Advances in Alzheimer's Disease
Vol.1 No.3(2012), Article ID:26145,9 pages DOI:10.4236/aad.2012.13012
Tyrosine kinase receptor B isoforms alter APP and BACE1 endogenous levels independently of BDNF
![]()
1Biology Department, Drexel University, Philadelphia, USA; *Corresponding Author: ansaloni@molgen.mpg.de
2Department of Vertebrate Genomics, Max Planck Institute for Molecular Genetics, Berlin, Germany
3Neuroscience Department, University of Southern California, Los Angeles, USA
4Department of Biochemistry and Molecular Biology, University of Medicine and Dentistry of New Jersey, Piscataway, USA
Received 1 November 2012; revised 2 December 2012; accepted 10 December 2012
Keywords: TrkB; Alzheimer; BACE1; SHC; NFAT
ABSTRACT
Brain derived neurotrophic factor (BDNF) levels and signaling via the tyrosine receptor kinase B (TrkB) have been shown to be altered in Alzheimer’s Disease. In addition, it has been reported that the isoforms of TrkB can differentially affect metabolism of amyloid precursor protein (APP). Conversely, A(, a neurotoxic cleavage product of APP, has been shown to impair TrkB/ BDNF signaling. Therefore, we investigated whether the changes observed in APP metabolism were due to the isoform-specific effects of TrkB on either APP expression, and/or on the expression and activity of ADAM10 and BACE1. Since BDNF levels are decreased in AD, we focused on BDNF independent effects of the TrkB isoforms. We found that TrkB FL increases endogenous APP levels in both HEK293 and SH-SY5Y naïve cells. We did not find an increase in ADAM10 activity in HEK293 cells, but an increase in BACE1 levels. Additionally, we have found that TrkB FL is able to increase NFAT3 mediated transcriptional activity and we suggest that this causes transcriptional activation of the BACE1 promoter.
1. INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease that mainly affects the elderly and causes memory impairment and cognitive deficits, thus leading to a dramatic reduction in quality of life. Because people aged 60 or greater are the fastest growing segment of the population in Western countries, the number of AD patients is expected to reach 16 million by 2050 in the USA alone [1]. AD is currently incurable, approved therapies only affect symptoms and do not alter the progression of the degeneration therefore new therapeutic approaches are needed [2]. Thus, AD continues to be one of the major healthcare challenges of this century. Some therapeutic approaches have focused on the elimination of the Ab peptide that is neurotoxic and accumulates in the brain of the patients. Ab is produced by cleavage of the amyloid precursor protein (APP) by b-secretase, BACE1, and then by g-secretase [3]. Other therapeutic approaches are aimed at restoring signaling pathways that are defective in AD [2]. One such signaling defect is that of the tyrosine kinase receptor B (TrkB) and its ligand, brain derived neurotrophic factor (BDNF) [4]. TrkB/BDNF are important factors for neuronal homeostasis [5] and enhancing their signaling in AD is accompanied by cognitive benefits [6]. Therefore BDNF mimetics may have therapeutic applications [7,8]. The efficacy of these therapies will depend on the ability of these compounds to bind to the variety of TrkB receptor isoforms that are expressed in neurons and on activation of the downstream signaling pathways.
In humans, TrkB is present in at least three isoforms that differ in their intracellular domain [9,10]. Full length TrkB (TrkB FL) includes a tyrosine kinase domain, a SHC binding domain and a PLC-g binding domain. TrkB SHC has a shorter intra-cellular portion that contains a SHC binding domain, TrkB SHC is present in humans but not in rodents [11] . TrkB T has a short intracellular portion that does not contain known functional domains [9,10]. There is evidence that TrkB truncated isoforms, TrkB SHC and T, are up-regulated in AD brains [12,13] and that Ab can disrupt TrkB/BDNF signaling [14,15]. Therefore the role of TrkB isoforms in the pathogenesis of AD is important if BDNF replacement therapies are to be implemented. In the context of AD, is fundamental to elucidate how TrkB isoforms affect Ab production/clearance.
It is known that BDNF/TrkB signaling can affect APP expression levels, glycosylation and cleavage [16-19]. We have found that TrkB isoforms can differentially affect the processing and the expression levels of over-expressed APP in SH-SY5Y neuroblastoma cells [20]. BDNF signaling, mediated by TrkB, is known to increase APP promoter activity [17], however in the AD brain BDNF levels are lower while TrkB T and TrkB SHC levels are higher compared to asymptomatic, age matched controls. Since TrkB FL mediated signaling can occur independently of BDNF [21,22], we wanted to test if TrkB FL and TrkB T could have BDNF independent effects on endogenously expressed APP.
TrkB FL also has an effect on APP processing that is dependent on the tyrosine kinase activity of the receptor. In SH-SY5Y cells over-expressing APP TrkB FL increased AICD (APP Intra Cellular Domain) mediated transcriptional activity and AICD levels [20]. These results suggest an effect of TrkB isoforms on the APP processsing enzymes, such as a-disintegrin metalloprotease 10 (ADAM10) and b-secretase (BACE1). Previous work showed increased a-secretase cleavage of APP in SHSY5Y cells stimulated by all-trans retinoic acid (RA), RA induces up-regulation of TrkB [16]. These studies did not investigate if RA was able to increase ADAM10 activity or levels but only measured the products of APP a-secretase cleavage, sAPP-a, as indirect evidence of increased ADAM activity. We therefore hypothesized that TrkB isoforms can directly affect the activity and the levels of ADAM10, the enzyme mainly responsible for a-secretase cleavage of APP, independent of BDNF.
BACE1 is responsible for b-secretase cleavage of APP and its levels are influenced by SHC levels and by NFAT mediated transcription [23,24]. Since both SHC and NFAT are mediators of TrkB FL signaling pathways, we tested the hypothesis that TrkB FL auto-activation in HEK293 cells might modulate BACE1 levels via NFAT mediated transcriptional regulation.
2. MATERIALS AND METHODS
2.1. Cell Lines and Stable Populations
SH-SY5Y Naïve and 293HEK cells were maintained in DMEM medium (Gibco) with 10% FBS (Gibco) and penicillin streptomycin. SH-SY5Y APP-Gal-4 cells were maintained in the same media supplemented with 200 mg/ml of G418 (Gibco).
