Open Journal of Rheumatology and Autoimmune Diseases
Vol.3 No.4(2013), Article ID:39731,15 pages DOI:10.4236/ojra.2013.34037
Epidemiology of Cancer in Systemic Sclerosis—Systematic Review and Meta-Analysis of Cancer Incidence, Predictors and Mortality*
![]()
1Toronto Scleroderma Program, Division of Rheumatology, Mount Sinai and Toronto Western Hospitals, University of Toronto, Toronto, Canada; 2Institute of Rheumatology, Russian Academy of Medical Sciences, Moscow, Russian.
Email: #Sindhu.Johnson@uhn.ca
Copyright © 2013 Tatiana Nevskaya et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received September 20th, 2013; revised October 20th, 2013; accepted October 28th, 2013
Keywords: Systemic Sclerosis; Neoplasms; Epidemiology; Risk Factors; Mortality
ABSTRACT
Objectives: The study was conducted to improve our understanding of the epidemiology of cancer in systemic sclerosis (SSc) by evaluating the incidence, prevalence, relative risk of overall and site-specific malignancies, predictors and cancer-attributable mortality. Methods: MEDLINE, CINAHL, EMBASE and Cochrane Library (inception-May 2012) were searched. Estimates were combined using a random effects model. Consistency was evaluated using the I2 statistic. Results: 4876 citations were searched to identify 60 articles. The average incidence of malignancy in SSc was 14 cases/1000 person-years; the prevalence ranged between 4% - 22%. Cancer was the leading cause of non-SSc related deaths with a mean of 38%. Overall SIR for all-site malignancy risk was 1.85 (95%CI 1.52, 2.25; I276%). There was a greater risk of lung (SIR 4.69, 95%CI 2.84, 7.75; I293%) and haematological (SIR 2.58, CI 95% 1.75, 3.81; I20%) malignancies, including non-Hodgkin’s lymphoma (SIR 2.55, 95%CI 1.40, 4.67; I20%). SSc patients were at a higher risk of leukemia (SIR 2.79, 95%CI 1.22, 6.37; I20%), malignant melanoma (SIR 2.92, 95%CI 1.76, 4.83; I235%), liver (SIR 4.75, 95%CI 3.09, 7.31; I20%), cervical (SIR 2.28, 95%CI 1.26, 4.09; I254%) and oropharyngeal (SIR 5.0, 95%CI 2.18, 11.47; I258%) cancers. Risk factors include a-RNAP I/III seropositivity, male sex, and late onset SSc. Smoking and longstanding interstitial lung disease increase the risk of lung cancer; Barrett’s esophagus and a positive family history of breast cancer, respectively, increase the risk of esophageal adenocarcinoma and breast cancer. Conclusions: SSc patients have a two-fold increase in all-site malignancy, and greater risk of lung and haematological malignancies that contribute significantly to mortality. Vigilance should be considered in SSc patients with risk factors for cancer.
1. Introduction
Systemic sclerosis (SSc) is a chronic multisystem autoimmune disorder characterized by vasculopathy and fibrosis of the skin and visceral organs, which is associated with substantially reduced survival [1,2]. The occurrence of cancer significantly complicates the disease course, burdens its management and contributes to mortality
[1-3]. Accumulating evidence from cohort studies has suggested an overall higher risk of cancer in SSc patients. However, data are still contradictory with some studies showing the risk comparable to the general populations [4,5], whereas others reporting a moderate [6-12] or significantly [13,14] increased risk of cancer in SSc. The absolute standardized incidence ratio (SIR) values for individual cancers were also variable in the different studies. Meanwhile, the characterization of malignancy risk in SSc has gained considerable importance with the recently published case reports of cancer occurrence in patients receiving novel therapeutic interventions like biologic therapy and intense immunosuppression for stemcell transplantation [15,16].
To interpret these findings correctly, it is necessary to determine the underlying risk of cancer development in SSc patients compared to the general population. The only systematic review of malignancy risk in SSc [17] was limited in its search scope. It calculated the overall relative risk of malignancy in SSc based on mixed prevalence and incidence ratios and summarized the results on malignancy risk in SSc patients and SSc risk in patients with certain types of cancer, creating a potential bias. Furthermore, the ability to predict cancer would fulfill the needs of clinical practitioners to better allocate efforts and resources towards those SSc patients who are at increased risk of malignancy resulting in earlier effective therapy, better survival, and ideally, primary prevention of cancer. This has prompted the search for cancer predictors in SSc.
However, not all reports agree that determinants suggested as risk factors are more common in SSc patients with cancer, [11-13,18,19] nor is there complete agreement regarding the factors that might increase the risk of site-specific malignancies [21,22].
All this emphasizes the need of systematic analysis to evaluate the risk of malignancy in SSc patients and to interpret the clinical value of characteristics suggested to identify high-risk individuals. Consequently, the aim of this study was to systematically review the incidence, prevalence and relative risk of overall and site-specific malignancies in SSc patients, as well as to synthesize the clinical evidence for the relationship between risk factors and malignancy. Additionally, we analysed the data from the studies examining the cancer-attributable mortality in SSc. Given our findings, we discuss the potential implications of these investigations and future directions involving a combination of population and molecular studies that are needed to predict malignancy in SSc patients.
2. Methods
Search strategy. MEDLINE, MEDLINE In-Process & Other Non-Indexed Citations, the Cumulative Index to Nursing and Allied Health Literature (CINAHL), EMBASE and the Cochrane Library were searched from inception to May 2012, using the terms “scleroderma” OR “systemic sclerosis” OR “CREST syndrome” or “calcinosis” or “Raynaud”s phenomenon” AND (“malignan- сy or malignancies” or “cancer” or “neoplasm(s)” or “tumor(s)” or “carcinoma(s)” or “lymphoma(s)” or “leukemia” or “sarcoma” or “melanoma” or “neoplastic” or “carcino” or “onco”) AND (“risk”, “mortality”, “cancer mortality”, standardized mortality ratio”, or “cause of death”, or “survival”, or “cancer specific survival”, or “life expectancy” or “overall survival”, or “morbidity”, or “comorbidity”, or “incidence”, or “prevalence”) AND (“epidemiolog(y)” or “case control study” or “population-based study” or “hospital based study”, or “retrospective study” or “prospective study” or “follow up”, or “longitudinal study”, or “cross sectional study”, or “observational study” or “cohort study”). The search was conducted independently by an investigator (T.N.) and a University Health Network Library Services Information Specialist. End-NoteX4 software was used to check for duplicate publications.
