International Journal of Clinical Medicine
Vol.3 No.3(2012), Article ID:19405,5 pages DOI:10.4236/ijcm.2012.33042
Different Clinical Phenotypes in Adams-Oliver Syndrome Conservative Approach to Aplasia Cutis: A Report of Two Cases
![]()
1Neonatology Unit, Mother and Child Department, University Hospital of Pisa, Pisa, Italy; 2Dermatology Unit, University Hospital of Pisa, Pisa, Italy; 3Scuola Superiore Sant’Anna, Pisa, Italy; 4Cytogenetics and Molecular Genetics Unit, University Hospital of Pisa, Pisa, Italy; 5Section of Neonatal Endocrinology and Dysmorphology, University Hospital of Pisa, Pisa, Italy.
Email: #pghirri@med.unipi.it
Received January 9th, 2012; revised February 24th, 2012; accepted March 18th, 2012
Keywords: Adams-Oliver Syndrome; Aplasia Cutis Congenita; Scalp and Skull Defects; Dermal Regeneration Template
ABSTRACT
Adams-Oliver Syndrome (AOS) is a rare genetic disease characterized by combination of aplasia cutis congenita (ACC) and terminal transverse limb defects (TTLD), often accompanied by defects in scalp and skull ossification. Different clinical phenotypes may be related to variable severity both of aplasia cutis and TTLD, and of minor clinical features as cutis marmorata telangiectatica congenita (CMTC), congenital cardiac defect and vascular anomalies. The treatment is multidisciplinary: dermatologic, orthopedic and surgical consult should be required. It still remains unclear how to treat patients with a large skin defect that can’t be closed primarly and may require both surgical and conservative management. We report two cases of AOS with typical limb defects and an area of aplasia cutis over vertex of the scalp managed conservatively with two different dermatologic devices.
1. Introduction
Adams-Oliver Syndrome (OMIM 100300) is a rare congenital disorder which includes congenital scalp and skull defects and terminal transverse limb malformations, but it may present a wide spectrum of physical anomalies [1]. Diagnosis is based on clinical criteria. Major features are aplasia cutis congenita (frequently localized over vertex of the scalp with more or less important involvement of the underlying bone), terminal transverse limb defects (commonly bilateral but often asymmetric and usually consisting in terminal reduction defects of fingers and toes) and family history of AOS. Minor features are cutis marmorata telangiectatica congenita (CMTC), congenital cardiac defect and vascular anomalies; this may be associated with a number of additional clinical defects including neurological abnormalities, growth retardation and skin tags. The presence of two major features is considered sufficient for diagnosis. The combination of one major and one minor feature should place AOS high in the differential diagnosis of such individuals [2].
The management consists in a difficult multi-disciplinary approach aimed primarily at the treatment of the aplasia cutis. Large skin defects that cannot be closed primarily present a management dilemma, and may require skin grafting or flaps, or a combination of both operative and conservative modalities [3].
The mortality rate reported in the literature, mainly attributed to infection, meningitis or bleeding from the sagittal sinus, is about 20%.
These two cases report are presented to describe two different methods of conservative managing of both small and large cutis aplasia.
2. Case Report 1
A full term male presented with aplasia cutis over vertex of the scalp limited to the skin (1.5 × 1 cm) (Figure 1), hypoplasia of bilateral fingers and toes, and dystrophy of all nails. He also had cleft lip and palate, hypertelorism, wide anterior fontanel and a cutaneous appendage in the back. Pregnancy was normal and the APGAR score at 5 minutes was 8. The child’s birth weight was 4700 gr with cranic circumference of 37.5 cm (both > 97˚ centile).
Family history was not significant and the parents were non-consanguineous. We performed genetic studies: the karyotype demonstrated a normal male (46, XY) and the patient and parents’ CGH-array revealed no microdeletion nor microduplication. The combination of ACC and TTLD allowed the diagnosis of AOS. The MRI revealed agenesis of the corpus callosum, absence of the pituitary and dysmorphism of the lateral ventricles and frontomesial cerebral convolutions. Assessment of serum levels of pituitary hormones was within normal range. To promote epithelialisation of the aplasia we used a dressing hydrocolloid (DuoDERM®). With this device, changed daily, we obtained a complete resolution of the aplasia in about 1 month. After the discharge, we programmed cleft lip and palate’s surgical correction during second month of life. The patient is currently being followed-up by a multidisciplinary team.
