American Journal of Analytical Chemistry
Vol. 3 No. 8 (2012) , Article ID: 21915 , 4 pages DOI:10.4236/ajac.2012.38075
Extraction Method and Determination of Sudan I Present in Sunset Yellow FCF by Isocratic High-Performance Liquid Chromatography
National Institute of Health and Sciences, Tokyo, Japan
Email: *c-sasaki@nihs.go.jp
Received June 15, 2012; revised July 19, 2012; accepted July 31, 2012
Keywords: Sudan I; Sunset Yellow FCF; HPLC
ABSTRACT
A method to extract and analyze Sudan I present in Sunset Yellow FCF (SYF) products was developed and validated. The method included the simple extraction of Sudan I from the SYF product using water, acetonitrile, and ethyl acetate and high-performance liquid chromatography (HPLC) analysis with isocratic elution using acetonitrile:water (7:3) with a photodiode array detector at 485 nm. This method was found to remove most of the excess SYF colorant and other impurities before injection to the HPLC instrument, making it easy to maintain precision control in routine laboratory tests for Sudan I in the SYF colorant. The detection limit of Sudan I in SYF products was 0.2 μg/g. A survey conducted to determine Sudan I in 13 commercial SYF samples from Japanese manufacturers from 1970 to 2010 showed that the levels of Sudan I ranged from 0.3 to 1.9 μg/g in products manufactured from 1970 to 1996 and were below the limit of detection in products manufactured after 2005.
1. Introduction
Sunset Yellow FCF (SYF) is a synthetic yellow azo dye that is allowed worldwide as a food coloring agent and is known by designations such as FD&C Yellow No. 6, Food Yellow No. 5, E110, and Color Index No.15985. SYF is a disulfo monoazo dye prepared by the coupling of diazotized 4-aminobenzenesulfonic acid with the sodium salt of 6-hydroxy-2-naphthalenesulfonic acid. During the manufacture of SYF, 1-(phenylazo)-2-naphthalenol (Sudan I) may be produced by the diazotization and coupling of aniline, an impurity in technical and refined sulfanic acid, with 2-naphthol [1]. Sudan I is an unauthorized colorant and an undesirable substance in food because of its reported possible carcinogenicity and genotoxicity in Japan, the United States, and the European Union (EU) [2]. In 2003, Sudan I was detected in chili products in France. After this report, in the EU and China, Sudan I and other unauthorized Sudan dyes (Sudan II, III, and IV) have been detected in samples of hot chili and other related foods. Consequently, various analytical methods have been established to determine and identify unauthorized Sudan dyes in foods [3-6].
In 2005, the EU adopted a limit of <0.5 mg/g for Sudan I in SYF [7]. The Joint FAO/WHO Expert Committee on Food Additives (JECFA) conducted a survey from 17 international manufacturers of the amount of Sudan I in 28 SYF samples. Seventy-five percent of SYF samples contained Sudan I at concentrations ≤ 1 mg/g. Based on these data, JECFA established a maximum limit of 1 mg/g for Sudan I in SYF [8,9]. The US Food and Drug Administration (FDA) developed an analytical method for the detection of Sudan I in SYF, which involves the direct injection of SYF by reversed-phase liquid chromatography [1]. This method was also adopted by JECFA in an SYF monograph for the determination of Sudan I [8]. Although the method is simple and rapid, high concentrations of an SYF solution have to be injected to determine low levels of Sudan I in the SYF. Since injecting high concentrations of the SYF solution can contaminate the line, sample loop, and column in the high-performance liquid chromatography (HPLC) instrument, maintaining precision control in routine SYF laboratory tests is difficult. To reduce the risk of contamination to the analytical equipment from SYF, a method for extracting Sudan I from SYF was developed for cosmetic use. In the method, Sudan I is extracted using chloroform as a solvent and analyzed by reversed-phase HPLC [10]. However, owing to the toxicity of chloroform, it needs to be replaced with a less toxic solvent to reduce health hazards and environmental contamination. In this study, we developed a simple analytical method for detecting Sudan I in SYF using a less toxic organic solvent. The method was validated by recovery testing using two different concentrations of Sudan I and was applied to the analysis of commercial SYF samples in Japan.