Studies were limited to human but without language restriction. Articles were translated by native-language speakers or machine translation software. Studies were included if they: 1) had observational study design: a) reported overall and site-specific malignancy rates in SSc, b) case-control studies where the characteristics of SSc patients with and without malignancies were compared to determine the risk factors of cancer development in SSc, c) studies on cancer-attributable SSc mortality; 2) contained original data; 3) reported an adult population. Studies were excluded if they 1) reported localized scleroderma, juvenile scleroderma, paraneoplastic scleroderma-like syndrome; 2) were abstracts, case reports, editorials and review articles; 3) were studies for which updated manuscripts were available. Searches were supplemented by hand-searching relevant articles (including citation searching), references of selected studies, guidelines and reviews. Eligible citations were assessed independently by two investigators (T.N., S.C.). Disagreements were resolved by consensus between the two investigators or by involving an arbitrator (S.R.J.).
Outcomes. The outcomes of interest were 1) the relative risk of cancer in SSc patients compared with the general population, estimated as the ageand sex-adjusted standardized incidence ratio (SIR), 2) risk factors for cancer development in SSc patients and 3) cancer mortality rate in SSc. Measures of relative effect included absolute risk, odds ratios (OR), hazard ratios (HR), standardized incidence ratio (SIR), standardized mortality ratio (SMR), with or without 95% confidence intervals (CI). The SIR is the ratio of the incident number of cases to the incident number that would be expected if the study population had the same incidence rate as a standard or other population for which the incidence rate is known. The SMR is the ratio of the number of deaths observed in the study group to the number that would be expected if the study population had the same specific rates as the standard population. Study characteristics and outcome data were independently abstracted by 2 investigators (T.N, S.C) using a standardized abstraction from. The abstracted data was independently verified against the source document by 2 different investigators (A.R, C.P.).