3. Case Report 2
C.D.R., female, was born at 33 weeks of gestational age for intrauterine growth retardation. At clinical evaluation the following were observed: complete aplasia cutis over vertex of the scalp (4.5 cm of maximum diameter) (Figure 2), which involved epidermis, dermis and subcutaneous tissues of the scalp with significant bone defect and exposition of dura, bilateral hypoplasia of the toes (Figure 3), syndactyly and brachydactyly of the right hand and single umbilical artery. No history of AOS, mental retardation, infections or maternal drugs intake during pregnancy was reported and the parents were nonconsanguineous. The transfontanellar ultrasonography revealed asimmetry of the ventricles and hyperechoic areas in the basal ganglia. Patient’s karyotype and patient and parents’ CGH-array resulted normal. Clinical features resulted in a diagnosis of AOS. The aplasia cutis was treated conservatively with a dermal regeneration template (INTEGRA®), because of the complete involvement of epidermis, derma and skull (Figure 4). In few days after the application of Integra, granulation tissue at the periphery of the lesion started to form, and the defect progressively and slowly reduced in following days as granulation tissue continued to migrate to the center of the defect. The clinical course was not complicated and the patient was discharged at 41 week of gestational age. She was inserted in a close follow-up that included the performance of magnetic resonance imaging (MRI) of the brain and a surgical consultation to treat the terminal transverse limb defects. The dermatologic control at 3 months of corrected age showed a reduction of the maximum diameter of the aplasia cutis (2.5 cm) (Figure 5). At the last dermatologic control (7 months of corrected age) was observed an almost complete resolution of the aplasia cutis (0.5 cm) (Figure 6). This experience in management of huge scalp and bone defect with a dermal regeneration template (INTEGRA®) showed good scalp repair and no complications attributed to this approach.
4. Discussion
AOS is a rare genetic condition characterized by aplasia
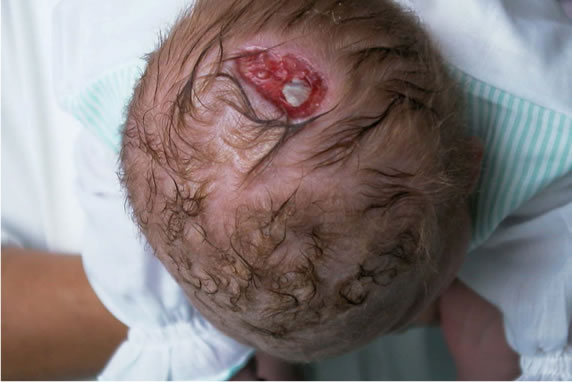
Figure 1. Patient n 1: aplasia cutis over vertex of the scalp limited to the skin.
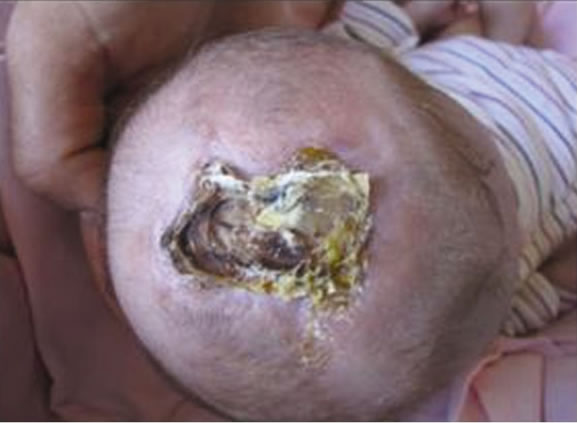
Figure 2. Patient n 2: aplasia cutis over vertex of the scalp at the birth.

Figure 3. Patient n 2: hypoplasia of the toes.