2. Experimental
2.1. Reagents and Equipment
Sudan I (Color Index No. 12055) from Kanto Chemical Co, Ltd. (special grade, Tokyo, Japan) and Wako Pure Chemical (standard for HPLC, Osaka, Japan) was used for an investigation of purity. The Sudan I from Kanto Chemical was recrystallized from ethanol (5 g/150 mL) to obtain pure Sudan I that was used as a standard for the analysis. Of 13 commercial SYF samples that were obtained from five Japanese companies for surveying the Sudan I content, one of them (No.10) did not contain Sudan I and was used for the recovery test of Sudan I.
The SYF sample for the recovery test was obtained from Daiwa Kasei Corporation (Saitama, Japan). The ammonium acetate and ethyl acetate were special grade reagents (Wako Pure Chemical Industries, Ltd.), the acetonitrile was liquid-chromatography (LC) grade (Merck KGaA, Darmstadt, Germany), and the ultrapure water used for LC was purified with the MilliQ® Gradient system equipped with a Millipak® 0.22 mm filter (Milliopore, Billerica, MA, USA). Parts of the solution for LC were filtered through a polytetrafluoroethylene (PTFE) membrane (Millex®-LH, 13 mm, 0.45 mm; Millipore, Billerica, MA, USA) using a 2.5 mL polypropylene/polyethylene syringe (Terumo syringe; Terumo Corp. Tokyo, Japan). Certified reference material 1,4-bis (trimethylsilyl) benzene-d4 (1,4-BTMSB-d4) (Code No. 024-17031, Lot. DCP5375, purity 99.8% ± 0.3%) was obtained from Wako Pure Chemical Industries, Ltd. Chloroform-d was purchased from Isotec Ltd. (Miamisburg, OH, USA).
2.2. qHNMR Measurement
First, 10 mg of Sudan I and 4 mg of 1,4-BTMSB-d4 were accurately weighed and then dissolved in 800 μL of chloroform-d. Next, approximately 600 μL of the solution was introduced into a nuclear magnetic resonance (NMR) tube with a 5 mm outer diameter (Kanto Chemical Co., Inc.) and subjected to a quantitative proton nuclear magnetic resonance (qHNMR) measurement using a JNM-ECA spectrometer (600 MHz; JEOL, Tokyo). The purity of the Sudan I was calculated by the equation
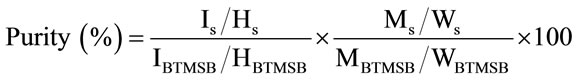 (1)
(1)
where IS and IBTMSB are the integrated signal intensity values of Sudan I and 1,4-BTMSB-d4, respectively; HS and HBTMSB are the number of proton signals from Sudan I and 1,4-BTMSB-d4, respectively; MS and MBTMSB are the molecular weights of Sudan I and 1,4-BTMSB-d4, respectively; and WS and WBTMSB are the weights of Sudan I and 1,4-BTMSB-d4, respectively. The qHNMR measurement was conducted with the following optimized parameters: irradiation frequency, 600 MHz; probe temperature, 25˚C; spinning, off; number of scans, eight; spectral width, 20 ppm; auto filter, on (eight times); acquisition time, 4 s; relaxation delay, 60 s; pulse angle, 90˚; pulse width, 12.2 μs; free induction decay (FID) data points, 49152; and 13C decoupling, multipulse decoupling with phase and frequency switching (MPF-8). The data were processed with qNMR analysis software, Alice 2 for qNMR “PURITY” (JEOL). The signal integral value calculated using the software was used for quantitative analysis. The chemical shift of all data was referenced to the 1,4-BTMSB-d4 resonance at δH 0.23.
