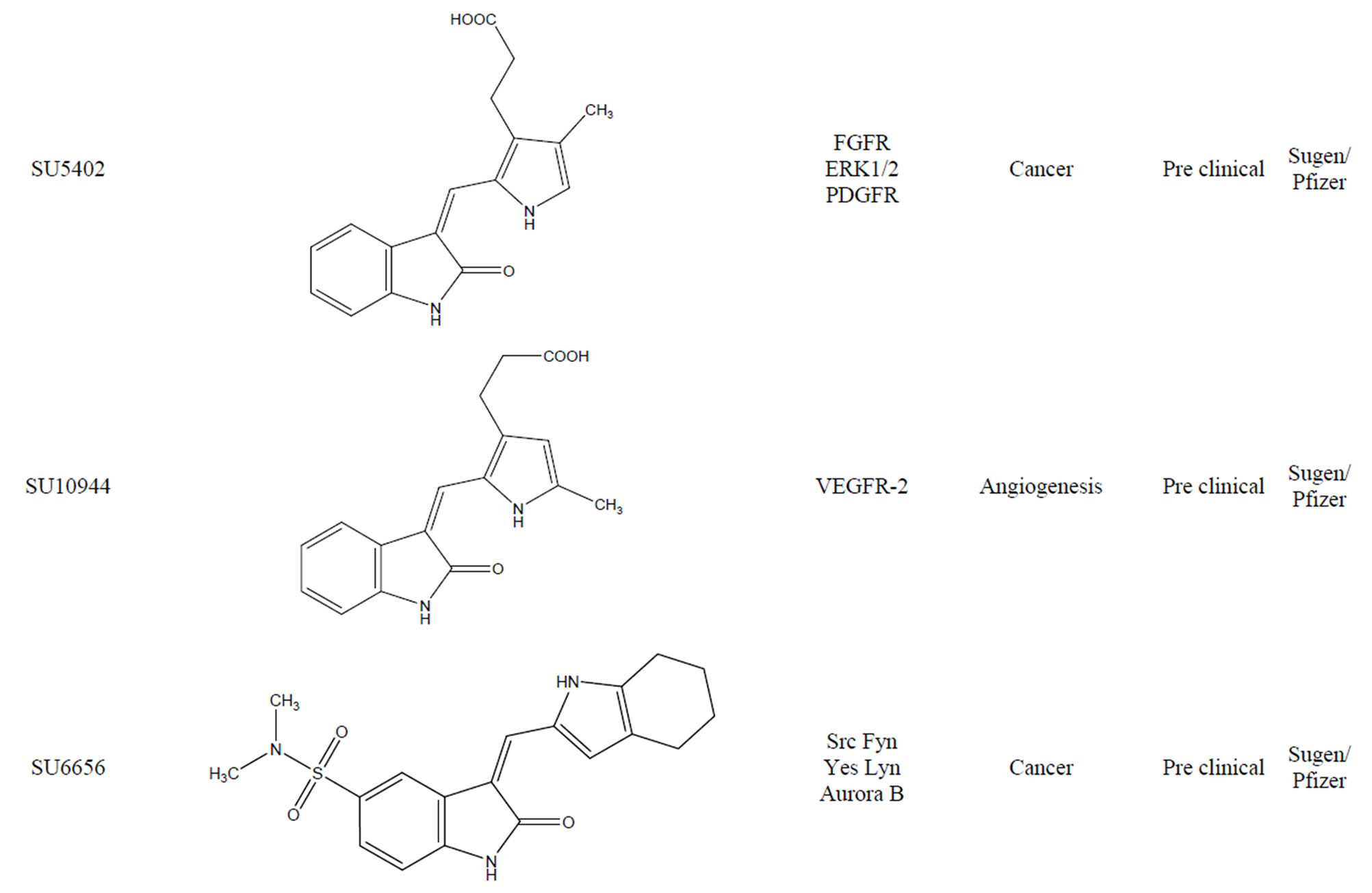
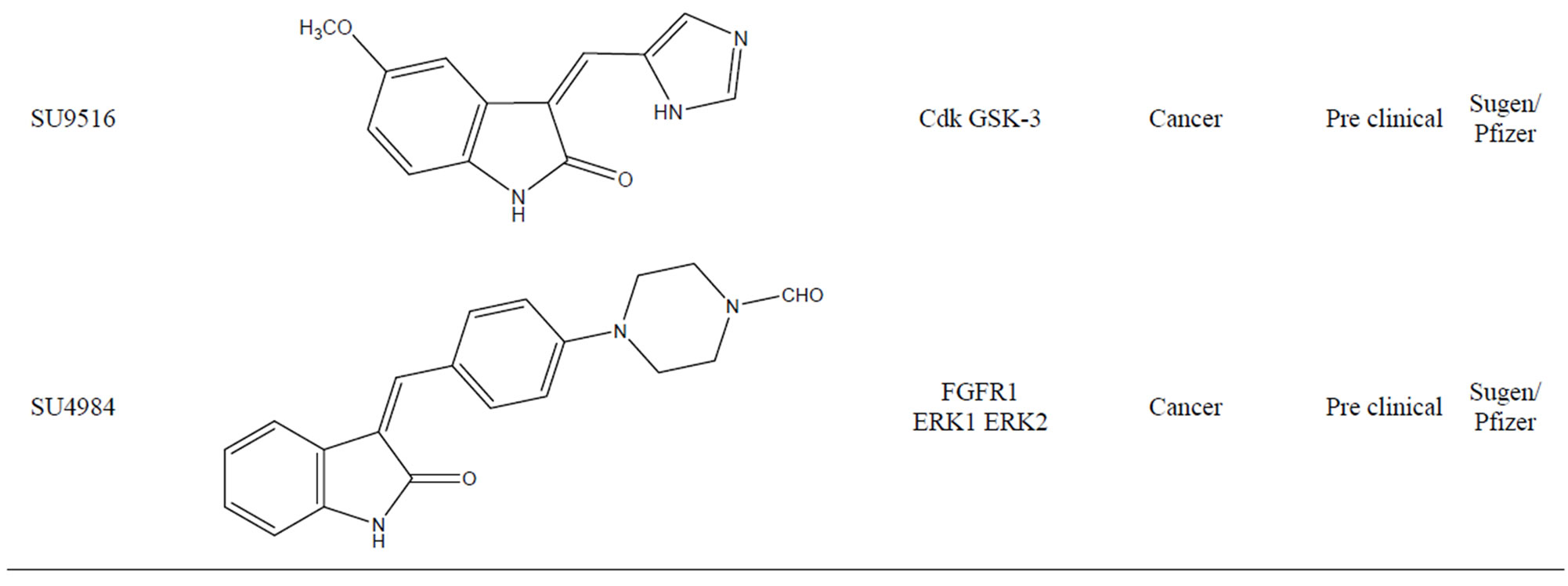
Table 1. Summary of indolinones as kinase inhibitor.