Analysis. Descriptive statistics were used to summarize the data. The proportions of deaths attributable to cancer were calculated separately among all causes of death in SSc patients, among all known causes of death and among SSc-unrelated (including possibly related) causes of deaths. Summary estimates of the relative risk of 1) overall malignancy, 2) site-specific malignancies, 3) overall malignancy by sex, 4) lung cancer by sex in SSc patients were calculated using Dersimonian and Laird inverse variance weighting random effect method. Analysis was conducted using SPSS v6.1 and RevMan 5.2 software. The systematic review and meta-analysis conform to the PRISMA statement [23].
3. Results
A total of 4876 citations (4203 English, 673 non-English) were identified using the search criteria, however 3767 citations were excluded (data on SSc but not malignancy n = 321, localized scleroderma n = 50, case reports n = 84, paraneoplastic syndrome n = 43, chemically induced SSc or Raynaud’s phenomenon n = 46, or sclerodermalike manifestations of graft versus host disease n = 34, malignancies associated with diseases other than SSc n = 3190). One hundred fifteen articles were further analyzed, and of these, 65 full-text papers were found to meet the inclusion criteria and not any of the exclusion criteria. However, 5 papers were excluded (up-dated manuscripts were available n = 1 [24], the data on the number or percent of deaths due to cancer were absent n = 2 [25,26], the total number of deceased SSc patients was too small to obtain reliable data on malignancy-attributable mortality n = 2 [27,28]). Three works of Derk et al. [6,29,30] were analysed as 1 study as they were based on the same SSc cohort. This left 60 papers reporting the results of 58 studies for inclusion in this review (Figure 1).
Twenty nine studies stated incidence or/and prevalence of malignancy in SSc patients [4-14,18,20,21,29- 5], of these 13 also analysed risk factors for cancer [4,8, 9,11-13,18,20,21,29,37,39,41] and 1-cancer-attributable mortality [14]. Five studies were directly addressed to the risk factors for cancer in SSc patients [19,22,42,46]. Seventeen studies reported cancer among the causes of SSc-unrelated deaths [1-3,43,49-61].