We also obtained stable cell populations expressing farnesylated GFP (GFP-F), TrkB T and TrkB FL both GFP tagged. Farnesylation on the GFP targets the GFP to the cell membrane making it a better control for surface receptors. Transfected cells were selected with 400 mg/ ml G418 from 48 hours after transfection up to one month. Liquid nitrogen stocks were obtained. Expression of the constructs was verified by fluorescence microscopy and Western blot.
For ADAM activity assays, cells were treated with 10 mM in 0.5% DMSO of the ADAM10 agonist PMA (phorbol 12-myristate 13-acetate, Sigma) or DMSO for 10 hours. We also treated cells with 20 mM of all transretinoic acid (ATRA or RA, Sigma) in 0.5% DMSO or 0.5% DMSO alone, for 48 hours prior to collection.
2.2. ADAM Activity Assay
Cell lysates were collected in extraction buffer provided with the R&D Systems ADAM activity kit and centrifuged at 4˚C. The supernatant was saved and combined with reaction buffer and substrate in a 96-well plate. The plate was incubated at 37˚C in the dark for 2 hours and then placed in a Tecan Infinite 200 plate reader controlled by the commercial software Magellan. An excitation wavelength of 355 nm and an absorption wavelength of 510 nm were used, the gain was automatically optimized and the integration time was set at 80 msec. Each well was read only once. As a positive control we used lysates from PMA treated cells. Each lysate was run in duplicates and readings were averaged. Background readings from wells containing reagents and substrate were divided by the experimental values and positive controls so that the ADAM10 activity is expressed in fold increase over background. The experiment was repeated twice independently for the cell lines and three times independently for the stable population of HEK293 cells transfected with TrkB constructs or GFP-F controls. Statistical analysis was conducted using the student t-test Bonferroni corrected for multiple comparison when applicable.
2.3. Western Blotting Procedure and Antibodies
Cells were collected 48 hours after all transfection procedures. Whole cell lysates were prepared by lysing cells in the plate with ice-cold radio immuno-precipitation buffer (150 mM NaCl, 1% NP40, 0.5% DOC, 1% SDS, 50 mM Tris, pH 8.0) supplemented with Halt cocktail of protease and phosphatase inhibitors (ThermoFisher). Cell lysates were sonicated in an ice-cold water bath sonicator for 6 minutes then centrifuged 20 minutes at 4˚C at 14,000 rpm. The resulting supernatants were collected and protein concentration measured with the BCA protein concentration kit (Pierce) according to manufacturer’s instructions. Western blot samples were prepared at a final concentration of 1 - 2 mg/ml in 4X reducing loading buffer (InVitrogen) and heated at 70˚C for 10 minutes. 15 - 25 mg of total protein/well from the cell lysates were separated on 4% - 12% Tris-Glycine midi gels (InVitrogen) in MES-SDS running buffer (InVitrogen) and run at 190 mVolts for 45 minutes. The separated proteins were transferred to PVDF FL membranes (Millipore) using a Semi-Dry transfer apparatus (AA Hoefer TE77X) for 3 hours at 125 mAmp per gel. Membranes were blocked one hour at room temperature using Licor blocking buffer then probed overnight with primary antibodies diluted in Licor blocking buffer at 4˚C - 25˚C. Membranes were then washed for 5 minutes 4 times with 0.1% Tween (Sigma) in PBS. After washing, membranes were incubated in the dark with the appropriate seconddary antibody IRDye (Licor) diluted in Licor blocking buffer for one hour. Again membranes were washed as above and finally rinsed with PBS. Membranes were scanned on an Odyssey InfraRed scanner (Licor) at appropriate intensities and images acquired at 159 mm resolution. Band intensities were quantified with the provided built-in software (Licor) and always normalized to the actin loading control.
APP was detected using a rabbit antibody A8717 (Sigma) at 1:2000 dilution; TrkB GFP tagged proteins were detected using a mouse GFP antibody (Clontech) at 1:500 dilution; TrkB was also detected with a pan-TrkB rabbit antibody (Santa Cruz Biotechnology); BACE1 was detected using a rabbit antibody ab2077 (Abcam) at 1:1000 dilution; ADAM10 was detected using a rabbit antibody (Chemicon); actin was detected using a mouse antibody (Sigma) at 1:15000 dilution. Secondary IRDye antibodies (anti rabbit IR800 and anti mouse IR700) were purchased from Licor and used at 1:15000 dilution.
2.4. Transfection and Luciferase Reporter Assays
Cells were transfected at approximately 50% - 60% confluency in 96-well plates using Arrest-In transfection reagent (ThermoScientific) according to manufacturer’s protocol. For the reporter experiments TrkB plasmids, the GFP-F (negative control, Clontech) and the NFAT3- GFP plasmids (positive control, a gift of Dr. Chris Norris) were co-transfected with the NFAT luciferase reporter (Promega) or the pGL3 luciferase reporter (Promega). Experimental or control plasmids were transfected in 1:1 molar ratio with the luciferase reporter plasmids. In all transfections a renilla luciferase reporter (pRLSV40 or pRLTK, Promega) was used as a transfection normalizetion control at a molar ratio of 1:40 to the luciferase plasmid.
Cells were collected in Dual Glo Lysis Buffer (E2920, Promega) 48 hours post transfection and the assay was carried out as per manufacturer’s instructions. Plates were read in a plate reader. Luciferase signal was normalized to renilla luciferase levels and data was analyzed using t-tests with Bonferroni correction for multiple comparisons.
3. RESULTS@NolistTemp#3.1. TrkB FL, Not TrkB T, Increases APP Endogenous Levels in HEK293 and SH-SY5Y Cells
TrkB FL has been reported to increase transcription of the APP promoter via IP3K and Akt mediated pathways in SH-SY5Y cells [19]. These cells express basal levels of the TrkB receptors and BDNF, which is the natural ligand for TrkB, and therefore can activate the receptor through the canonical pathway. It is known that TrkB receptors can also autoactivate in absence of BDNF [21, 22]. Thus, we hypothesized that, in both HEK293 naive cells, which do not express BDNF or TrkB and SHSY5Y naive cells, TrkB FL transfection increases APP transcription. In fact, the signaling pathways triggered by TrkB FL engage molecules that are ubiquitously expressed in all cell lines.