2.3. Sample Solutions and Calibration Curve
After 500 mg of the SYF product was dissolved in 10 mL of water by ultrasonic wave in a 50 mL glass centrifuge tube, 5 mL of acetonitrile was added and mixed well, followed by the addition and mixing of 30 mL of ethyl acetate. The centrifuge tube was centrifuged for 1 min at 3000 rpm, and the ethyl acetate layer was collected in a round flask; another 30 mL of ethyl acetate was added to the residue, and the procedure was repeated. The collected ethyl acetate layer was evaporated at a reduced pressure at 40˚C. The residue was dissolved in a 2 mL volumetric flask by sonication and was diluted with acetonitrile: water (7:3) up to the 2 mL mark. A portion of the sample solution was filtered into a vial using a PTFE membrane syringe filter.
2.4. Standard Stock Solution
To obtain a standard stock solution containing 1 mg/mL of Sudan I, recrystallized Sudan I was dried in a desiccator for 24 h under reduced pressure, and 50 mg of Sudan I was weighed and dissolved completely with 45 mL of acetonitrile by ultrasonication in a 50 mL volumetric flask.
2.5. Standard Calibration Curve
The Sudan I standard stock solution was diluted with acetonitrile:water (7:3), and 0.05 - 50 mg/mL standard solutions of Sudan I were prepared. These standard solutions were used to check the linearity of the calibration curve. One microgram of Sudan I in 1 mL of a standard solution was pipetted into 1, 2, 4, and 10 mL of appropriate standard solutions in a series of volumetric flasks and diluted to the 20 mL mark with acetonitrile:water (7:3). The standard solutions contained 0.05, 0.1, 0.2, and 0.5 mg of Sudan I/mL, and the calibration curve was used for the recovery test and the Sudan I content survey of commercial SYF products.
2.6. Recovery Test and Validation
To evaluate the accuracy of the developed method, recovery tests were performed. A 0.5 mL quantity of the 0.5 and 0.3 mg/mL Sudan I standard solutions was added to 500 mg of a commercial SYF product in the absence of Sudan I. The sample was left at room temperature overnight and then was treated as described above.
Calibration curves were prepared with the Sudan I standard solutions at concentration levels of 0.05 - 50 mg/mL to check the linearity of the calibration curve. Intra-day and inter-day precisions were assessed by analyzing five replicates of a sample during a day and five replicates on three different days, respectively. The limit of quantification of Sudan I was estimated to the limit of confirmatory detection allowed by the PDA detector.
2.7. Sudan I Content Survey of Commercial SYF Products
The amounts of Sudan I in 13 commercial SYF products produced as food additives in Japan from 1970 to 2010 were analyzed by the method described above in Sample solution.
2.8. HPLC
The LC system consisted of a Hewlett Packard 1100 series, G1315A photodiode array detector (PDA; monitored at 485 nm for Sudan I), L-column octadecylsilane (ODS; 150 × 4.6 mm id, particle size 5 mm, pore size 12 nm; Chemicals Evaluation and Research Institute, Tokyo, Japan), and a column heater set at 40˚C. The eluent was acetonitrile: water (7:3) in the isocratic mode. The injection volume was 20 mL, and the flow rate was 1.0 mL/ min. The apparatus was controlled and data were collected and analyzed by use of Agilent Chemstation software.
3. Results and Discussion
3.1. Purity Determination by qHNMR Measurement
To investigate the purity of Sudan I, we analyzed the commercial Sudan I standard by HPLC and the special grade Sudan I by HPLC at 230 nm to see the colorless impurities, previously reported. As shown in Figure 1(a) and (b), some unknown interference peaks between 2 and 5 min were detected in addition to a main peak at 12.5 min, which corresponds to Sudan I on the HPLC chromatogram. To remove impurities from the special grade Sudan I, recrystallization was performed in ethanol, and
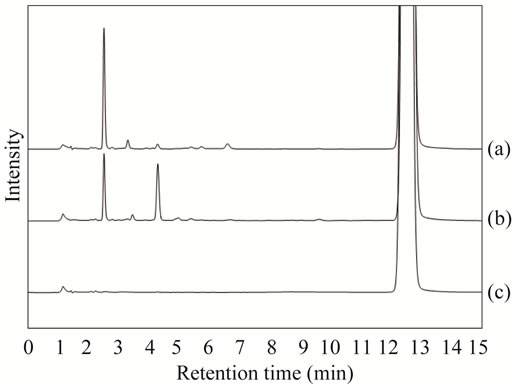
Figure 1. HPLC chromatogram of Sudan I and recrystallized Sudan I at 230 nm. (a) Commercial Sudan I standard for HPLC (Wako Pure Chemical Co.); (b) Special grade Sudan I (Kanto Chemical Co.); (c) Recrystallized Sudan I from Kanto Chemical Co.
