American Journal of Analytical Chemistry
Vol.06 No.04(2015), Article ID:55069,7 pages
10.4236/ajac.2015.64033
From Where Did the Water Come That Filled the Earth’s Oceans? A Widely Overlooked Redox Reaction
Friedemann T. Freund1,2,3, Minoru M. Freund1*
1NASA Ames Research Center, Moffett Field, CA, USA
2Department of Physics, San Jose State University, San Jose, CA, USA
3Senior Scientist, SETI Institute, Mountain View, CA, USA
Email: friedemann.t.freund@nasa.gov
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 February 2015; accepted 23 March 2015; published 26 March 2015
ABSTRACT
Though two-thirds of Earth’s surface is covered by oceans, measurements of hydroxyl concentrations in upper mantle minerals, specifically in olivine, reportedly provide surprisingly low values. This has been interpreted to mean that there is little dissolved H2O in the Earth’s mantle. By inference, when Earth formed, there might not have been able enough water to fill the oceans through volcanic degassing. It has therefore been proposed that the missing water was delivered to Earth from space, through comets and other impacting bodies. However, the reported low hydroxyl concentrations in olivine and similar mineralsis probably based on a profound misunderstanding of a solid state reaction that converts hydroxyls into something more difficult to detect. There is indeed a redox reaction that converts, during cooling, solute hydroxyls in the matrix of minerals into peroxy plus H2. This widely overlooked redox conversion takes place under thermodynamic non-equilibrium conditions. Its significance is that any mineral and any rock available for collection at the Earth surface has gone through a process that causes hydroxyls, the telltale sign of dissolved H2O, to change into peroxyplusH2. The H2 molecules are diffusively mobile and may leave even structurally dense mineral grains. The remaining peroxy thus become the memory of the “true” solute H2O content, besides a few residual hydroxyls. Though first described over 30 years ago, this redox conversion has been largely ignored. As a result it is unknown how much H2O is contained in the Earth’s upper mantle but it is certainly much more than has been assumed until now on the basis of analysis of residual hydroxyls.
Keywords:
Redox Conversion, Hydroxyls, Peroxy, Water, Earth’s Upper Mantle

1. Introduction
For decades astronomers and others have argued about how planet Earth got its water. Some believe the water to be as old as the Earth itself [1] -[4] , others think that, because Earth’s surface was once deeply molten, the planet could not retain its primordial water. Therefore, much of the water available now must have arrived later, presumably through collisions with comets [5] -[7] . The latter belief is based on observations that today’s upper mantle appears to be relatively “dry”.
Laboratory measurements of olivine crystals and other minerals, brought up from the upper mantle by volcanic actions, suggest surprisingly low to very low “water” concentration in form of solute hydroxyls, 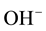 or O3Si-OH. Using mostly infrared (IR) spectroscopy as diagnostic tool, the minerals retrieved from upper mantle sources were indeed found to be generally low in hydroxyl, implying low H2O contents in the source region [8] -[10] . Since the water in the oceans must have degassed from the Earth’s mantle, if upper mantle rocks were low in solute H2O, there might not have been enough water to fill the oceans. This led to the idea that the only way to supply enough water to the Earth would have been by massive cometary impacts [5] [11] [12] .
or O3Si-OH. Using mostly infrared (IR) spectroscopy as diagnostic tool, the minerals retrieved from upper mantle sources were indeed found to be generally low in hydroxyl, implying low H2O contents in the source region [8] -[10] . Since the water in the oceans must have degassed from the Earth’s mantle, if upper mantle rocks were low in solute H2O, there might not have been enough water to fill the oceans. This led to the idea that the only way to supply enough water to the Earth would have been by massive cometary impacts [5] [11] [12] .
However, the argument is fallacious that low hydroxyl contents in upper mantle minerals collected at the Earth surface are a proof of low solute H2O contents in the source region. There is a way how minerals, which started out with fairly high concentrations of solute hydroxyls in the hot regions of the Earth’s deeper crust and upper mantle, can end up at the surface of the Earth with much lower solute hydroxyl concentrations. The reason is a redox conversion that consumes solute hydroxyls in the matrix of minerals by converting them into peroxy plus molecular H2.