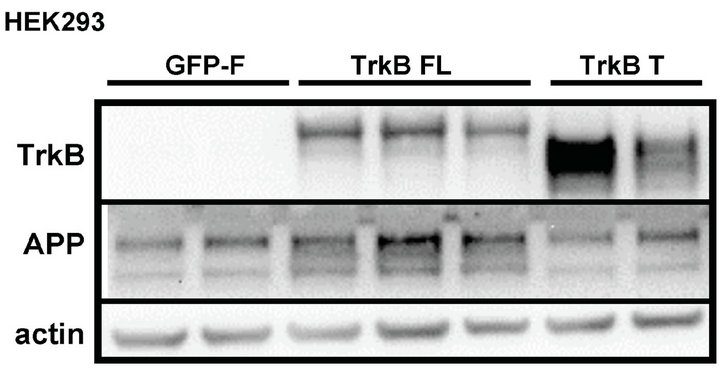
We transiently transfected plasmids encoding farnesylated GFP (GFP-F), TrkB T and TrkB FL into HEK293 cells and measured APP levels 48 hours post-transfection.
We found that TrkB FL transfection significantly increased APP levels compared to GFP-F or TrkB T transfection (Figures 1(a) and (c)).
The extent of the increase, though, was modest. We then measured APP in a stable population of cells that had been selected for TrkB or GFP-F expression with antibiotic treatment for at least one month. We again found that cells expressing TrkB FL displayed higher levels of APP compared to GFP-F and TrkB T expressing cells (Figure 1(b)).
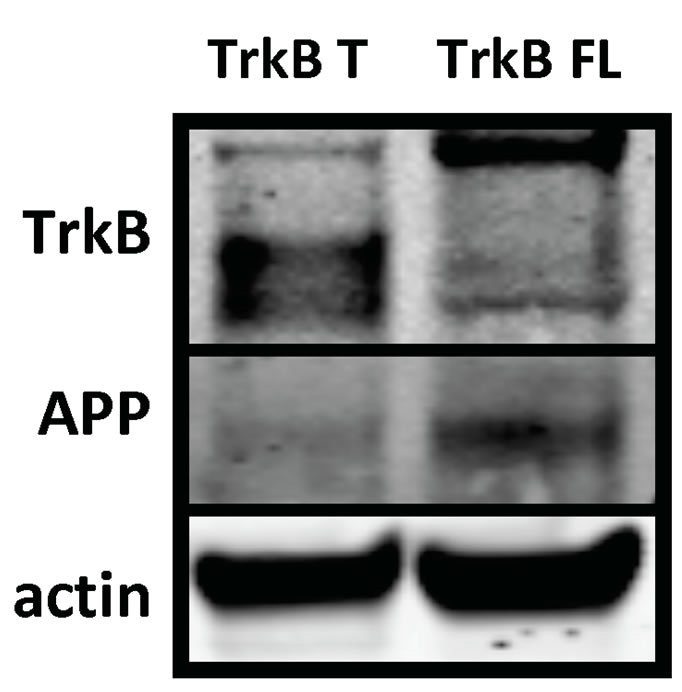
We transiently transfected TrkB T, TrkB FL and the GFP-F plasmid control in SH-SY5Y naïve cells and found similar results (Figures 2(a) and (b)).
We conclude that TrkB FL expression increases endogenous, full-length APP levels independent of BDNF while TrkB T does not.
3.2. ADAM10 Activity Is Not Affected by TrkB FL or TrkB T
TrkB FL has been shown to increase APP promoter transcription but also to increase sAPP-a levels in the media of SH-SY5Y cells [16]. We hypothesized that the increase in sAPP-a might be due to TrkB FL mediated PLC-g activation, since PLC-g produces diacylglycerol (DAG), an ADAM activity stimulator. Therefore we employed an ADAM activity assay to measure ADAM activity in cells that had been transfected with TrkB T or FL.
We initially checked for the assay reliability and the baseline ADAM activity of three different cell lines. A neuroblastoma cell line SH-SY5Y and the same cell line stably expressing APP fused to Gal4. We also used HEK293 cells. We collected lysates of cells treated with vehicle dimethyl sulfoxide (DMSO) or phorbol myristate acetate (PMA), a phorbol ester that mimics DAG activity.
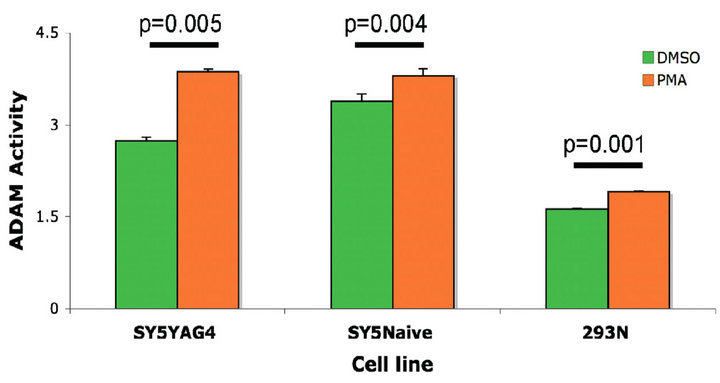
SH-SY5Y cells showed a higher ADAM basal activity compared to HEK293 cells. PMA was able to increase the ADAM activity signal in both cell lines (Figure 3 (a)).