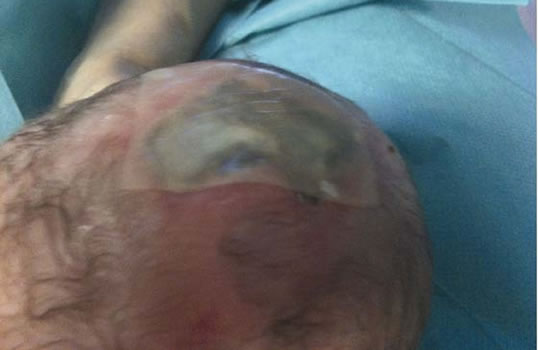
Figure 4. Patient n 2: application of a dermal regeneration template.
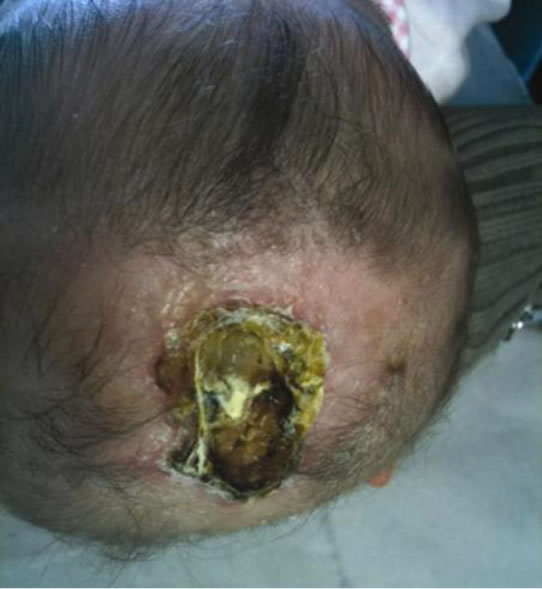
Figure 5. Patient n 2: aplasia cutis over vertex of the scalp at 3 months of corrected age.
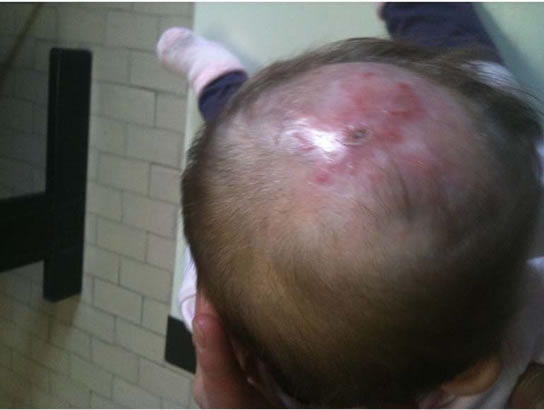
Figure 6. Patient n 2: aplasia cutis over vertex of the scalp at 7 months of corrected age.
cutis congenita with a wide range of scalp defects and transverse limb malformations.
The disease has been described for the first time by F. Adams and C. P. Oliver in 1945, in 8 members of the same family highlighting an autosomal dominant mode of inheritance with variable expressivity, although recessive patterns as well as sporadic forms often occour. Although the molecular basis of AOS remains unknown, an alteration of embryonic vascularization has been hypothesized as a possible pathophysiological mechanism. CGH-array of our patients and their parents didn‘t show any abnormalities. Neither of our patients had family history of AOS and results of their chromosomal study were unremarkable, then we can suppose a sporadic form.
Diagnosis is based on clinical criteria: 2 major features are sufficient. Our first patient had typical limb defects and an area of aplasia cutis over vertex of the scalp, limited to skin. As additional features he exhibited central nervous system defects (agenesis of the corpus callosum and absence of the pituitary) and a severe cleft lip and palate. Near total agenesis or hypoplasia of corpus callosum in AOS have been described, but our patient is the first one with a complete agenesia [2]. Gingival cleft was reported in different members of the same family [4] and right cleft lip and palate in only one case [5]. In the same way the second patient presented typical limb defects, but an area of aplasia cutis larger than the first one, which involved even the underlying structures with exposition of dura, and less important neurologic anomalies.