Here we present this still widely unknown solid state redox conversion, which is fundamentally important to understand the history and evolution of planet Earth.
2. Thermodynamics of Solid Solutions and Supersaturated Solid Solutions
To explain the redox conversion that changes hydroxyls into peroxy plus H2 we can neglect the chemical complexity of mantle minerals such as olivine, ideally 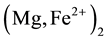 SiO4. Instead we focus on the simplest binary oxide, MgO and ask: what happens when MgO incorporates small amounts of dissolved H2O in its crystal structure?
SiO4. Instead we focus on the simplest binary oxide, MgO and ask: what happens when MgO incorporates small amounts of dissolved H2O in its crystal structure?
MgO single crystals are routinely grown from the melt in a carbon-arc-fusion furnace [13] . Though the procedure is typically done in air, the MgO melt produced by the carbon arc plasma is extremely reducing. Hence, the MgO crystals, which grow from the melt, should be highly reduced.
Paradoxically, by the time the carbon-arc-fusion grown MgO crystals have cooled to room temperature, they contain perox defects. However, peroxy defects are the hallmark of highly oxidizing conditions. This raises the question: Does the presence of peroxy defects in MgO crystals that have been grown under extremely reducing conditions violate the laws of thermodynamics? The answer is “no”, because the peroxy defects are introduced under non-equilibrium conditions.
If peroxy defects in carbon arc fusion grown MgO crystals are the result of a non-equilibrium reaction in the solid state, what is this reaction and how might it shed light on processes that happen in Nature with minerals that have crystallized in the reducing environment of the Earth’s upper mantle and were subsequently brought to the surface of the Earth?
Figure 1 shows the AO-rich side of a binary phase diagram for a high melting oxide material AO with H2O as gas/fluid phase component. AO can also be any silicate mineral. Tmelt indicates the melting temperature of pure AO, which is “nominally anhydrous”, meaning that its structure does not have any regular lattice sites to accommodate solute H2O. However, the presence of H2O in the AO melt causes Tmelt to decrease to Tcryst and a finite concentration of the H2O component to become incorporated into the AO matrix in form of hydroxyls, OH−. The result is an AO-H2O solid solution (ss).
Thermodynamics mandates that, with decreasing T, the width of the AO-H2O ss field must decrease. If it were possible to maintain thermodynamic equilibrium throughout cooling, the solute  would continuously segregate, until the width of the ss field shrinks to zero at 0 K.
would continuously segregate, until the width of the ss field shrinks to zero at 0 K.
As long as thermodynamic equilibrium is maintained during cooling, the MgO-H2O solid solution will adjust to the narrowing of the ss field. However, “exsolution” can only be achieved, if solute  plus
plus 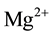 vacancies are able to diffuse from inside the grains to their surfaces or boundaries.
vacancies are able to diffuse from inside the grains to their surfaces or boundaries.
Segregation is a process that is diffusion-controlled. Thus, inextricably, the MgO-H2O system will reach a

Figure 1. AO-side of binary AO-H2O phase diagram showing the solid solution and supersaturated solid solution fields. AO can be an oxide or silicate mineral.
temperature Tfreez’g, below which segregation becomes too sluggish. Therefore, below Tfreez’g, the solid solution (ss) leaves thermodynamic equilibrium and turns into a supersaturated solid solution (sss). Though Tfreez’g depends on the cooling rate, it typically falls into the 500˚C - 600˚C range [14] .
Since no changes in the overall composition of the sss system are supposed to take place.
Below Tfreez’g, we write the dissolution of 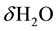 in the MgO matrix simply as:
in the MgO matrix simply as:
 (1)
(1)
Equation (1) is mass-balanced. It states that 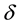 are formed by introducing
are formed by introducing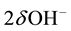 . In other words, in the MgO-H2O ss,
. In other words, in the MgO-H2O ss, 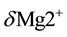 are substituted by
are substituted by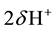 .
.