 (a)
(a)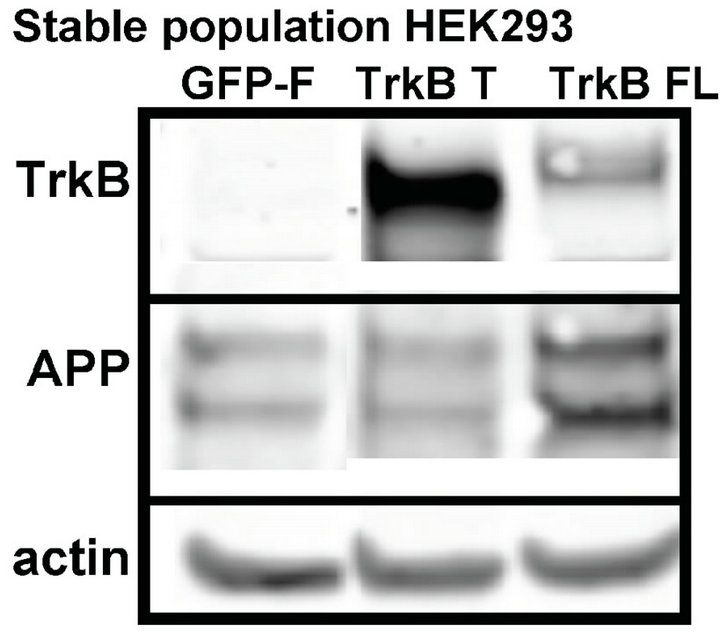 (b)
(b)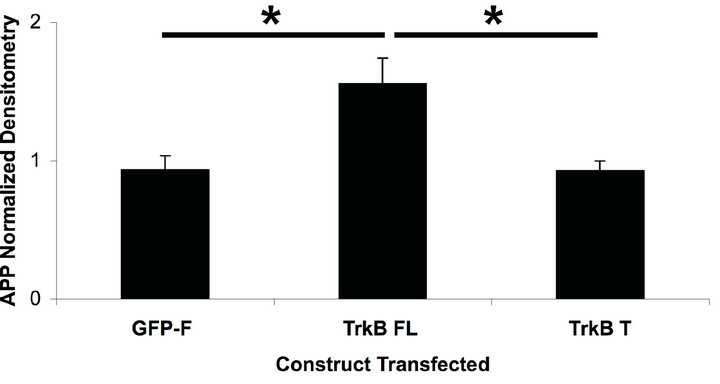 (c)
(c)
Figure 1. TrkB FL transfection increases endogenous fulllength APP levels in HEK293 cells. (a) Transient transfection of TrkB FL induces an increase in APP full-length levels while TrkB T does not affect APP levels compared to GFP-F control; (b) Stable transfection of TrkB FL also induces an increase in APP full-length levels compared to both TrkB T and GF-P-F stably transfected cells; (c) Quantification of APP FL levels in cells transfected with TrkB FL shows a statistically significant increase in APP levels over both TrkB T and GFP-F. Error bars represent standard deviation; n = 3.
SH-SY5Y cells have been shown to display accumulation of sAPP-a when treated with retinoic acid [16]. The ADAM10 promoter contains retinoic acid responsive elements (RARE), so we hypothesized that this increase sAPP-a release is mediated by an increased transcription and maybe activity of ADAM10 in this cell line. To verify this, we treated SH-SY5Y cells for 48 hours with 20 mM RA or DMSO and measured ADAM activity in the cell lysates. We also treated cells for 10 hours with PMA as a positive control. Both RA and PMA treatments resulted in a detectable increase in ADAM10 activity, the increase induced by PMA was higher (Figure 3(b)). To verify that the increase in ADAM activity observed after RA treatment, was due to an increase in ADAM levels, we performed a Western blot analysis on the same cell lysates used for the ADAM activity assay. We found that there was an increase in ADAM10 levels in the cells trea-
 (a)
(a)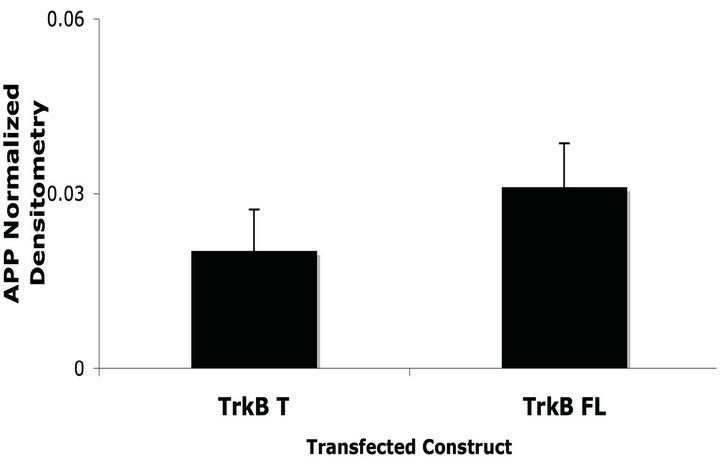 (b)
(b)
Figure 2. TrkB FL transfection increases endogenous fulllenght APP levels in SH-SY5Y Naïve cells. (a) Representative western blot of SH-SY5Y Naïve cells lysates transiently transfected with TrkB T or FL; (b) TrkB FL transfection induces an increase in APP full-length levels compared to TrkB T transfection. Error bars represent standard deviation; n = 3.
ted with RA compared to the cells treated with vehicle only (DMSO) (Figures 3(c) and (d)).
These experiments show that the ADAM assay is able to detect differences in ADAM activity and validate its use in our in vitro system. They also show that RA causes increased ADAM activity by up-regulating the protein levels.
We then applied this assay to test the hypothesis that TrkB FL activates ADAM independently of BDNF. We obtained populations of HEK293 cells stably transfected with a GFP-F, TrkB T or TrkB FL. We measured ADAM activity of the cells stably transfected with the different constructs and compared them (Figure 4). There was no statistical difference between the ADAM activity levels detected in TrkB FL or TrkB T transfected cells compared to the GFP-F transfected cells (Figure 4).
3.3. BACE1 Levels Are Increased by TrkB FL and TrkB SHC but Not by TrkB T
We did not observe altered ADAM activity when transfecting TrkB FL or TrkB T in HEK293 cells. ADAMs are responsible for a-secretase cleavage of APP. Because we observed an increased APP processing upon TrkB FL transfection [20], we hypothesized that BACE1, responsible for b-secretase cleavage of APP, levels might be
 (a)
(a) (b)
(b)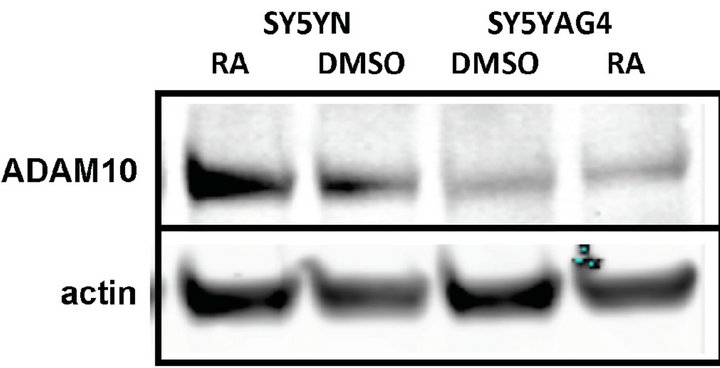 (c)
(c)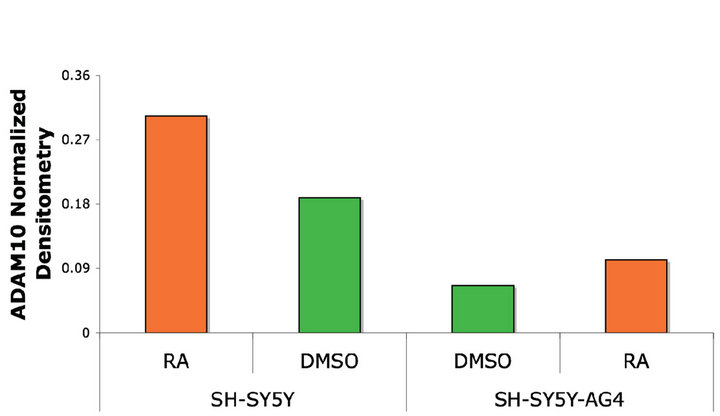 (d)
(d)
Figure 3. ADAM10 activity/expression in cell lines treated with PMA or stably transfected with TrkB. (a) PMA treatment of three different cell lines induces a significant increase in ADAM activity compared to the vehicle treated controls; (b) RA treatment of SH-SY5Y cells induces an increase in ADAM activity compared to vehicle control treated cells but not compared to PMA treated cells; (c) Representative Western blot for detection of ADAM10 levels in RA treated SH-SY5Y and SH-SY5Y-APP-Gal4 cells; (d) Quantification of the Western blot in (c) showing that ADAM10 levels are increased by RA treatment of the SH-SY5Y neuroblastoma cell line. Error bars represent standard deviation; n = 3.