recrystallized Sudan I was obtained. As shown in Figure 1(c), a few impurity peaks were detected on the HPLC chromatogram of Sudan I after the recrystallization.
To determine the absolute purity of recrystallized Sudan I, Sudan I was analyzed by qHNMR. As shown in Figure 2, the 1H NMR spectrum exhibited signals characteristic of the aromatic protons of Sudan I in δH 6.80 - 8.60. Among them, the doublet signals at δH 6.87 and 8.57 were assigned to H-3 and H-8, respectively. In qHNMR, it is necessary that the signal for quantification should separate the other intramolecular signals in order to accurately determine the purity of the analyte. Both signals were applied to qHNMR measurement because these were well resolved from the other signals. The absolute purity of Sudan I was calibrated from the ratio of the integrated signal intensity values (signal area values) of each signal to that of 1,4-BTMSB-d4 at δH 0.23. The mean purity value was calculated to be 98.9% ± 0.1% (mean ± SD). These results indicate that the purity of the recrystallized Sudan I is higher than those of the commercial Sudan I standard and special grade Sudan I.
3.2. Extraction of Sudan I from SYF
The self-regulation method of the Japan Cosmetic Industry Association, which uses chloroform to extract Sudan I from SYF followed by reversed-phase HPLC analysis [10], was used. In an effort to replace chloroform with non-toxic solvents, we examined the pretreatment procedure of Sudan I from commercial SYF using methanol and ethyl acetate. The recovery tests were conducted using commercial SYF spiked with the recrystallized Sudan I. Since SYF is soluble in water and Sudan I is only negligibly soluble in water, 0.5 g of commercial SYF spiked with the recrystallized Sudan I (0.5 mg/g of SYF) was dissolved in 10 mL of water, and 10 mL of methanol and 20 mL of ethyl acetate were added. The
 (a)
(a)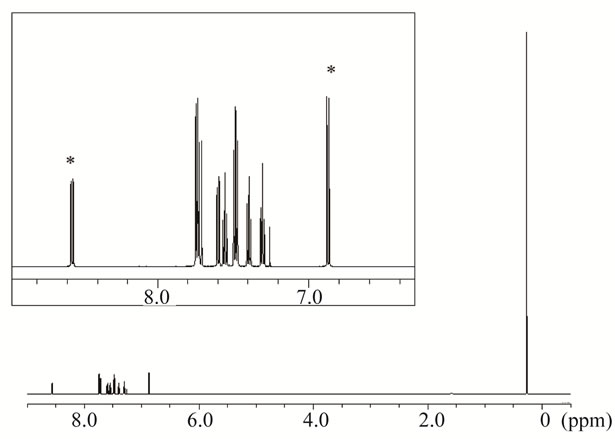 (b)
(b)
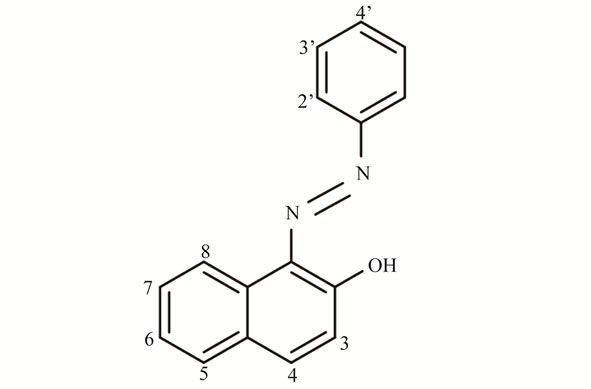
Figure 2. (a) Chemical structure of Sudan I and (b) 1H NMR spectrum of recrystallized Sudan I in chloroform-d containing 1,4-BTMSB-d4. The signals of Sudan I shown on the top are highlighted. Signals marked with asterisks were used for quantification, and the purities were calculated.