3. Solute OH− in Solid Solution
To more fully understand the complexity of what can happen in the sss field, we need to learn what kind of “impurity”  are part of the solid solution, more specifically
are part of the solid solution, more specifically
1) what types of 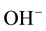 -bearing defects are formed;
-bearing defects are formed;
2) where the 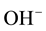 reside in the crystal matrix and;
reside in the crystal matrix and;
3) what happens to them during further cooling through the sss field.
The substitutional mode given by Equation [1] predicts 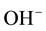 pairs at
pairs at  vacancy sites as the main defect type, here labeled I. Using the Kröger-Vinck point defect designation1 defect I is written as
vacancy sites as the main defect type, here labeled I. Using the Kröger-Vinck point defect designation1 defect I is written as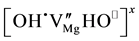 , where two
, where two 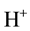 compensate for the missing
compensate for the missing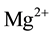 . It will dissociate according to:
. It will dissociate according to:
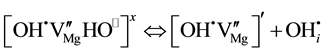 (2)
(2)
where defect II is a single  at an
at an 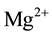 vacancy and defect III is an interstitial
vacancy and defect III is an interstitial  with its proton at any
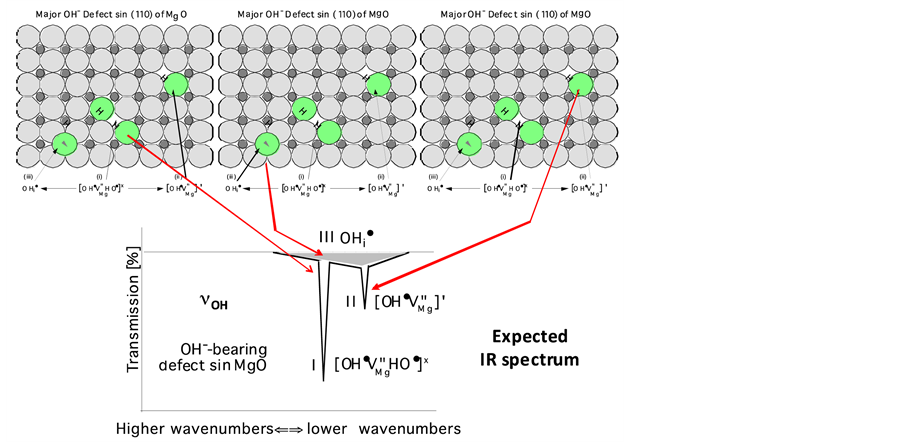
with its proton at any 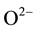 in the MgO structure. The three defects are depicted in Figure 2(a) along with the expected relative intensities of their O-H stretching bands,
in the MgO structure. The three defects are depicted in Figure 2(a) along with the expected relative intensities of their O-H stretching bands,  , according to Equation [2] .
, according to Equation [2] .
Figure 2(b) shows the measured IR spectrum with the assignment of the 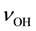 bands to the three types of
bands to the three types of  in the MgO matrix [16] . There is significant disagreement between the “expected” and the “observed” spectra. The
in the MgO matrix [16] . There is significant disagreement between the “expected” and the “observed” spectra. The  band at 3560 cm−1, due to OH− pairs, defect I, does not show up with the highest intensity but with the lowest, leaving the
band at 3560 cm−1, due to OH− pairs, defect I, does not show up with the highest intensity but with the lowest, leaving the 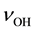 band at 3700 cm−1, due to single
band at 3700 cm−1, due to single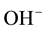 , to dominate the spectrum.
, to dominate the spectrum.