Population level associations between SSc and malignnancies have been evaluated by two study designs which we analysed separately considering the different methodological approaches and outcome measures (SIR, odds ratios). A majority of studies have assembled cohorts of SSc patients followed-up for the occurrence of cancer to calculate SIRs for malignancies in SSc patients compared to the general population (Table 1). In contrast, seven studies used the case-control design based on the largesized samples of patients with a certain site-specific malignancy [62-68]. Among these cancer patents, those who
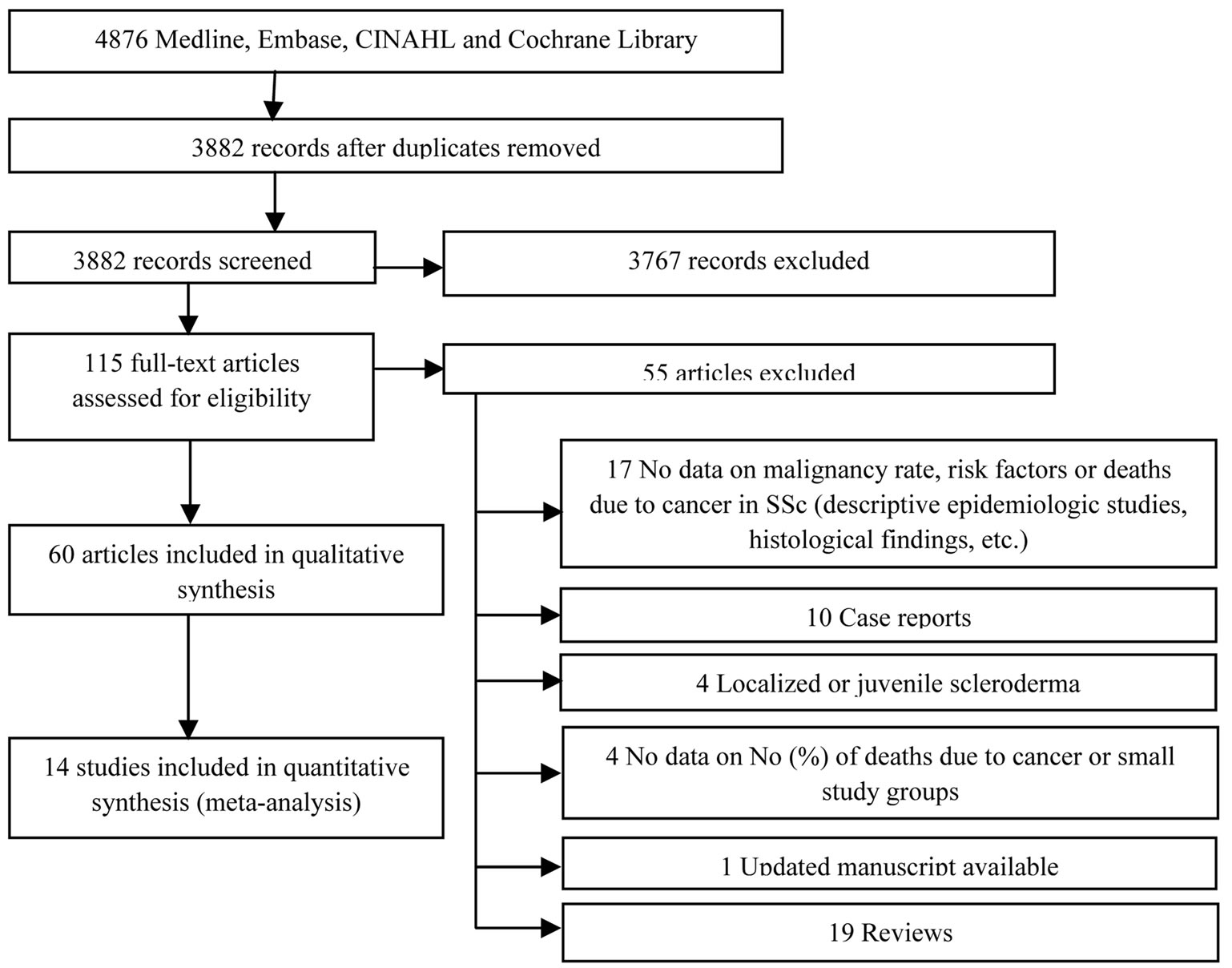
Figure 1. Flow diagram of search results.
 (a)
(a) 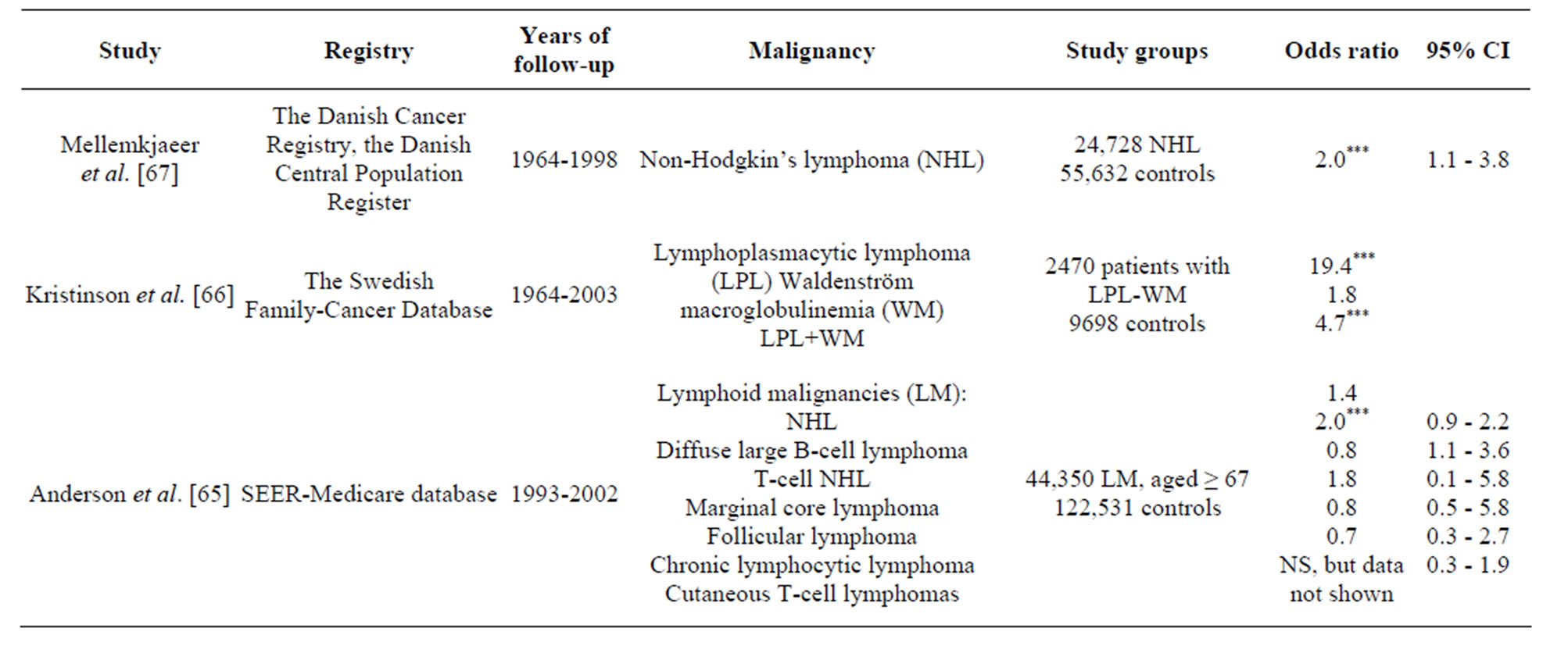 (b)
(b)
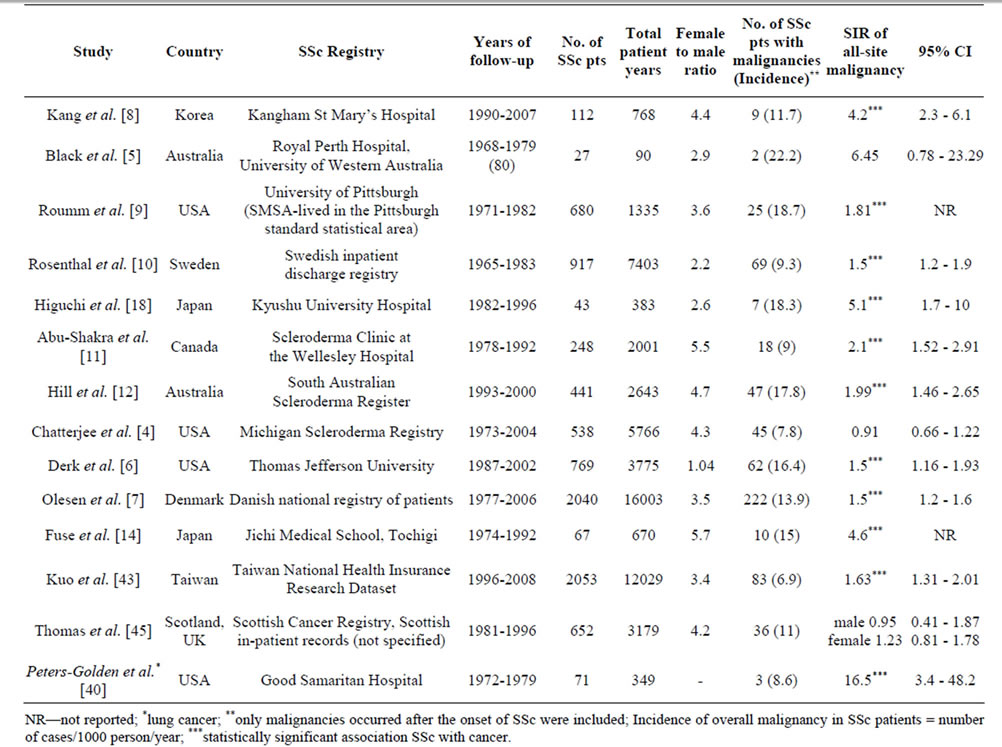
Table 1. Characteristics of the population-based studies reporting relative risk of malignancy in systemic sclerosis. a) Population-based studies of SSc patients followed-up for malignancy risk assessment, b) Case-control population-based studies of cancer patients aimed to detect an association of a certain cancer with SSc.
Continued reported a prior history of SSc were detected to estimate relative risks for the association between certain types of cancer and SSc.
Prevalence and incidence of malignancy in SSc. The prevalence of overall malignancy ranged from 4% to 22% considering wide variation in follow-up time and different timeframes chosen for the inclusion of malignnancy cases. The average incidence of overall malignnancy was 14 cases/1000 person-years, with most commonly seen lung (3.3), breast (1.6) and haematological (1.6) malignancies, including Non-Hogkin’s lymphoma (NHL) (1.0) (Table 2).
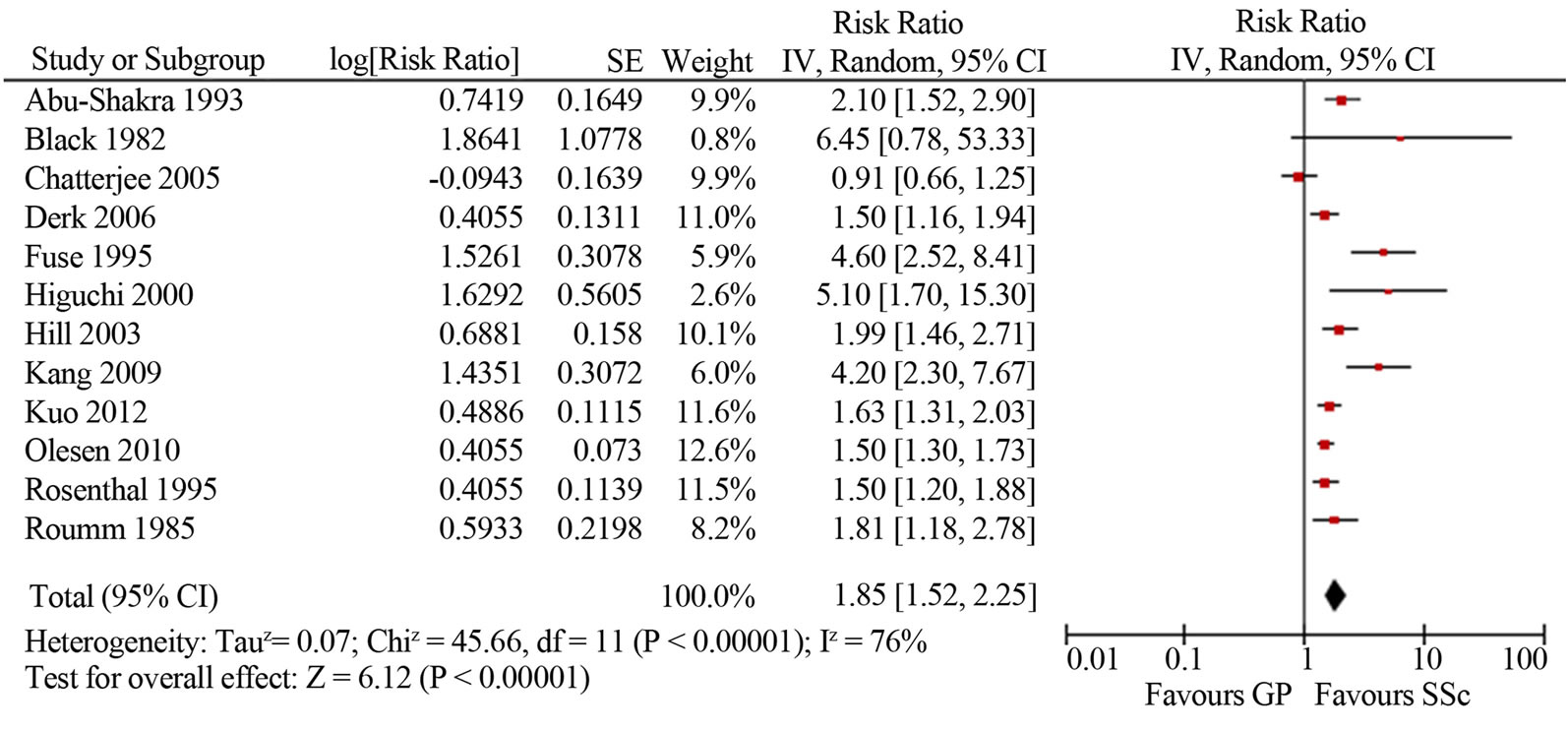