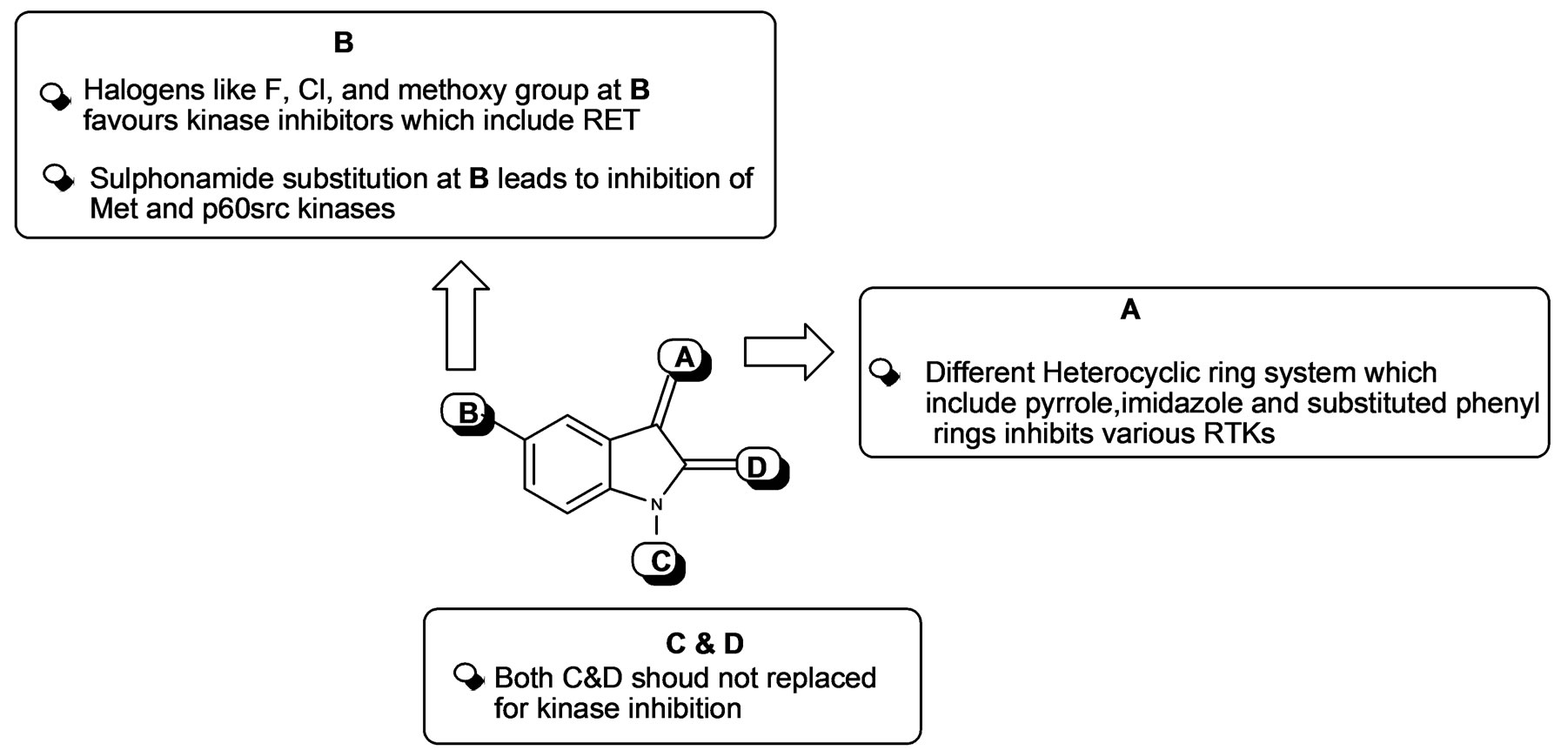
Figure 15. Structural activity summary of indolinones derivatives discussed in this review as kinase inhibitors.
induced phosphorylation of ERK1 and ERK2 and tyrosine phosphorylation of PDGF and insulin receptors. SU4984 also effective against KIT with juxtamembrane activating mutations [24].
4. Conclusion
In less than a two decade, the field of chemical inhibitors of receptor tyrosine kinases has seen spectacular developments. Starting from “low throughput screens” the search for selective inhibitors has progressed to massive industrial High Throughput Screens (HTS). It has been accompanied by a large variety of studies ranging from kinase inhibitor interaction at the atomic level (crystallography), molecular modeling, in silico screening, thorough investigation of the inhibitor’s selectivity and cellular effects, and pre-clinical studies. These efforts have provided very useful pharmacological tools that can be used, amongst other applications, to synchronize cells, to scrutinize cell cycle phases, to link physiological/biochemical events with kinase activities. More challenging is the development of the application of these pharmacological compounds to multiple areas the scope of which is much wider than initially expected. So far, various kinase inhibitors have been tested in clinical trials resulting in great survival benefit for patients for whom no effective therapy was available. Indolinone a class of compounds displays very promising anti-tumor properties by inhibiting various kinase families. The antikinase activity of the indolinone set of kinase inhibitors was first reported in 1997 by Mohammadi et al. [25]. These small molecules have a low molecular weight and most of them bind to protein kinases competing with ATP for the ATP binding site. Moreover the apparent selectivity of the compound, its potency and favorable physico-chemical properties render this molecule as an interesting lead structure for development of new kinase inhibitors. Because of that above mentioned reason a number of indolinone based anti angiogenic TKIs currently under development may provide additional treatment options for patients in the future (A summary of indolinones as kinase inhibitors are shown in Figure 15). Preclinical as well as preliminary results of clinical trials suggest that this class of agent is promising for cancer treatment and may offer a promising alternative to classical anticancer drugs. Table 1 summarizes the current protein kinase inhibitors in the clinic and in development.