The most unusual feature, however, is the very weak band at 4150 cm−1. It is diagnostically distinct, unambi
 (a)
(a)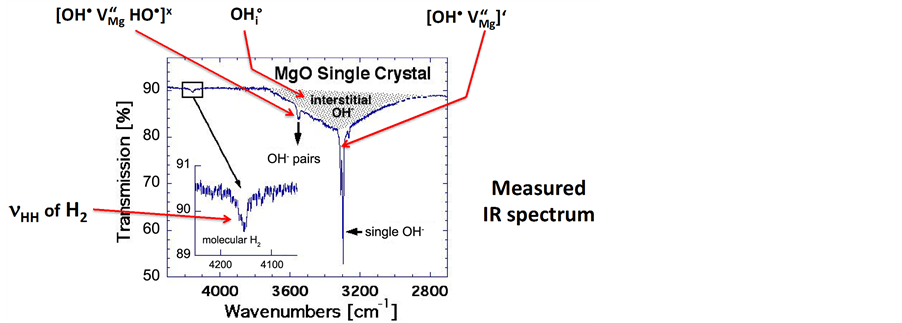 (b)
(b)
Figure 2. (a) OH−-bearing point defects derived from the dissolution of H2O in MgO and expected relative intensities of the νOH bands due to OH− on the three dominant OH−-bearing defect sites. Of those three expected νOH bands the one labeled I, due to OH− pairs at Mg2+ vacancies, should be most intense (after [Freund and Wengeler, 1982] [16] ). The νOH band arising from defect III, interstitial OH−, is broadened and intensified due to H-bonding [17] ; (b) Measured IR spectrum: contrary to expectation the νOH band due to OH− pairs at Mg2+ vacancy sites has the lowest intensity, allowing the νOH band due to single OH− to dominate the spectrum. The band at 4150 cm−1 is due to νHH indicating the presence of H2 molecules in the MgO matrix (after [Freund and Wengeler, 1982] [16] ).
guously identifiable as due to lattice-bound H2 through the combination of the H-H stretching mode 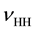 with a phonon mode [18] . This assignment has been confirmed through the
with a phonon mode [18] . This assignment has been confirmed through the 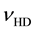 band in MgO crystals grown from a melt containing dissolved D2O [19] .
band in MgO crystals grown from a melt containing dissolved D2O [19] .
4. Redox Conversion of OH− Pairs to Peroxy plus H2
First indication for molecular H2 was obtained nearly 40 years ago during a mass spectroscopic study of gases released from ultrahigh purity  -rich nano-sized MgO produced by the thermal decomposition of Mg(OH)2 [20] . Copious amounts of H2 were found to evolve from the MgO crystallites, followed by atomic O with a sharp on-set at 600˚C. The release of atomic O and sharp on-set at 600˚C points to a on-set at disproportionation of peroxy:
-rich nano-sized MgO produced by the thermal decomposition of Mg(OH)2 [20] . Copious amounts of H2 were found to evolve from the MgO crystallites, followed by atomic O with a sharp on-set at 600˚C. The release of atomic O and sharp on-set at 600˚C points to a on-set at disproportionation of peroxy:
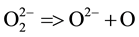 (3)
(3)
Forming H2 from  requires a redox reaction. Since Mg is a main group metal with only one chemically accessible valence state, 2+, in order to reduce hydroxyl protons to H2 something else must be oxidized. The only possibility is the hydroxyl oxygen changing its valence from
requires a redox reaction. Since Mg is a main group metal with only one chemically accessible valence state, 2+, in order to reduce hydroxyl protons to H2 something else must be oxidized. The only possibility is the hydroxyl oxygen changing its valence from  to
to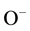 .
.
This turned out be true, involving the 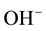 -pairs of defect I in Figures 2(a) and Figure 2(b). In the process the oxygens of the two
-pairs of defect I in Figures 2(a) and Figure 2(b). In the process the oxygens of the two 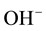 transfer an electron to their respect protons [16] , thereby oxidizing to the 1− valence state. The two
transfer an electron to their respect protons [16] , thereby oxidizing to the 1− valence state. The two 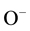 coupleto form a peroxy anion,
coupleto form a peroxy anion,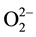 . The two H atoms bind to form an H2 molecule at the
. The two H atoms bind to form an H2 molecule at the  vacancy site:
vacancy site:
 (4a)
(4a)
As a redox conversion this process requires nothing more than a local rearrangement of electrons and a slight shift of atomic positions. It can take place perfectly well in the sss state, under non-equilibrium conditions, in the region marked in gray in Figure 1, around or below 500˚C. As a result the concentration of 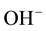 detectable via their
detectable via their 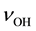 bands must decrease significantly, suggesting a low solute H2O content.
bands must decrease significantly, suggesting a low solute H2O content.