Malignancy risk in SSc. The mean SIR for all-site malignancy was 1.85 (95%CI 1.52, 2.25; I2 76%) based on the results of 12 studies [4-12,14,18,43]; for males 1.99 (95%CI 1.61, 2.35; I2 12%) and for females 1.38 (95%CI 1.22, 1.55; I2 14%) (Figure 2). Most studies based on Caucasian SSc cohorts showed similar slightly increased (1.5-2-fold) risk of malignancy in SSc, whereas three studies from Asia indicated a cancer risk that was significantly (4-5-fold) higher [8,14,18]. Kang et al. reported that the incidence of cancer among patients with SSc was 4.2 times higher than in the Korean general population (95%CI 2.3, 6.1). Fuse et al. reported malignnancy occurred in ten out of sixty seven SSc patients (14.9%), and that was 4.6 times higher than the incidence of malignancy in the general population of Japan. Higuchi et al. found the highest among all published works cancer rate in SSc patients (16.3%), with an estimated SIR of 5.1. Similar to the study of Kang and Fuse, SSc group was small and included only 43 patients, 7 of whom developed malignancies during a follow-up period of 6 years at average. In two larger studies of SSc cohorts in Japan (8327 patients) and Taiwan (2053 patients) the relative risks of malignancy were analogous to those reported for Caucasian SSc cohorts: 1.6 and 2.3 times higher compared to the general populations [39,43]. Three studies did not reveal an increased risk of cancer in SSc patients [4,5,45].
Site-specific malignancies. There was a greater risk of lung cancer (SIR 4.69, 95%CI 2.84, 7.75; I2 93%) [4, 6-12,40,43], and haematological (SIR 2.58, 95%CI 1.75, 3.81; I2 0%) malignancies [7,10,12,43], including non-Hodgkin’s lymphoma (SIR 2.55, 95%CI 1.40, 4.67; I2 0%) [4,6,7,10]. SSc patients may be also at a higher risk of leukemia (SIR 2.79, 95%CI 1.22, 6.37; I2 0%) [7,10], malignant melanoma (SIR 2.92, 95%CI 1.76, 4.83; I2 35%) [7,11], liver (SIR 4.75, 95%CI 3.09, 7.31; I20%) [4,8,10], cervical (SIR 1.89, 95%CI 1.01, 3.54; I2 62%) [4,7,11,43] and oropharyngeal (SIR 3.97, 95%CI 3.02, 5.21; I2 16%) [11,30,43] cancer; but this was only supported by ≤4 studies. In contrast, the risk of breast and esophageal cancers associated with SSc appeared to be comparable with that in the general population: SIR 1.3, 95%CI 0.51, 3.33 [4,6,7,9-12,43] and SIR 5.26, 95%CI 0.36, 77.28 [6-9], respectively. However, the inconsistency (I2) between the results of these studies was very high —98% - 99%.