REFERENCES
- P. Cohen, “Protein Kinases the Major Drug Targets of the Twenty First Century?” Nature Reviews Drug Discovery, Vol. 1, No. 4, 2002, pp. 309-315. doi:10.1038/nrd773
- M. Krug and A. Hilgeroth, “Recent Advances in the Development of Multi Kinase Inhibitors,” Mini-Reviews in Medicinal Chemistry, Vol. 8, No. 13, 2008, pp. 1312- 1327. doi:10.2174/138955708786369591
- T. L. Underiner, B. Ruggeri and D. E. Gingrich, “Development of Vascular Endothelial Growth Factor Receptor (VEGFR) Kinase Inhibitors as Anti-Angiogenic Agents in Cancer Therapy,” Current Medicinal Chemistry, Vol. 11, No. 6, 2004, pp. 731-745. doi:10.2174/0929867043455756
- L. Q. Chow and S. Gail Eckhardt, “Sunitinib: From Rational Design to Clinical Efficacy,” Journal of Clinical Oncology, Vol. 25, No. 19, 2007, p. 2858. doi:10.1200/JCO.2006.06.3602
- F. Sandrine, D. George, S. William and R. Eric, “Molecular Basis for Sunitinib Efficacy and Future Clinical Development,” Nature Reviews Drug Discovery, Vol. 6, No. 9, 2007, pp. 734-745. doi:10.1038/nrd2380
- A. L. Cheryl, B. M. Phyllis, L. W. F. Stacey, F. B. Joseph, W. R. Anthony, M. P. Rosenberg, C. J. Henry, K. L. Mitchener, M. K. Klein, J. G. Hintermeister, P. J. Bergman, G. C. Couto, G. N. Mauldin and G. M. Michels, “Multi-Center, Placebo-Controlled, Double-Blind, Randomized Study of Oral Toceranib Phosphate (su11654), a Receptor Tyrosine Kinase Inhibitor, for the Treatment of Dogs with Recurrent (either Local or Distant) Mast Cell Tumor Following Surgical Excision,” Clinical Cancer Research, Vol. 15, No. 11, 2009, pp. 3856-3865. doi:10.1158/1078-0432.CCR-08-1860
- D. M. McLean, “Methisazone Therapy in Vaccinia Complications,” Annals of the New York Academy of Sciences, Vol. 284, 1977, pp. 118-121. doi:10.1111/j.1749-6632.1977.tb21942.x
- A. O. Donnell, A. Padhani, C. Hayes, A. J. Kakkar, M. Leach, J. M. Trigo, M. Scurr, F. Raynaud, S. Phillips, W. Aherne, A. Hardcastle, P. Workman, A. Hannah and I. Judson, “A Phase I Study of the Angiogenesis Inhibitor SU5416 (Semaxanib) in Solid Tumors, Incorporating Dynamic Contrast MR Pharmacodynamic End Points,” British Journal of Cancer, Vol. 93, No. 8, 2005, pp. 876- 883. doi:10.1038/sj.bjc.6602797
- G. F. Matthew, Z. Andrew, W. Richard, V. Ennapadam, L. Eric, H. Lucy, A. Timothy, G. William, F. Martin and G. P. David, “A Phase II Study of SU5416 in Patients with Advanced or Recurrent Head and Neck Cancers,” Invest New Drugs, Vol. 25, 2007, pp. 162-172.