Since H2 molecules are mobile, even in densely packed MgO, they can diffuse away from the 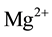 vacancy site leaving behind an
vacancy site leaving behind an 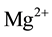 vacancy charge wise compensated by a peroxy anion,
vacancy charge wise compensated by a peroxy anion,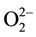 :
:
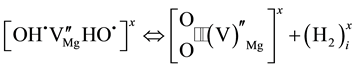 (4b)
(4b)
Equation (4b) indicates that, when the H2 molecules may move away from the 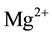 vacancy site, they become unavailable to convert back to
vacancy site, they become unavailable to convert back to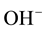 . The process continues by H2 molecules diffusing out:
. The process continues by H2 molecules diffusing out:
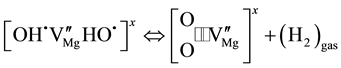 (4c)
(4c)
Equation (4c) marks the transition to irreversibility with H2 molecules having left the solid state. Thus, Equation (4c) describes the formation of a cation-deficient MgO, . If
. If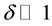 , we may approximate its composition as MgO with excess-oxygen,
, we may approximate its composition as MgO with excess-oxygen,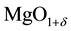 .
.
Equations (4a/c) introduce peroxy, even though there was no need for the MgO-H2O system to have ever experienced oxidizing conditions. The peroxy enters under non-equilibrium conditions via the above-mentioned redox conversion.
Other techniques in addition to mass spectrometry and IR spectroscopy have been employed to study the MgO-H2O sss system, specifically the electrical conductivity [21] , thermal expansion [22] , magnetic susceptibility [23] , electron spin resonance [24] , dielectric polarization [25] , refractive index [26] , and most recently muon spin relaxation [unpublished results] of MgO single crystals. These additional investigations have provided irrefutable evidence that, despite their provenance from the extremely highly reducing conditions of a carbon-arc-fusion melt [13] [27] , melt-grown MgO crystals contain peroxy defects.
5. Peroxy as the Memory of a Former Dissolved “Water” Content
In plain chemical language the redox conversion of hydroxyl pairs can be written as:
 (5)
(5)
This is such a fundamental equation that it would be surprising, if the reaction were unique to the MgO-H2O system. Indeed, this redox conversion appears to be universal, applicable as well as to silicates, where solute hydroxyls can be written as O3X-OH, with , Al3+ etc. Hydroxyl pairs, O3X-OH OH-XO3, whenever they exist, seem to be subject to the same redox conversion:
, Al3+ etc. Hydroxyl pairs, O3X-OH OH-XO3, whenever they exist, seem to be subject to the same redox conversion:
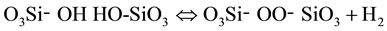 (6)
(6)
In fact there is evidence that every igneous rock that crystallized from a magma, every high-grade metamorphic rock originating from a high temperature, fluid-rich environment, and even every sedimentary rock that contains detrital grains of (mostly) quartz washed down from the mountains during weathering, will have H2 and peroxy defects [28] -[30] . Thus, the peroxy defects become a “memory”, a universal “memory” of former solute H2O contents, along with residual hydroxyls that have not undergone this redox conversion.
The said argument also applies to upper mantle rocks, e.g. peridotites, and to olivine single crystals that have been brought up from mantle depth. As they cooled to Earth surface temperatures, they underwent the same redox conversion, changing their existing hydroxyls into peroxy plus H2.
While it is true that upper mantle-derived olivine come from highly reducing environment, olivine single crystals have also provided strong evidence for the presence of peroxy plus H2 [28] [31] . This implies that, while the olivine crystals resided in the upper mantle, they were significantly richer in hydroxyls, i.e. solute H2O, than suggested by the low to very low residual hydroxyl contents, that has been provided by different types of chemical and spectroscopic of analyses of specimens collected at the Earth’s surface. This implies that most of solute hydroxyls that there once part of the olivine-H2O solid solution at upper mantle depth have been lost in the temperature window marked in gray in Figure 1, due to their redox conversion to peroxy plus H2.