Lung cancer was the most frequent malignancy in SSc patients comprising from 14% - 44% of all SSc cancer cases [4,6-12,18,40]. The risk of lung cancer was higher in men (SIR 5.03, 95%CI 2.8, 9.04; I2 66%) than in women SIR 4.24, 95%CI 2.49, 7.24; I2 78%) [6-8,10,11, 43]. Tissue types included adenocarcinoma (from 20% to 50%, mean ± SD 40% ± 20%), squamous cell cancer (from 0% to 47%, mean ± SD 21% ± 17%), oat cell cancer (0% - 14%), mixed histology (0% - 27%), alveolar cell carcinoma (0% - 14%) and undifferentiated (0% - 50%) lung cancer [4,8-12,20,41].
Haematological malignancies have been reported to be associated with systemic sclerosis in several studies. In a cross-sectional study of Siau et al. the highest risk among
 (a)
(a)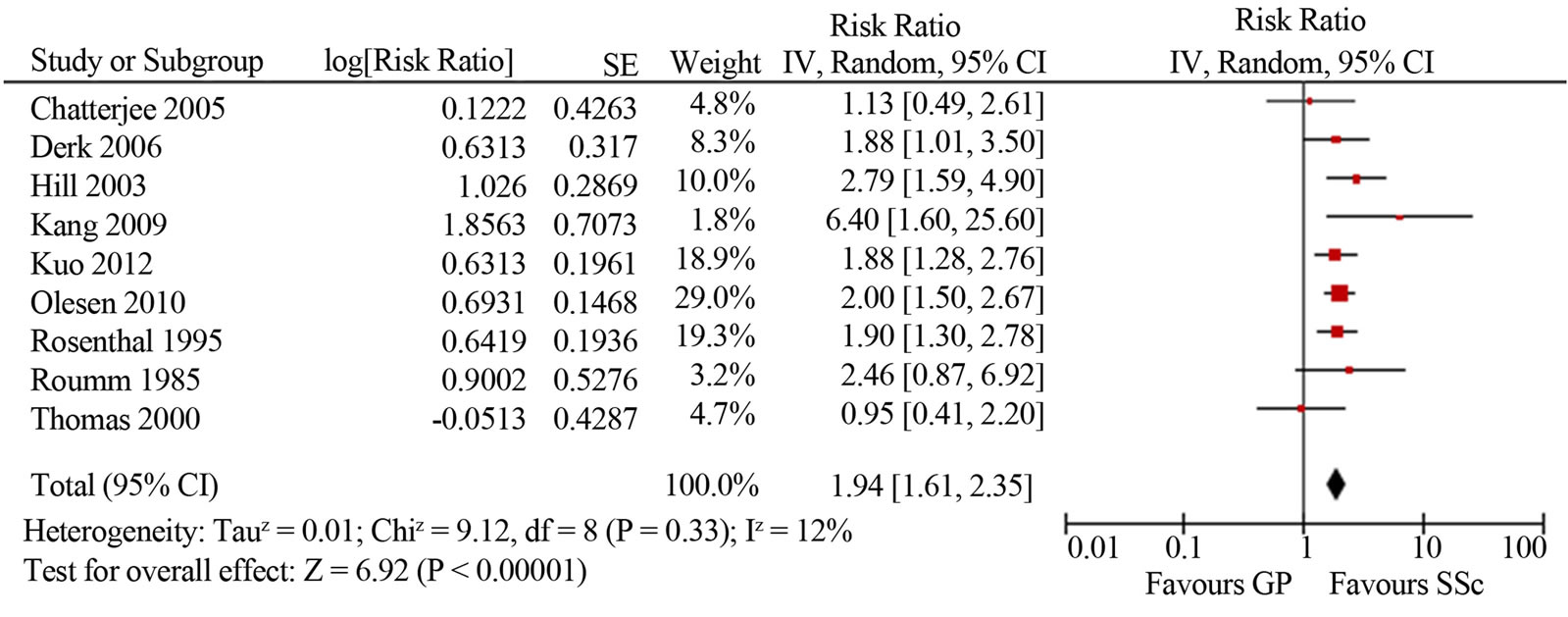 (b)
(b)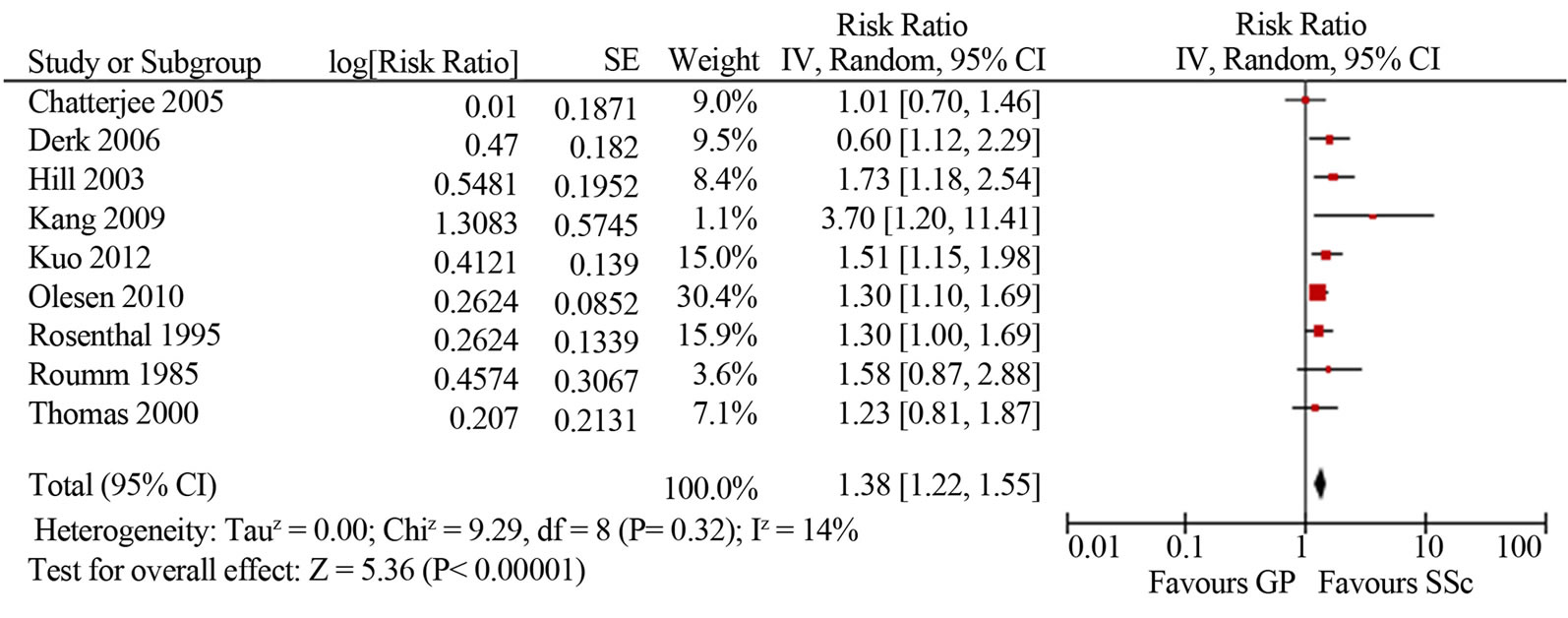 (c)
(c)
Figure 2. Relative risk of overall malignancies in patients with systemic sclerosis (total (a), male (b) and female (c)) compared with the general population. CI, confidence interval.
all site-specific cancers was found for haematological malignancies (RR18.5, 95%CI 6, 43, p = 0.03) [13]. Olesen et al. showed a 2.5-fold increased risk of haematological malignancies and non-Hodgkin’s lymphoma (NHL) and 2.9-fold increased risk of leukemia [7]. Rosenthal et al. reported an increased risk of non-Hodgkin lymphoma in women and an increased risk of leukaemia in men [10]. These finding were confirmed by the series of studies used the extended case-control approach based on the large-sized samples of patients with a certain sitespecific malignancy. Mellemkjaer et al. demonstrate that SSc was linked with an increased risk of NHL which was most evident in the period 1 - 4 years subsequent to the diagnosis of SSc (RR 3.8, 95%CI 1.1, 13) [68]. In two population-based studies of haematological malignancies in elderly adults, Andersen et al. reported the association of SSc with diffuse large B-cell lymphoma, but not hairy cell leukemia [65,66]. Kristinsson et al. found a strong association between SSc and increased risk of subsequent lymphoplasmacytic lymphoma [67]. In the study of Brown et al., based on a large male population, SSc and multiple myeloma were significantly linked [63].