resulting solution was mixed using a shaker for 1 min, after which 10 mL of water was added. After centrifuging the solution for 5 min at 3000 rpm, it separated into two layers, and we visually considered the upper layer to contain Sudan I and the lower layer to contain SYF. After taking the upper layer and washing it twice with water, the solution was concentrated and desiccated under a reduced pressure at 40˚C, dissolved and made up to a 2 mL volume with acetonitrile:water (7:3), and analyzed with HPLC. The recovery of Sudan I was 89%. To further improve the recovery of Sudan I, 5 mL of acetonitrile was added to the solution instead of 5 mL of methanol, and when extracted with 20 mL of ethyl acetate two times in the same manner, the recovery of Sudan I was 94%. These results suggest that the latter preparation is an improvement over the former preparation in terms of recovery and procedure.
3.3. HPLC Conditions
To determine Sudan I using HPLC, we optimized the HPLC conditions. Tsuji et al. [11] reported HPLC conditions to determine raw materials, intermediates, and subsidiary colors in SYF using a gradient system of 20 mmol/L ammonium acetate and acetonitrile:water (7:3).
We utilized the HPLC conditions reported by Tsuji et al., since this gradient system using these solvents will be adopted for the examination of raw materials, intermediates, and subsidiary colors in food colors other than SYF in Japan’s Specifications and Standards of Food Additives. Sudan I could not be eluted until 50 min had elapsed because Sudan I appeared to be retained on the reversed-phase column. Upon examination, we found that the use of the acetonitrile:water (7:3) mobile phase led to the efficient separation and elution of Sudan I at 12.5 min under isocratic conditions (Figure 1). For the detection of Sudan I, we compared the sensitivity and separation pattern at 230 nm, which is thought to be suitable for the detection of aromatic compounds, and at 485 nm, which is used in the methods reported by Petigara et al. [1]. Consequently, the separation pattern of the 485 nm detection had fewer impurity peaks than that at 230 nm, although the sensitivity of the 230 nm detection was higher than that at 485 nm detection (Figure 3). Therefore, we chose 485 nm for the detection of Sudan I.
3.4. Method Validation and Recovery Test of Sudan I from SYF
To assess the linearity, the calibration curve for Sudan I
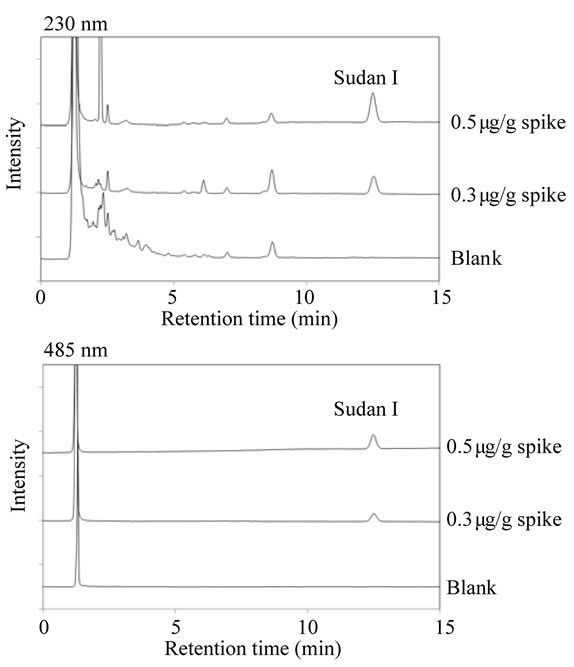
Figure 3. HPLC chromatogram of sample solutions from Sunset Yellow FCF product (blank) and Sunset Yellow FCF product spiked with Sudan I (0.3 and 0.5 μg/g) at 230 nm and 485 nm.
was constructed using concentrations from 0.05 to 50 mg/mL. As shown in Figure 4, good linearity was achieved over Sudan I concentrations of 0.05 - 50 mg/mL. The correlation coefficient for Sudan I was R2 = 0.9999.