6. Discussion
The redox conversion of solute H2O to peroxy plus H2 has all the bearings of a universal redox reaction. It is expected to apply to all rocks residing in the Earth’s rock column at temperatures below ~500˚C. Regardless of the environment from where these rocks come, from a high temperature portion of the crust or from the upper mantle, whenever they crossed―as part of their geological history―the temperature window marked in gray in Figure 1, much of their original solute hydroxyl content would have converted to H2 plus peroxy according to Equations (5) and (6).
This leads us to the rather daring conclusion that only a small fraction of the “true” solute H2O content once contained, for instance, in the upper mantle will remain in form of residual OH− or O3X-OH, where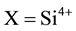 ,
,  , etc. This does not mean that those minerals suddenly became “dry” during transport to the Earth’s surface but only that their solute H2O content is no longer expressed in form of hydroxyls. This conclusion, however, also implies that IR spectroscopy is ill-suited to assess the true solute H2O content. Hence, publications that rely on IR spectroscopy as analytical tool [9] [10] [32] [33] probably severely underestimated the “true” solute H2O content in Earth’s upper mantle. Even techniques that probe H as an element such as SIMS and Ion Probes [34] [35] will not have been able to provide the “true” solute H2O contents, if some or most of the H2 molecules formed by the redox conversion had already out-diffused from the crystals under study.
, etc. This does not mean that those minerals suddenly became “dry” during transport to the Earth’s surface but only that their solute H2O content is no longer expressed in form of hydroxyls. This conclusion, however, also implies that IR spectroscopy is ill-suited to assess the true solute H2O content. Hence, publications that rely on IR spectroscopy as analytical tool [9] [10] [32] [33] probably severely underestimated the “true” solute H2O content in Earth’s upper mantle. Even techniques that probe H as an element such as SIMS and Ion Probes [34] [35] will not have been able to provide the “true” solute H2O contents, if some or most of the H2 molecules formed by the redox conversion had already out-diffused from the crystals under study.
The only way forward is to develop techniques that also probe peroxy concentrations. The reason is that, though the redox conversion of hydroxyl pairs into peroxy plus H2 has been described more than 30 years ago [16] and affirmed in the intervening years, the reality of peroxy in minerals and their potential role a memory of “true” H2O contents have not yet found sufficiently wide recognition within the community. An effort to develop the necessary analytical techniques to quantify peroxy contents is needed [25] .
In summary, on the basis of available data, it can be stated with a high degree of confidence that the Earth’s upper mantle has most likely contained in the past―and continues to contain today―plenty of “water” in the form of solute hydroxyls, probably enough to fill the worlds oceans. Cometary impacts, which attracted so much attentin over the years [5] [6] , and are still widely discussed today, may not have been needed to “help out”.
Acknowledgements
The results reported in this paper evolved over many years, starting with early studies supported in part by the Deutsche Forschungsgemeinschaft and continuing with support by the NASA Ames Research Center Director’s Discretionary Fund and the NASA Earth Surface and Interior (ESI) program under grant # NNX12AL71G.