Risk factors for malignancy in SSc. RNP I/III positivity as a risk factor of malignancy developed temporally close to SSc onset was demonstrated across all studies where it was evaluated [19,41,42], whereas other purported characteristics showed inconsistent results (Table 3). Male sex and late onset disease were most often reported as risk factors [6,8,11,12,22,39]. Smoking and duration of interstitial lung disease (ILD) increased the risk of lung cancer [9,13,20,22]; a positive family history of breast cancer increased the risk of breast cancer [21,22]. Male sex and longstanding gastroesophageal reflux disease with Barrett’s esophagus independently increased the risk of esophageal adenocarcinama in SSc patients [6, 46,64].
Immunosupressive therapy and SSc duration were not associated with cancer risk [8,21,22,30,37]. The SIRs for cancer were analysed in dSSc and lSSc patients in 2 papers [4,12] showing inconsistent results: no difference between SSc subtypes [4] and increased SIR for cancer in dSSc patients [12]. All other studies comparing SSc patients with and without cancer did not find any differences in cancer risk in respect to SSc subtype [6,8,9,11, 18,20-22,37] (Table 3).
Attributable risk of cancer to mortality yin systemic sclerosis. The mean (±standard deviation) percentage of deaths attributable to cancer among all SSc patients who had died was 14.5% ± 6.2% (median 13.8%), and among only known causes of deaths was 15.7% ± 6.6% (median 14.7%) (Table 4). Malignancy was the leading cause of non-SSc related mortality (37.8% ± 22.6%, median 31.3%) followed by infection or atherosclerosis [1,2,3,7,50-53,60]. Lung cancer was the most frequently seen site-specific malignancy [1-3,49,50,59] accounting for 23% - 58% of all fatal malignancy cases, followed by breast and hematological malignancies (Table 4). Two studies were population-based and reported the SMRs for specific causes of death [25,36]. Kuo et al. reported a slightly increased cancer-attributable mortality (SMR 1.5, 95%CI 1.03, 2.11) compared to the general population with the diagnosis of cancer being a risk factor for death (HR 2.71, 95%CI 1.27, 5.76) [48]. Hashimoto et al. found no demonstrable excess mortality from malignancy (SMR 1.7, 95%CI 0.94, 2.47) [58].
4. Discussion
Synthesis of the published literature suggests a twofold increased risk of all-site malignancy in SSc patients compared to the general population, with men being at a higher risk than women. The average incidence of all-site malignancy was 14 cases per 1000 person-years and the prevalence ranged from 4% to 22%. A recent systematic review showed similar findings of 1.75-fold increase in malignancy risk in SSc patients [17]. The strengths of the present work are an increased scope of literature search resulting in additional articles captured for meta-analysis [14,45,65,66,68], evaluation of SIRs by sex and estimation of overall SIRs for all site-specific malignancies reported in the published literature.