Figure 4. TrkB FL and TrkB T stable transfection does not increase ADAM activity compared to GFP-F transfected cells. Error bars: standard deviation; n = 3.
affected upon TrkB transfection. To test this hypothesis, we measured BACE1 levels in cells transfected with the TrkB isoforms. We also employed TrkB FL mutants to identify the important functional domain mediating an effect on BACE1 (Figure 5(a)).
We found that TrkB FL significantly increased BACE1 levels compared to TrkB T. Transfection of the TrkB SHC isoform yielded BACE1 levels similar to TrkB FL. TrkB FL Y515F, mutated on the SHC binding site, decreased BACE1 levels compared to TrkB FL, suggesting a role of SHC binding to TrkB in mediating the effect. The tyrosine kinase inactive mutant, K571M, did not alter BACE1 levels compared to TrkB FL (Figure 5 (b)) suggesting that receptor phosphorylation is not required.
3.4. TrkB FL Activates NFAT3 Mediated Transcription
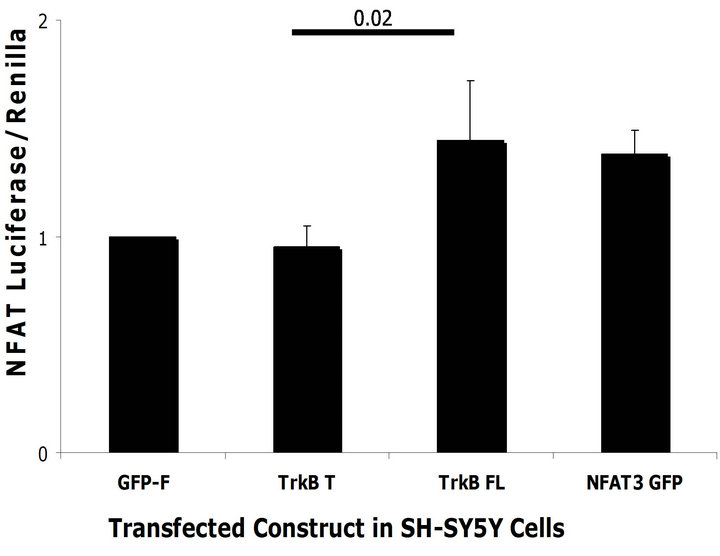
We observed an increase in BACE1 levels upon TrkB FL transfection. BACE1 levels can be modulated by the Ca2+ activated transcription factor NFAT1 [25]. Because TrkB FL can activate NFAT3, the neuronal specific form of NFAT, we hypothesized that TrkB FL might mediate activation of NFAT3 and thus increase BACE1 transcription. To test whether TrkB FL transfection was able to activate NFAT3, we transfected a NFAT luciferase reporter construct together with TrkB FL or TrkB T. We used a GFP tagged NFAT3 over-expression construct as a positive control and a renilla luciferase reporter to normalize transfection efficiency. In both SH-SY5Y naïve and HEK293 naïve cells, we found that TrkB FL transfection increased NFAT3 mediated luciferase activity compared to TrkB T and GFP-F control but not compared to the NFAT3 over-expression construct (Figures 6 (a) and (b)).
TrkB FL can activate signaling pathways that increase release of Ca2+ from intracellular storage, therefore the
 (a)
(a)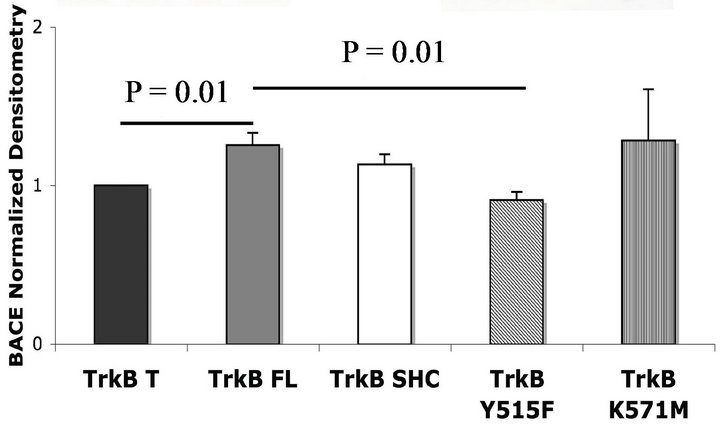 (b)
(b)
Figure 5. TrkB transfection affects BACE1 levels in HEK293 cells. (a) Representatives Western blots (WB) of BACE1 in cells transfected with TrkB isoforms and mutants; (b) WB Quantification. TrkB FL significantly increases BACE1 levels compared to TrkB T. Transfection of the mutant on the SHC binding site of TrkB FL, Y515F, decreases BACE1 levels compared to TrkB FL. Error bars: SEM; n = 6 for TrkB T, FL, SHC; n = 4 for TrkB Y515F; n = 2 for K571M.
effects that we observe might be due to Ca2+ mediated unspecific increase in transcription. To rule out this possibility we co-transfected the TrkB constructs, the control constructs and the NFAT3 construct together with a pGL3 luciferase reporter that does not respond to NFAT specific activation but would be subjected to general increases in transcription activity within the cells. We did not observe any differences in luciferase activity between TrkB T and TrkB FL when utilizing pGL3 as a reporter construct (Figures 6(c) and (d)) in both cell lines. This demonstrates that TrkB transcriptional effects are specific to NFAT. The increase in luciferase activity observed when transfecting NFAT3 and pGL3 luciferase is due to NFAT binding to the SV40 promoter, enhancing its transcription [26].