- S. Karsten, F. Florian, V. Robert and H. Wolfgang, “The Protein Tyrosine Kinase Inhibitor SU5614 Inhibits VEGF-Induced Endothelial Cell Sprouting and Induces Growth Arrest and Apoptosis by Inhibition of c-kit in AML Cells,” Experimental Hematology, Vol. 30, No. 7, 2002, pp. 767-773. doi:10.1016/S0301-472X(02)00837-8
- S. Cristiana, V. Lucia, G. Giacomo, T. Jose, M. Irene, L. Anna, B. Alberta, I. Nicoletta, M. Silvia and G. Luca, “Phase I Clinical and Pharmacological Evaluation of the Multi Tyrosine Kinase Inhibitor SU006668 by Chronic Oral Dosing,” European Journal of Cancer, Vol. 42, No. 2, 2006, pp. 171-178. doi:10.1016/j.ejca.2005.09.033
- A. D. Laird, P. Vajkoczy, L. K. Shawver, A. Thurnher, C. Liang, M. Mohammadi, J. Schles Singer, A. Ullrich, S. R. Hubbard, R. A. Blake, T. A. Fong, L. M. Strawn, L. Sun, C. Tang, R. Hawtin, F. Tang, N. Shenoy, K. P. Hirth, G. McMahon and J. M. Cherrington, “SU6668 Is a Potent Antiangiogenic and Antitumor Agent That Induces Regression of Established Tumor,” Cancer Research, Vol. 60, 2000, pp. 4152-4160.
- W. Fiedler, G. Giaccone, P. Lasch, I. V. Horst, N. Brega, R. Courtney, A. Abbattista, D. R. Shalinsky, C. Bokemeyer and E. Boven, “Phase I Trial of SU14813 in Patients with Advanced Solid Malignancies,” Annals of Oncology, Vol. 22, No. 1, 2011, pp. 195-201. doi:10.1093/annonc/mdq313
- P. Shem, A. D. Laird and D. B. Mendel, “SU14813: A Novel Multiple Receptor Tyrosine Kinase Inhibitor with Potent Antiangiogenic and Antitumor Activity,” Molecular Cancer Therapeutics, Vol. 5, No. 7, 2006, pp. 1774- 1782. doi:10.1158/1535-7163.MCT-05-0333
- Z. Yitzhak, V. V. Angelina, M. Michaela, S. Bruno, B. L. Wieslawa, H. G. Richard, S. Nikolaus and M. A. Daniel, “Differential Inhibition Sensitivities of MET Mutants to the Small Molecule Inhibitor SU11274,” Cancer Letters, Vol. 289, No. 2, 2010, pp. 228-236. doi:10.1016/j.canlet.2009.08.017
- W. Xueyan, L. Phuong and L. Congxin, “Potent and Selective Inhibitors of the Met [Hepatocyte Growth Factor/Scatter Factor (HGF/SF) Receptor] Tyrosine Kinase Block HGF/ SF-Induced Tumor Cell Growth and Invasion,” Molecular Cancer Therapeutics, Vol. 2, No. 11, 2003, pp. 1085-1092.
- E. H. Brett, L. B. Carlo and K. Dongwoo, “A Population Pharmacokinetic Meta-Analysis of Sunitinib Malate (SU11248) and Its Primary Metabolite (SU12662) in Healthy Volunteers and Oncology Patients,” Clinical Cancer Research, Vol. 15, No. 7, 2009, pp. 2497-2506. doi:10.1158/1078-0432.CCR-08-1893
- E. K. Grand, A. J. Chase, C. Heath, A. Rahemtulla and N. C. P. Cross, “Targeting FGFR3 in Multiple Myeloma: Inhibition of t(4;14)-Positive Cells by SU5402 and PD173074,” Leukemia, Vol. 18, No. 5, 2004, pp. 962-966. doi:10.1038/sj.leu.2403347
- P. Neela, S. Li, M. Deborah, C. Hui, M. L. Kathleen, L. Phuong, G. M. Katherine, W. Xueyan, R. Audie, T. Danny, A. D. Laird, X. M. Yu, Z. Qingling, T. Cho, M. M. Gerald and H. Anthony, “A Selective and Oral Small Molecule Inhibitor of VEGFR-2 and VEGFR-1 Inhibits Neovascularization and Vascular Permeability,” Journal of Pharmacology and Experimental Therapeutics, Vol. 306, No. 3, 2003, pp. 838-845.