References
- Fassett, C.I. and Minton, D.A. (2013) Impact Bombardment of the Terrestrial Planets and the Early History of the Solar System. Nature Geoscience, 6, 520-524. http://dx.doi.org/10.1038/ngeo1841
- Morbidelli, A., Chambers, J., Lunine, J.I., Petit1, J.M., Robert, F., Valsecchi, G.B. and Cyr, K.E. (2000) Source Regions and Timescales for the Delivery of Water to the Earth. Meteoritics and Planetary Science, 35, 1309-1320. http://dx.doi.org/10.1111/j.1945-5100.2000.tb01518.x
- Sarafian, A.R., Nielsen, S.G., Marschall, H.R., McCubbin, F.M. and Monteleone, B.D. (2014) Early Accretion of Water in the Inner Solar System from a Carbonaceous Chondrite-Like Source. Science, 346, 623-626. http://dx.doi.org/10.1126/science.1256717
- Shiga, D. (2010) Earth May Have Had Water from Day One. The New Scientist, 208, 12. http://dx.doi.org/10.1016/S0262-4079(10)62726-1
- Chyba, C.F. (1987) The Cometary Contribution to the Oceans of Primitive Earth. Nature, 330, 632-635. http://dx.doi.org/10.1038/330632a0
- Oberbeck, V.R. and Aggarwal, H. (1993) Comet Impacts and Chemical Evolution on the Bombarded Earth. Origins of Life and Evolution of the Biosphere, 21, 317-338. http://dx.doi.org/10.1007/BF01808305
- Robert, F. (2001) The Origin of Water on Earth. Science, 293, 1056-1058. http://dx.doi.org/10.1126/science.1064051
- Bell, D.R. and Rossman, G.R. (1992) Water in Earth’s Mantle: The Role of Nominally Anhydrous Minerals. Science, 255, 1391-1397. http://dx.doi.org/10.1126/science.255.5050.1391
- Bell, D.R., Rossman, G.R., Maldener, J., Endisch, D. and Rauch, F. (2003) Hydroxide in Olivine: A Quantitative Determination of the Absolute Amount and Calibration of the IR Spectrum. Journal of Geophysical Research (Solid Earth), 108, ECV 8-1. http://dx.doi.org/10.1029/2001JB000679
- Miller, G.H., Rossman, G.R. and Harlow, G.E. (1987) The Natural Occurrence of Hydroxide in Olivine. Physics and Chemistry of Minerals, 14, 461-472. http://dx.doi.org/10.1007/BF00628824
- Bolfan-Casanova, N. (2005) Water in the Earth’s Mantle. Mineralogical Magazine, 69, 229-257. http://dx.doi.org/10.1180/0026461056930248
- Owen, C.T. (1999) What Do We Know about the Origin of the Earth’s Oceans? Scientific American, 21 October 1999.
- Abraham, M.M., Butler, C.T. and Chen, Y. (1971) Growth of High-Purity and Doped Alkaline Earth Oxides, Part I: Magnesium Oxide and Calcium Oxide. The Journal of Chemical Physics, 55, 3752-3756. http://dx.doi.org/10.1063/1.1676658
- Nesse, W.D. (2000) Introduction to Mineralogy. Oxford University Press, New York.
- Kröger, F.A. (1964) The Chemistry of Imperfect Crystals. North-Holland, Amsterdam.
- Freund, F. and Wengeler, H. (1982) The Infrared Spectrum of OH-Compensated Defect Sites in C-Doped MgO and CaO Single Crystals. Journal of Physics and Chemistry of Solids, 43, 129-145. http://dx.doi.org/10.1016/0022-3697(82)90131-7
- Steiner, T. (2002) The Hydrogen Bond in the Solid State. Angewandte Chemie International Edition, 41, 48-76. http://dx.doi.org/10.1002/1521-3773(20020104)41:1<48::AID-ANIE48>3.0.CO;2-U
- Kriegler, R.J. and Welch, H.L. (1968) Induced Infrared Fundamental Band of Hydrogen Dissolved in Solid Argon. Canadian Journal of Physics, 46, 1181-1189. http://dx.doi.org/10.1139/p68-151
- Freund, F., Wengeler, H. and Martens, R. (1982) A Hydrogen-Deuterium Fractionation Mechanism in Magnesium Oxide. Geochimica et Cosmochimica Acta, 46, 1821-1829. http://dx.doi.org/10.1016/0016-7037(82)90121-1
- Martens, R., Gentsch, H. and Freund, F. (1976) Hydrogen Release during the Thermal Decomposition of Magnesium Hydroxide to Magnesium Oxide. Journal of Catalysis, 44, 366-372. http://dx.doi.org/10.1016/0021-9517(76)90413-9