4. DISCUSSION
In this study, we have investigated TrkB mediated modulation of endogenous APP and of the APP processing enzymes ADAM10 and BACE1.
APP levels are increased by TrkB FL auto-activation and the increase is more dramatic in the HEK293 cells line than in the SH-SY5Y cell line. This might be due to the presence of all TrkB isoforms and BDNF in the latter cell line that might contribute to regulate levels of the TrkB FL/T exogenously expressed. APP promoter activation mediated by TrkB FL had been previously shown through reporter assays in SH-SY5Y cells [17], here we show that APP protein levels are increased by TrkB FL transfection compared to TrkB T and that this increase is apparent even in absence of exogenous BDNF. We observe the same increase in APP levels in HEK293 cells that do not express BDNF/TrkB endogenously. Therefore, over-expression of the TrkB FL receptor probably causes auto-activation and signaling through the IP3K pathway that has been shown to mediate APP transcription [17].
The fact that we observe a TrkB FL mediated increase in APP levels in both cell lines suggests that the pathways engaged are common among the two. Interestingly, TrkB FL/BDNF signaling is associated with beneficial effects in the context of AD while any factor that increases APP levels is generally associated with worse degeneration. This apparent contradiction can be resolved by several considerations. First, increased APP levels are not necessarily associated with more degeneration since APP is important in cell-matrix interaction and cell-cell interactions, APP is also expressed on dendritic spines and plays important roles in synaptic regulation [27,28]. Secondly, increased APP levels do not necessarily correlate with increased production of neurotoxic fragments. The outcome of APP proteolytic cleavage is central for the pathogenesis of AD. If APP is cleaved by a-secretases, it generates fragments that have growthfactor-like characteristics, such as sAPP-a [29]. Finally, TrkB FL activates many signaling pathways that are antiapoptotic and favor synaptic transmission and LTP [5]. In the broader context of all these interactions and balance, TrkB FL beneficial effects can be associated even with over-all increased APP levels. Very recently it has been shown that sAPP-a, the fragment generated by ADAMs from APP, can inhibit BACE1 activity [30] therefore TrkB FL could be involved in this mechanism by promoting sAPP-a production and increasing BACE1 levels.
The hypothesis that TrkB FL might favor a-secretase cleavage of APP has been addressed before. In previous experiments, RA treatment of cells had been showed to promote accumulation of sAPP-a. Even if the authors show that BDNF stimulation of the cells increases sAPPa accumulation, it is difficult to unequivocally determine if this effect is only, or at least mainly, mediated by TrkB. In fact RA can activate ADAM10 transcription independently of TrkB [31]. This work did not determine if the effect was specifically due to the tyrosine kinase activity of the full-length receptor. We could not detect an ADAM activity increase in HEK293 cells transfected with TrkB T or TrkB FL. This could be due to the fact that HEK293 cells have lower basal ADAM activity, lowering the sensitivity and dynamic range of the assay for this particular cell line. We could also speculate that auto-activation of the TrkB FL receptor differs from the
 (a)
(a)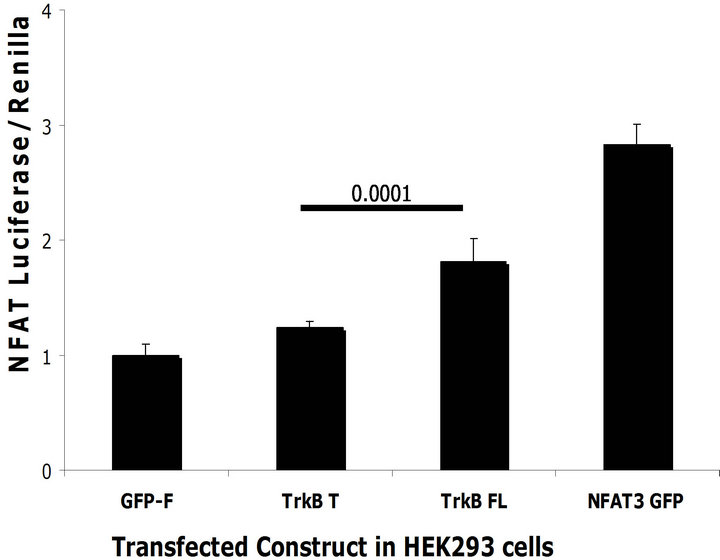 (b)
(b)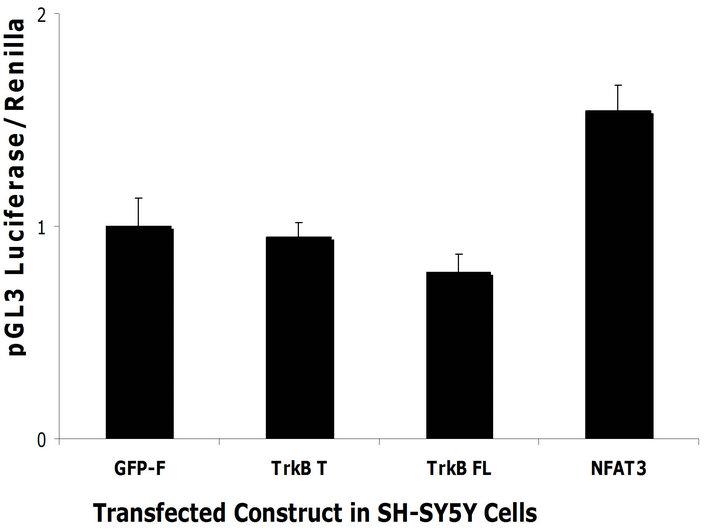 (c)
(c)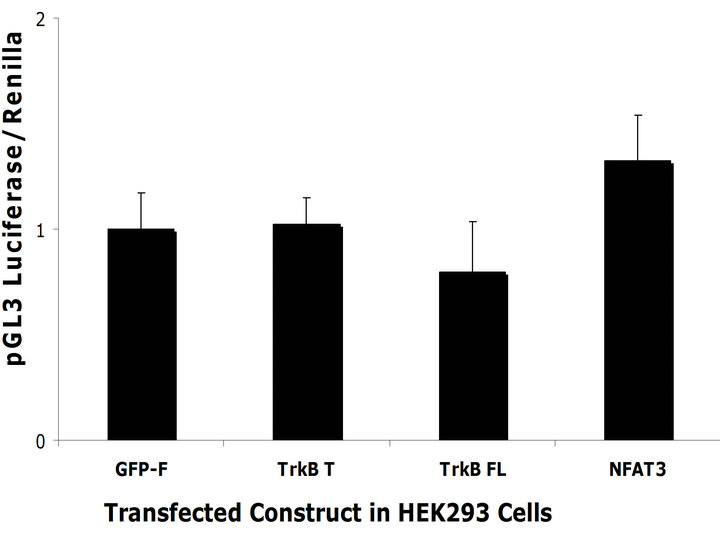 (d)
(d)
Figure 6. TrkB FL increases NFAT mediated transcription. (a) TrkB FL and TrkB T were transfected in SH-SY5Y Naïve cells with a NFAT luciferase reporter construct. TrkB FL significantly increased NFAT mediated luciferase compared to TrkB T (p = 0.02). GFP-F was used as a negative control while NFAT3 GFP tagged was used as a positive control; (b) TrkB FL and TrkB T were transfected in HEK293 naïve cells with a NFAT luciferase reporter constructs. TrkB FL significantly increased NFAT mediated luciferase compared to TrkB T (p = 0.0001); (c) TrkB FL did not increase transcription of a pGL3 reporter construct co-transfected in SH-SY5Y naïve cells. NFAT3 increased transcription of pGL3 because it can bind to its SV40 viral promoter; (d) TrkB FL did not increase transcription of a pGL3 reporter construct co-transfected in HEK293 Naïve cells. NFAT3 increased transcription of pGL3 because it can bind to its SV40 viral promoter. Error bars: standard deviation; n = 3 for SH-SY5Y cells with 6 replicates, n = 2 for HEK293 cells with 6 replicates.
BDNF dependent activation and is not efficient at mediating PLC-g binding and therefore at increasing ADAM activity.