- R. J. Robert, “Sunitinib: A VEGF and PDGF Receptor Protein Kinase and Angiogenesis Inhibitor,” Biochemical and Biophysical Research Communications, Vol. 356, No. 2, 2007, pp. 323-328. doi:10.1016/j.bbrc.2007.02.156
- D. Nathalie, S. Carl, N. Sonia and C. Serge, “Human B Lymphocytes and Non-Hodgkin’s Lymphoma Cells Become Polyploid in Response to the Protein Kinase Inhibitor SU6656,” Blood Cells, Molecules, and Diseases, Vol. 39, No. 1, 2007, pp. 130-134. doi:10.1016/j.bcmd.2007.02.010
- J. M. Deborah, R. B. Cornelia, C. B. David, U. Ping, H. J. J. Terence, H. K. Sung and G. R. Audie, “SU9516: Biochemical Analysis of cdk Inhibition and Crystal Structure in Complex with cdk2,” Biochemical and Biophysical Research Communications, Vol. 310, No. 3, 2003, pp. 1026-1031. doi:10.1016/j.bbrc.2003.09.114
- M. E. Lane, B. Yu, A. Rice, K. E. Lipson, C. Liang, L. Sun, C. Tang, G. McMahon, R. G. Pestell and S. Wadler, “A Novel cdk2-Selective Inhibitor, SU9516, Induces Apoptosis in Colon Carcinoma Cells,” Cancer Research, Vol. 61, No. 16, 2001, pp. 6170-6177.
- M. Yongsheng, C. Eric, W. Xiaomei, S. Chang, M. M. Gerald and B. J. Longley, “Indolinone Derivative Inhibit Constitutively Activated KIT Mutants and Kill Neoplastic Mast Cells,” Journal of Investigative Dermatology, Vol. 114, 2000, pp. 392-394. doi:10.1046/j.1523-1747.2000.00888.x
- M. Mohammadi, G. McMahon, L. Sun, C. Tang, P. Hirth, B. K. Yeh, S. R. Hubbard and J. Schlessinger, “Structures of the Tyrosine Kinase Domain of Fibroblast Growth Factor Receptor in Complex with Inhibitors,” Science, Vol. 276, No. 5314, 1997, pp. 955-960. doi:10.1126/science.276.5314.955
List of Abbreviations
AKT = AKR mouse thymoma kinase
AML = Acute myeloid leukemia
ATP = Adenosine tri phosphate
c-Kit = Stem cell factor receptor tyrosine kinase
CDK = Cyclin dependent kinase
CSF1R = Colony-stimulating factor 1 receptor
EGFR = Epidermal growth factor receptor
ERK = Extracellular signal regulated kinases
FDA = Food and Drug Administration
FGF = Fibroblast growth factor
FGFR = Fibroblast growth factor receptor
FLT3 = fms-related tyrosine kinase 3
GIST = Gastrointestinal stromal tumor
GSK = Glycogen synthase kinase
HGF = Hepatocyte growth factor
IC50 = Concentration required for 50% inhibition of a biochemical process in vitro
KIT = Stem-cell growth factor receptor
MAPK = Mitogen-activated protein kinase
PDGF = Platelet-derived growth factor
PDGFR = Platelet-derived growth factor receptor
RTKs = Receptor tyrosine kinases
RTKIs = Receptor tyrosine kinases inhibitors
SAR = Structure-activity relationship
Src = v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homologue
STAT = Signal transducer and activator of transcription
TKI = Tyrosine kinase inhibitor
VEGF = Vascular endothelial growth factor
VEGFR = Vascular endothelial growth factor receptor
NOTES
*Corresponding author.