- Kathrein, H. and Freund, F. (1983) Electrical Conductivity of Magnesium Oxide Single Crystal below 1200 K. Journal of Physics and Chemistry of Solids, 44, 177-186. http://dx.doi.org/10.1016/0022-3697(83)90052-5
- Wengeler, H. and Freund, F. (1980) Atomic Carbon in Magnesium Oxide, Part III: Anomalous Thermal Expansion Behavior. Materials Research Bulletin, 15, 1241-1245. http://dx.doi.org/10.1016/0025-5408(80)90026-4
- Batllo, F., LeRoy, R.C., Parvin, K., Freund, F. and Freund, M.M. (1991) Positive Hole Centers in Magnesium Oxide― Correlation between Magnetic Susceptibility, Dielectric Anomalies and Electric Conductivity. Journal of Applied Physics, 69, 6031-6033. http://dx.doi.org/10.1063/1.347807
- Kathrein, H., Freund, F. and Nagy, J. (1984) O−-Ions and Their Relation to Traces of H2O and CO2 in Magnesium Oxide: An EPR Study. Journal of Physics and Chemistry of Solids, 45, 1155-1163. http://dx.doi.org/10.1016/0022-3697(84)90011-8
- Freund, F., Freund, M.M. and Batllo, F. (1993) Critical Review of Electrical Conductivity Measurements and Charge Distribution Analysis of MgO. Journal of Geophysical Research, 98, 22209-22229. http://dx.doi.org/10.1029/93JB01327
- Freund, F., Whang, E.-J., Batllo, F., Desgranges, L., Desgranges, C. and Freund, M.M. (1994) Positive Hole-Type Charge Carriers in Oxide Materials. In: Levinson, L.M., Ed., Grain Boundaries and Interfacial Phenomena in Electronic Ceramics, American Ceramic Society, Cincinnati, 263-278.
- Butler, C.T., Sturm, B.J. and Quincy, R.B. (1971) Arc Fusion Growth and Characterization of High-Purity Magnesium Oxide Single Crystals. Journal of Crystal Growth, 8, 197-204. http://dx.doi.org/10.1016/0022-0248(71)90142-4
- Freund, F., Dickinson, J.T. and Cash, M. (2002) Hydrogen in Rocks: An Energy Source for Deep Microbial Communities. Astrobiology, 2, 83-92. http://dx.doi.org/10.1089/153110702753621367
- Freund, F. (2011) Pre-Earthquake Signals: Underlying Physical Processes. Journal of Asian Earth Sciences, 41, 383- 400. http://dx.doi.org/10.1016/j.jseaes.2010.03.009
- Freund, F.T. (2013) Earthquake Forewarning―A Multidisciplinary Challenge from the Ground up to Space. Acta Geophysica, 61, 775-807. http://dx.doi.org/10.2478/s11600-013-0130-4
- Freund, F.T. (2003) On the Electrical Conductivity Structure of the Stable Continental Crust. Journal of Geodynamics, 35, 353-388. http://dx.doi.org/10.1016/S0264-3707(02)00154-0
- Aines, R.D. and Rossman, G.R. (1984) Water in Minerals―A Peak in the Infrared? Journal of Geophysical Research, 89, 4059-4071. http://dx.doi.org/10.1029/JB089iB06p04059
- Rossman, G.R. (1990) Hydrogen in “Anhydrous” Minerals. Nuclear Instruments and Methods in Physics Research, B45, 41-44. http://dx.doi.org/10.1016/0168-583X(90)90780-X
- Koga, K., Hari, E., Hirschmann, M. and Bell, D. (2003) Hydrogen Concentration Analyses Using SIMS and FTIR: Comparison and Calibration for Nominally Anhydrous Minerals. Geochemistry, Geophysics, Geosystems, 4, 1-20. http://dx.doi.org/10.1029/2002GC000378
- Stephant, A., Remusat, L., Thomena, A. and Robert, F. (2014) Reduction of OH Contamination in Quantification of Water Contents Using NanoSIMS Imaging. Chemical Geology, 380, 20-26. http://dx.doi.org/10.1016/j.chemgeo.2014.04.018
NOTES

*Unfortunately, this author was deceased.

1V stands for vacancy; subscripts identify the site (except for oxygen sites, where subscripts are omitted); superscript prime, dot, and x designate single negative, positive and neutral charges, respectively, double prime and double dot designate double negative and positive charges; subscript i means interstitial; square brackets outline the essential parts of any given point defect [Kröger, F. A. (1964), The Chemistry of Imperfect Crystals, North-Holland, Amsterdam.] [15] .