BACE1 is responsible for the b-secretase cleavage of APP that initiates generation of the toxic Ab fragment. We observed that TrkB FL was able to increase APP processing in SH-SY5Y cells [20], therefore we tested if BACE1 levels were affected by TrkB FL or its isoforms. We found that BACE1 levels were increased by TrkB FL but not by TrkB T. Interestingly, TrkB FL Y515F, mutated on the SHC binding site, also decreases BACE1 levels compared to TrkB FL; while TrkB K571M, a tyrosine kinase deficient mutant, does not alter BACE1 levels compared to TrkB FL. The TrkB SHC isoform also displays BACE1 levels similar to TrkB FL. The knockdown of the adaptor protein SHC has been shown to decrease BACE1 levels and therefore Ab production [24]. SHC mediates downstream signaling of TrkB FL and it acts upstream of the NFAT/AP-1 activation pathway [32] . Moreover, NFAT1 activation has been shown to increase BACE1 levels [25]. NFAT3 is activated by TrkB FL/ BDNF signaling [23] and we observe a specific increase in NFAT3 mediated luciferase transcription when overexpressing TrkB FL but not TrkB T. We then suggest a mechanism for BACE1 transcriptional regulation mediated by TrkB FL/SHC and NFAT. The finding that BACE1 levels are increased by TrkB FL would suggest a negative effect of TrkB FL in AD and support the hypothesis that TrkB truncated isoforms are protective in neurodegeneration [12,33]. It remains to be determined if TrkB/FL signaling is impaired as a consequence of the neurodegeneration or if decreased TrkB/BDNF signaling due to disease state, aging or life style can be a participating factor in the initiation of AD neurodegeneration cascade.
5. CONCLUSION
In conclusion we find that TrkB FL over-expression increases APP levels even in absence of exogenous BDNF, suggesting that auto-activation of the receptor can mediate APP transcription. We also find that BACE1 levels are increased by TrkB FL auto-activation in HEK293 cells. BDNF levels are decreased in AD brains and TrkB FL activation independent of BDNF could be a mechanism of action in the pathogenesis. We finally identify SHC as a possible mediator of the BACE1 up-regulation since mutation of the SHC binding site was sufficient to eliminate the TrkB FL mediated BACE1 level increase. We finally suggest that NFAT mediated transcriptional activation could be the mechanism of BACE1 transcription up-regulation downstream of TrkB signaling. NFAT nuclear translocation is in fact observed in AD [34] and that could correlate to increased BACE1 levels in AD [35].
6. ACKNOWLEDGEMENTS
The authors are grateful for the Mozino Scholarship and the MaxNet Aging Post Doctoral fellowship provided to S.A. This work was funded by Drexel University, Commonwealth of Pennsylvania, the National Institute of Health (NS057295) and the Alzheimer’s Association. This paper is subject to the NIH Public Access Policy.
REFERENCES
- Alzheimer’s Association (2012) Alzheimer’s disease facts and figures. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 8, 131-168.
- Corbett, A., Smith, J. and Ballard, C. (2012) New and emerging treatments for Alzheimer’s disease. Expert Review of Neurotherapeutics, 12, 535-543. doi:10.1586/ern.12.43
- Benilova, I., Karran, E. and De Strooper, B. (2012) The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nature Neuroscience, 15, 349- 357. doi:10.1038/nn.3028
- Zhang, F., et al. (2012) Roles of brain-derived neurotrophic factor/tropomyosin-related kinase B (BDNF/TrkB) signalling in Alzheimer’s disease. Journal of Clinical Neuroscience, 19, 946-949. doi:10.1016/j.jocn.2011.12.022
- Minichiello, L. (2009) TrkB signalling pathways in LTP and learning. Nature Reviews Neuroscience, 10, 850-860. doi:10.1038/nrn2738
- Rothman, S.M., et al. (2012) Brain-derived neurotrophic factor as a regulator of systemic and brain energy metabolism and cardiovascular health. Annals of the New York Academy of Sciences, 1264, 49-63. doi:10.1111/j.1749-6632.2012.06525.x
- Nagahara, A.H. and Tuszynski, M.H. (2011) Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nature Reviews Drug Discovery, 10, 209-219. doi:10.1038/nrd3366
- Longo, F.M., et al. (2007) Small molecule neurotrophin receptor ligands: Novel strategies for targeting Alzheimer’s disease mechanisms. Current Alzheimer Research, 4, 503-506. doi:10.2174/156720507783018316
- Luberg, K., et al. (2010) Human TrkB gene: Novel alternative transcripts, protein isoforms and expression pattern in the prefrontal cerebral cortex during postnatal development. Journal of Neurochemistry, 113, 952-964. doi:10.1111/j.1471-4159.2010.06662.x
- Stoilov, P., Castren, E. and Stamm, S. (2002) Analysis of the human TrkB gene genomic organization reveals novel TrkB isoforms, unusual gene length, and splicing mechanism. Biochemical and Biophysical Research Communications, 290, 1054-1065. doi:10.1006/bbrc.2001.6301
- Baxter, G.T., et al. (1997) Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB. T2. The Journal of Neuroscience, 17, 2683-2690.