The treatment of cutis aplasia remains controversial because AOS is a rare disease and there is a lack of experience related to outcomes of different therapeutic methods.
The goal of treatment is to achieve complete closure of the defect, avoiding major complications such as meningitis, hemorrhages, and trauma to the brain [6-7]. Reported treatments have included surgical closure, conservative management, or a combination of the two [6-8]. Determination of appropriate clinical management during the early stages of life improves survival and is based on the size of the defect, presence of an underlying skull defect, the child’s general condition, and his/her associated life expectancy [7-9].
Minor lesions can be controlled with conservative therapy, like dressing regimens (daily changing of moist sterile gauze and topical and systemic antibiotic therapy) [7,9,10], hydrocolloids or devices that stimulate tissue repair processes. This use of nonadherent, atraumatic, and relatively cheap dressings has been reportedly successful for the initial conservative treatment of large defects [11] because it may avoid the potential failure of skin flap or heterologous graft coverage [12] and it may maintains dural induction of osseous regeneration [13]. Some authors recommend a combined regimen of conservative and operative treatment in patients with large defects to decrease the chance of unsuccessful surgical management [7,8,11,14].
Large lesions with an underlying skull defect and uncovered brain require early surgery to prevent massive hemorrhage, infections and sinus thrombosis. There is still no agreement about the size of scalp lesion appropriate for surgery: some suggest surgery for lesions >1 cm, others use conservative approach with excellent outcome for defects >2 cm and others advise skin grafts for sizes >4 - 5 cm. Surgical regimens include primary closure, splitor full-thickness skin graft, scalp rotation flaps, pericranial flaps [9], split rib grafts with a latissimus dorsi muscle flap [7] and a 1stage operation of free flap performed using microsurgical vascular anastomosis [7-13]. Surgery-related risks are hemorrhage, infections, partial or total graft failure because of the size of the defect or the associated abnormality of the adjacent skin [9] and inhibition of the osteogenic potential of the dura mater which results in poor regrowth of bone [13,14].
To avoid plastic surgery, in selected cases it’s possible to try a dermal regeneration template (INTEGRA®). Integra is a two-layer skin regeneration system: the outer is made of silicon film, that protects from infections and controls both heat and moisture loss; the inner is a complex matrix of cross-linked fibers that acts as a scaffold to regenerate dermal skin cells.
This device can be used when the cutis aplasia involves a large portion of the dermis and prevents selfhealing of the skin. It can also be a good choice in newborns where it’s difficult to obtain a sufficient amount of skin for autologus grafting.
There is only one case of AOS in which the area of aplasia cutis was treated successfully with a dermal regeneration template (INTEGRA®). In this patient the skull defect was also associated with ruptured dura mater, closed primarily with braided nylon [3].
In our first patient the exclusive skin involvement allowed to use a simple dressing hydrocolloid. The second one, instead, required a skin regeneration system because of the greater defect; this approach showed good scalp repair and no complications, with a reduction of the size of the defect from 4.5 cm to 0.5 cm.
Any plastic surgery that might be done to complete resolution of the lesion, should be easier with less complications.
A dermal regeneration template can be used successfully as initial treatment in full-thickness aplasia cutis at least to reduce the size of defect and to avoid or postpone plastic surgery and make it easier.
REFERENCES
- M. Mempel, D. Abeck, I. Lange, K. Strom, A. Caliebe, A.Beham, et al., “The Wide Spectrum of Clinical Expression in Adams-Oliver Syndrome: A Report of Two Cases,” British Journal of Dermatology, Vol. 140, No. 6, 1999, pp. 1157-1160. doi:10.1046/j.1365-2133.1999.02881.x
- K. M. G. Snape, D. Ruddy, M. Zenker, W. Wuyts, M. Whiteford, D. Johnson, W. Lam and R.C. Trembath, “The Spectra of Clinical Phenotypes in Aplasia Cutis Congenital and Terminal Transverse Limb Defects,” American Journal of Medical Genetics, Vol. 149A, No. 8, 2009, pp. 1860-1881. doi:10.1002/ajmg.a.32708
- M. El Khashab, S. T. Rhee, S. D. Pierce, Y. El Khashab, F. Nejat and A. Fried, “Management of Large Scalp and Skull Defects in a Severe Case of Adams-Oliver Syndrome. Case Report,” Journal of Neurosurgery: Pediatrics, Vol. 4, No. 6, 2009, pp. 523-527.