To assess the accuracy of the developed method, we conducted recovery tests using the SYF product spiked with Sudan I at two levels—0.5 and 0.3 mg—in 1 g of SYF. As shown in Table 1, the recovery rates were within the 95.5% - 97.9% range. To assess the precision of the method, intra-day and inter-day precisions were within the 0.4% - 1.8% and 1.1% - 3.8% ranges, respectively, all expressed as relative standard deviations (RSDs) (Table 1). The limit of detection of Sudan I was 0.2 mg/g, calculated from the signal-to-noise ratio (S/N > 10) of the peak intensity, and the limit of identification of Sudan I was estimated to be 0.4 mg/g, which was considered to be the limit of confirmatory detection using the PDA detector.
These data clearly demonstrate acceptable linearity, recovery rates, and RSDs, suggesting that the developed method is reliable for the accurate quantitative determination of Sudan I in SYF products.
3.5. Quantitative Survey of Sudan I in Manufactured SYF
A survey determination of Sudan I in commercial SYF products manufactured in Japan from 1970 to 2010 was conducted to confirm the application of the developed method. The results are summarized in Table 2. The levels of Sudan I in SYF products ranged from 0.3 to 1.9 mg/g in SYF products manufactured from 1970 to 1996. Using the PDA detector, specific spectra of Sudan I could be confirmed from the peaks of Sudan I in all SYF products except for Sample No. 3 SYF products. Sudan I was below the limit of detection in commercial SYF products manufactured after 2005, which implies that the SYF products manufactured in Japan from 2005 to 2010 contained marginal amounts of Sudan I.
The low level of detection of Sudan I in commercial SYF products manufactured in recent years appears to be attributable to self-regulation by each manufacturer, considering the low limit on Sudan I (<0.5 mg/g) that has been specified by the EU.
4. Conclusion
A rapid, useful analytical method for monitoring Sudan I in SYF products has been developed. The method includes a simple procedure for extracting Sudan I from SYF products and analyzing it by HPLC with isocratic elution using a PDA detector. The method can prevent the contamination of the HPLC instrument and column with excess SYF colorant and other impurities. This method permits the determination of 0.2 mg of Sudan I in
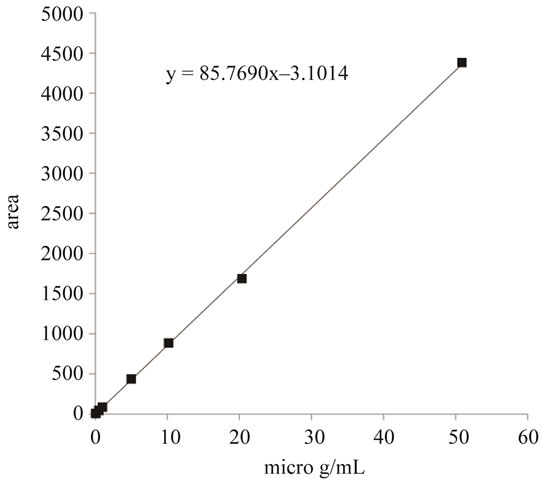
Figure 4. Calibration curve of Sudan I.
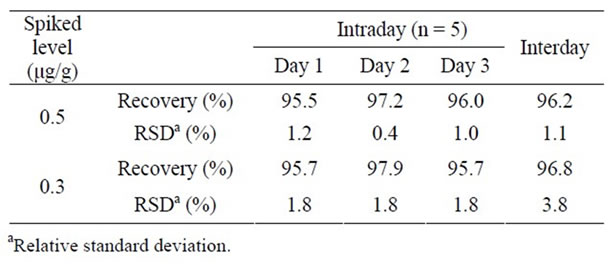
Table 1. Intra-day and inter-day precisions in the recovery test of Sudan I in Sunset Yellow FCF.