- Wong, J., et al. (2012) Amyloid beta selectively modulates neuronal TrkB alternative transcript expression with implications for Alzheimer’s disease. Neuroscience, 210, 363-374. doi:10.1016/j.neuroscience.2012.02.037
- Ginsberg, S.D., et al. (2006) Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. Journal of Neurochemistry, 97, 475-487. doi:10.1111/j.1471-4159.2006.03764.x
- Poon, W.W., et al. (2009) β-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiology Aging, 32, 821-833.
- Tong, L., et al. (2004) Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. Journal of Neuroscience, 24, 6799-6809. doi:10.1523/JNEUROSCI.5463-03.2004
- Holback, S., Adlerz, L. and Iverfeldt, K. (2005) Increased processing of APLP2 and APP with concomitant formation of APP intracellular domains in BDNF and retinoic acid-differentiated human neuroblastoma cells. Journal of Neurochemistry, 95, 1059-1068. doi:10.1111/j.1471-4159.2005.03440.x
- Ruiz-Leon, Y. and Pascual, A. (2001) Brain-derived neurotrophic factor stimulates beta-amyloid gene promoter activity by a Ras-dependent/AP-1-independent mechanism in SH-SY5Y neuroblastoma cells. Journal of Neurochemistry, 79, 278-285. doi:10.1046/j.1471-4159.2001.00547.x
- Ruiz-Leon, Y. and Pascual, A. (2003) Induction of tyrosine kinase receptor b by retinoic acid allows brain-derived neurotrophic factor-induced amyloid precursor protein gene expression in human SH-SY5Y neuroblastoma cells. Neuroscience, 120, 1019-1026. doi:10.1016/S0306-4522(03)00391-9
- Ruiz-Leon, Y. and Pascual, A. (2004) Regulation of betaamyloid precursor protein expression by brain-derived neurotrophic factor involves activation of both the Ras and phosphatidylinositide 3-kinase signalling pathways. Journal of Neurochemistry, 88, 1010-1018. doi:10.1046/j.1471-4159.2003.02226.x
- Ansaloni, S., et al. (2011) TrkB isoforms differentially affect AICD production though their intracellular functional domains. International Journal of Alzheimer’s Disease, 2011, Article ID: 729382.
- Rajagopal, R., et al., (2004) Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. Journal of Neuroscience, 24, 6650-6658. doi:10.1523/JNEUROSCI.0010-04.2004
- Schecterson, L.C., et al. (2010) Trk activation in the secretory pathway promotes Golgi fragmentation. Molecular and Cellular Neuroscience, 43, 403-413. doi:10.1016/j.mcn.2010.01.007
- Groth, R.D. and Mermelstein, P.G. (2003) Brain-derived neurotrophic factor activation of NFAT (nuclear factor of activated T-cells)-dependent transcription: A role for the transcription factor NFATc4 in neurotrophin-mediated gene expression. Journal of Neuroscience, 23, 8125-8134.
- Xie, Z., et al. (2007) RNA interference silencing of the adaptor molecules ShcC and Fe65 differentially affect amyloid precursor protein processing and Abeta generation. The Journal of Biological Chemistry, 282, 4318- 4325. doi:10.1074/jbc.M609293200
- Cho, H.J., et al. (2008) Disrupted intracellular calcium regulates BACE1 gene expression via nuclear factor of activated T cells 1 (NFAT 1) signaling. Aging Cell, 7, 137- 147. doi:10.1111/j.1474-9726.2007.00360.x
- Manley, K., O’Hara, B.A. and Atwood, W.J. (2008) Nuclear factor of activated T-cells (NFAT) plays a role in SV40 infection. Virology, 372, 48-55. doi:10.1016/j.virol.2007.10.029
- Kamenetz, F., et al. (2003) APP processing and synaptic function. Neuron, 37, 925-937. doi:10.1016/S0896-6273(03)00124-7
- Hoe, H.S., et al. (2009) The effects of amyloid precursor protein on postsynaptic composition and activity. The Journal of Biological Chemistry, 284, 8495-8506. doi:10.1074/jbc.M900141200
- Caille, I., et al. (2004) Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development, 131, 2173-2181. doi:10.1242/dev.01103
- Obregon, D., et al. (2012) Soluble amyloid precursor protein-alpha modulates beta-secretase activity and amyloid-beta generation. Nature Communications, 3, 777. doi:10.1038/ncomms1781
- Postina, R. (2012) Activation of alpha-secretase cleavage. Journal of Neurochemistry, 120, 46-54. doi:10.1111/j.1471-4159.2011.07459.x
- Turner, S.D., et al. (2007) The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell Signal, 19, 740-747. doi:10.1016/j.cellsig.2006.09.007
- Gomes, J.R., et al. (2012) Excitotoxicity downregulates TrkB.FL signaling and upregulates the neuroprotective truncated TrkB receptors in cultured hippocampal and striatal neurons. Journal of Neuroscience, 32, 4610-4622. doi:10.1523/JNEUROSCI.0374-12.2012
- Abdul, H.M., et al. (2009) Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. Journal of Neuroscience, 29, 12957-12969. doi:10.1523/JNEUROSCI.1064-09.2009
- Holler, C.J., et al. (2012) BACE2 expression increases in human neurodegenerative disease. The American Journal of Pathology, 180, 337-350. doi:10.1016/j.ajpath.2011.09.034