- R. G. Rodrigues, “Aplasia Cutis Congenita, Congenital Heart Lesions, and Frontonasal Cysts in Four Successive Generations,” Clinical Genetics, Vol. 71, No. 6, 2007, pp. 558-560. doi:10.1111/j.1399-0004.2007.00806.x
- K. Keymolen, L. De Smet, P. Bracke and J. P. Fryns, “The Concurrence of Ring Constrictions in Adams-Oliver Syndrome: Additional Evidence for Vascular Disruption as Common Pathogenetic Mechanism,” Clinical Genetics, Vol. 10, No. 3, 1999, pp. 295-300.
- J. F. Martínez-Lage, M. J. Almagro, F. López Hernández and M. Poza, “Aplasia Cutis Congenita of the Scalp,” Child’s Nervous System, Vol. 18, No. 1, 2002, pp. 634- 637. doi:10.1007/s00381-002-0654-4
- R. Santos de Oliveira, C. E. Barros Jucá, A. Lopes LinsNeto, M. Aparecida do Carmo Rego, J. Farina and H. R. Machado, “Aplasia Cutis Congenita of the Scalp: Is There a Better Treatment Strategy?” Child’s Nervous System, Vol. 22, No. 9, 2006, pp. 1072-1079. doi:10.1007/s00381-006-0074-y
- D. A. Ross, S. W. Laurie, C. J. Coombs and K. L. Mutimer, “Aplasia Cutis Congenita: Failed Conservative Treatment,” Plastic and Reconstructive Surgery, Vol. 95, No. 1, 1995, pp. 124-129. doi:10.1097/00006534-199501000-00020
- B. Bernbeck, J. Schwabe, A. Groninger, J. Schaper, H. Messing-Junger, E. Mayatepek, et al., “Aplasia Cutis Congenita of the Scalp: How Much Therapy Is Necessary in Large Defects?” Acta Paediatrica, Vol. 94, No. 6, 2005, pp. 758-765. doi:10.1080/08035250410023395
- K. Islamoglu and E. Ozgentas, “Aplasia Cutis Congenita of the Scalp: Excessive Bleeding and Reconstructive Problems,” Annals of Plastic Surgery, Vol. 47, No. 2, 2001, pp. 213-214. doi:10.1097/00000637-200108000-00022
- P. Steinbok, “Repair of a Congenital Cranial Defect in a Newborn with Autologous Calvarial Bone,” Child’s Nervous System, Vol. 16, No. 4, 2000, pp. 247-250. doi:10.1007/s003810050506
- M. Bajpai and K. Pal, “Aplasia Cutis Cerebri with Partial Acrania—Total Reconstruction in a Severe Case and Review of the Literature,” Journal of Pediatric Surgery, Vol. 38, No. 2, 2003, p. e4. doi:10.1053/jpsu.2003.50064
- S. T. Rhee, C. Colville, S. R. Buchman and K. Muraszko, “Complete Osseous Regeneration of a Large Skull Defect in a Patient with Cutis Aplasia: A Conservative Approach,” The Journal of Craniofacial Surgery, Vol. 13, No. 4, 2002, pp. 497-500. doi:10.1097/00001665-200207000-00003
- S. J. Beekmans and M. J. Wiebe, “Surgical Treatment of Aplasia Cutis in the Adams-Oliver Syndrome,” The Journal of Craniofacial Surgery, Vol. 12, No. 6, 2001, pp. 569-572. doi:10.1097/00001665-200111000-00014
NOTES
*Dini F. and Tuoni C. must be considered both as first authors.
#Corresponding author