Table 2. Results of the survey determination of Sudan I in commercial Sunset Yellow FCF.
1 g of SYF; thus, it can readily detect the maximum limit of Sudan I in SYF as defined by the EU (0.5 mg/g).
5. Acknowledgements
We are grateful to San-Ei Gen F.F.I., Inc. for providing commercial SYF products.
REFERENCES
- B. R. Petigara and A. L. Scher, “Direct Method for Determination of Sudan I in FD&C Yellow No.6 and D&C Orange No.4 by Reversed-Phase Liquid Chromatography,” Journal of AOAC International, Vol. 90, No. 5, 2007, pp. 1373-1378.
- International Agency for Research on Cancer, “IARC Monographs on the Evaluation of Carcinogenic Risks to Humans,” Vol. 8, 1975, pp. 225-231. http://monographs.iarc.fr/ENG/Monographs/vol8/volume8.pdf
- Y. Uematsu, M. Ogimoto, J. Kabashima, K. Suzuki and K. Ito, “Fast Cleanup Method for the Analysis of Sudan I-IV and Para Red in Various Food and Paprika Color (Oleoresin) by High-Performance Liquid Chromatography/ Diode Array Detection: Focus on Removal of Fat and Oil as Fatty Acid Methyl Esters Prepared by Transesterification of Acylglycerols,” Journal of AOAC International, Vol. 90, No. 2, 2007, pp. 437-445.
- C. Ju, Y. Tang, H. Fan and J. Chen, “Enzyme-Linked Immunosorbent Assay (ELISA) Using a Specific Monoclonal Antibody as a New Tool to Detect Sudan Dye and Para Red,” Analytica Chimica Acta, Vol. 621, No. 2, 2008, pp. 200-206. doi:10.1016/j.aca.2008.05.055
- Y. Fan, M. Chen, C. Shentu, F. El-Sepai, K. Wang, Y. Zhu and M. Ye, “Ionic Liquid Extraction of Para Red and Sudan dyes from Chili Powder, Chili Oil and Food Additive Combined with High Performance Liquid Chromatography,” Analytica Chimica Acta, Vol. 650, No. 1, 2009, pp. 65-69. doi:10.1016/j.aca.2009.03.025
- C. Zhao, T. Zhao, X. Liu and H. Zhang, “A Novel Molecularly Imprinted Polymer for Simultaneous Extraction and Determination of Sudan Dyes by On-Line Solid Phase Extraction and High Performance Liquid Chromatography,” Journal of Chromatography A, Vol. 1217, No. 45, 2010, pp. 6995-7002. doi:10.1016/j.chroma.2010.09.005
- EU Commission Directive 2006/33/EC Official Journal of European Union, L82/10-13, 2006. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2006:082:0010:0013:en:pdf
- Joint FAO/WHO Expert Committee on Food Additives (JECFA), “Evaluation of Certain Food Additives, SixtyNine Report of the Joint FAO/WHO Expert Committee on Food Additives,” 2008, pp. 68-69. http://whqlibdoc.who.int/trs/WHO_TRS_952_eng.pdf
- Online Edition, “Combined Compendium of Food Additive Specifications”. http://www.fao.org/ag/agn/jecfa-additives/specs/monograph11/additive-450-m11.pdf
- Japan Cosmetic Industry Association, “Houtei Shikiso Handbook (Certified Colorants Handbook),” Tokyo, 2004, pp. 489-495.
- S. Tsuji, I. Matsumura, Y. Nakamura and Y. Tonogai, “Studies on Separation and Determination of Subsidiary Colors, Raw Materials and Intermediates in Food Yellow No.5 (Sunset Yellow FCF) by HPLC,” Shokuhin Eiseigaku Zasshi (Food Hygiene and Safety Science), Vol. 41, No. 6, 2000, pp. 357-363. doi:10.3358/shokueishi.41.357
NOTES
*Corresponding author.