The majority of studies, based on predominantly Caucasian cohorts, reported comparable results on a slightly increased (1.5-2-fold), but statistically significant risk of malignancy in SSc. The exceptionally high rise (4-5-fold) in cancer risk was found in Asian SSc population [8,14, 18]. The bias in these three studies from Japan and South Korea might relate to a small sized groups and a short observation period. Indeed, a recent large cross sectional study based on National annual survey conducted by the Ministry of Health, Labour and Welfare of Japan has revealed that the risk of malignancy in SSc was only two times higher than in the general population, in accordance with the slightly increased risk of malignnancy shown for the European and American cohorts of SSc patients [39]. Similar findings of a 1.6-fold increase in the observed number of cancers in SSc patients were reported in the study from Taiwan based on 2053 SSc patients [43].
Although most studies seemed to follow the trends seen in our meta-analysis, three studies did not show an increased risk of cancer in SSc patients [4,5,45]. Although, the overall cancer rate in SSc found by Chatterjee et al. was even higher (84%) than that in the majority of other studies, but similarly to the studies of Black et al. and Thomas et al. the difference did not reach statistical significance compared to the general population of metropolitan Detroit which is itself characterized by an increased cancer rate [69]. On the other hand, as these studies were based only on medical records, the local population of SSc patients might be overestimated that can potentially result in lower SIR.
Data also substantiate an increased risk of certain sitespecific cancers in SSc. Lung cancer was associated with the highest relative risk which was more than four-fold higher than in the general population. It often complicated the course of long-standing SSc with pulmonary fibrosis. Pontifex et al. showed that bronchogenic (small cell and squamous) lung cancer developed 25 years after SSc onset, while peripheral (large cell, broncho-alveolar
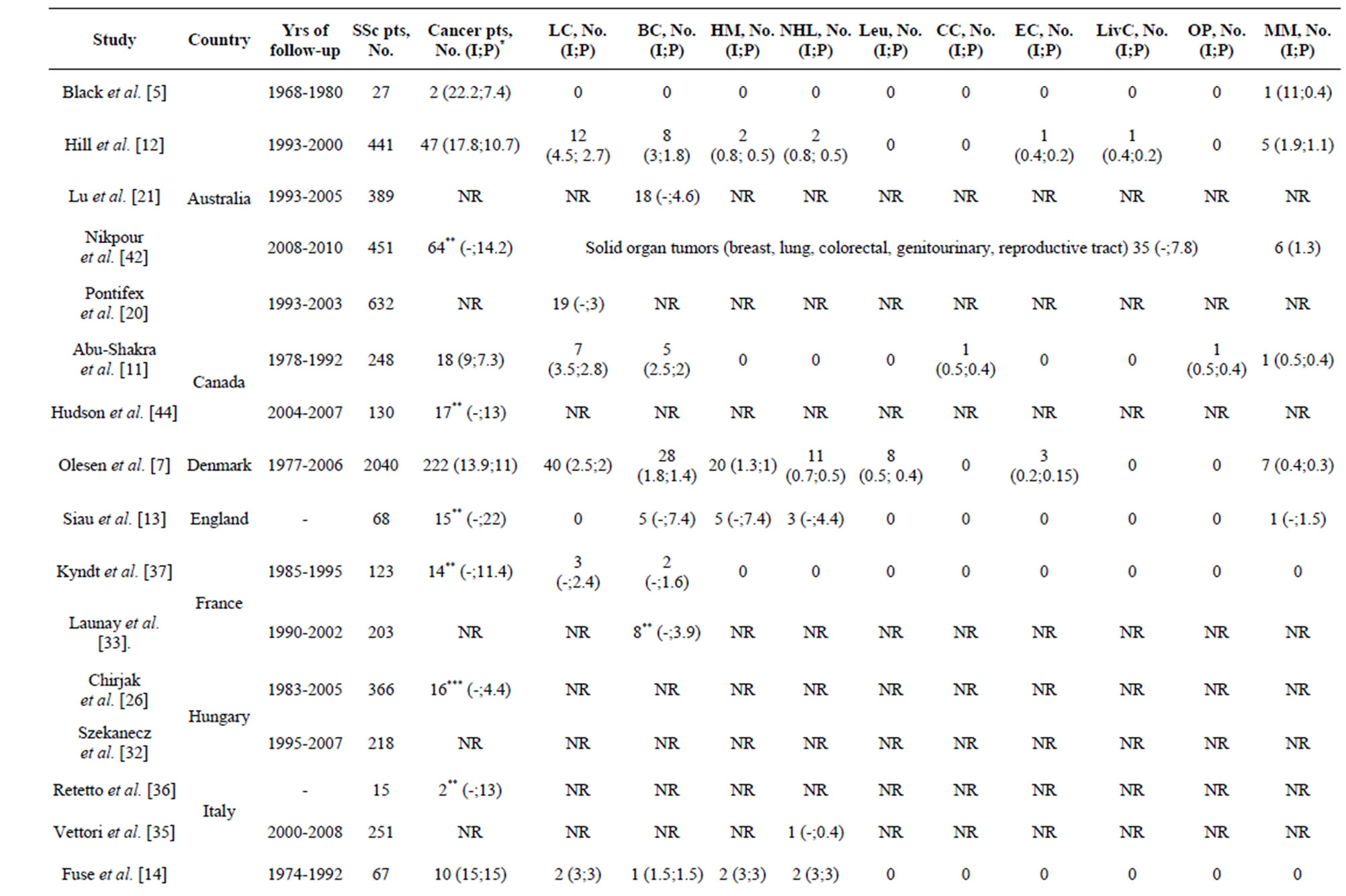
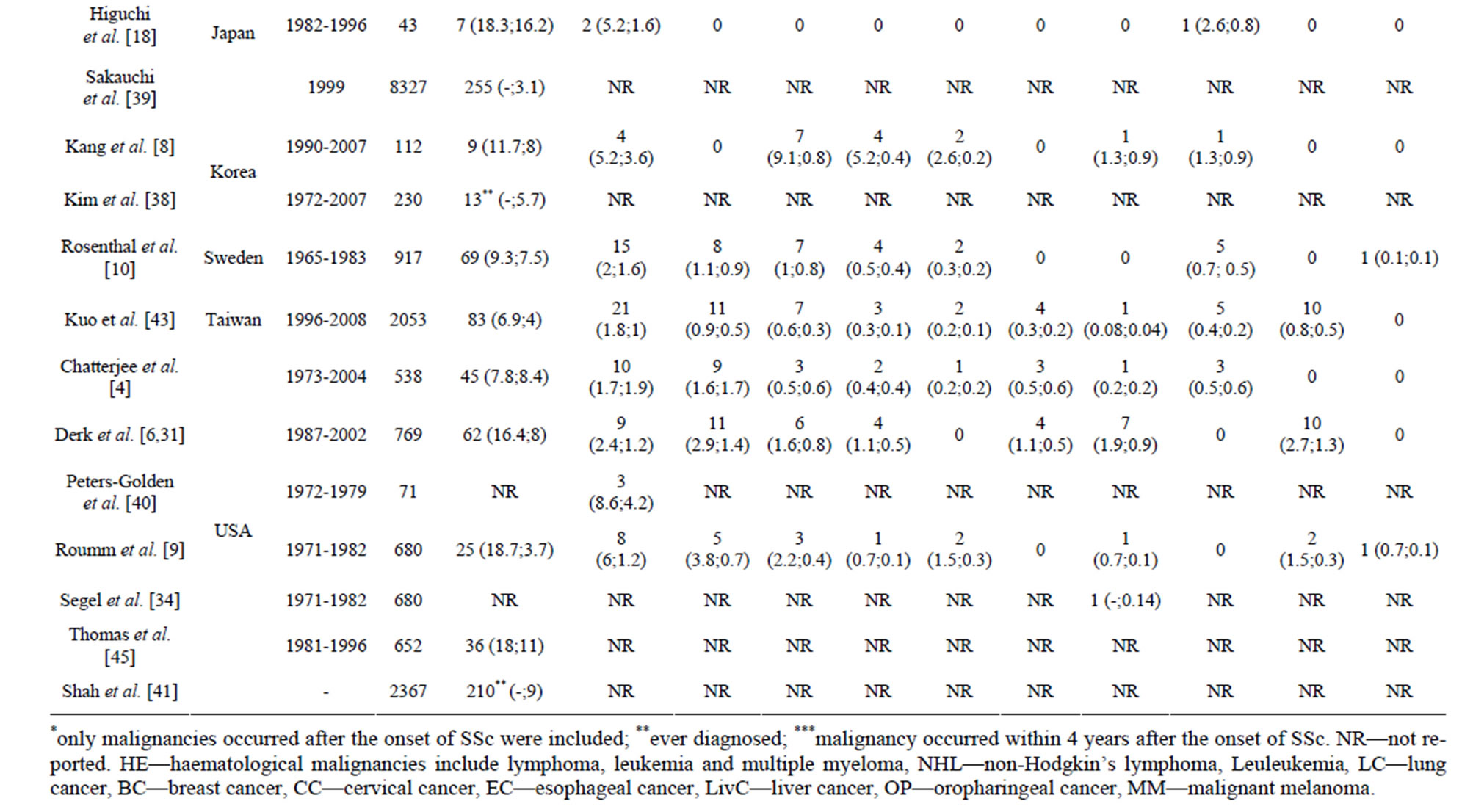
Table 2. Incidence (I, %) and prevalence (P, number of cases/1000 person/year) of the overall and site-specific malignancies in patients with systemic sclerosis.
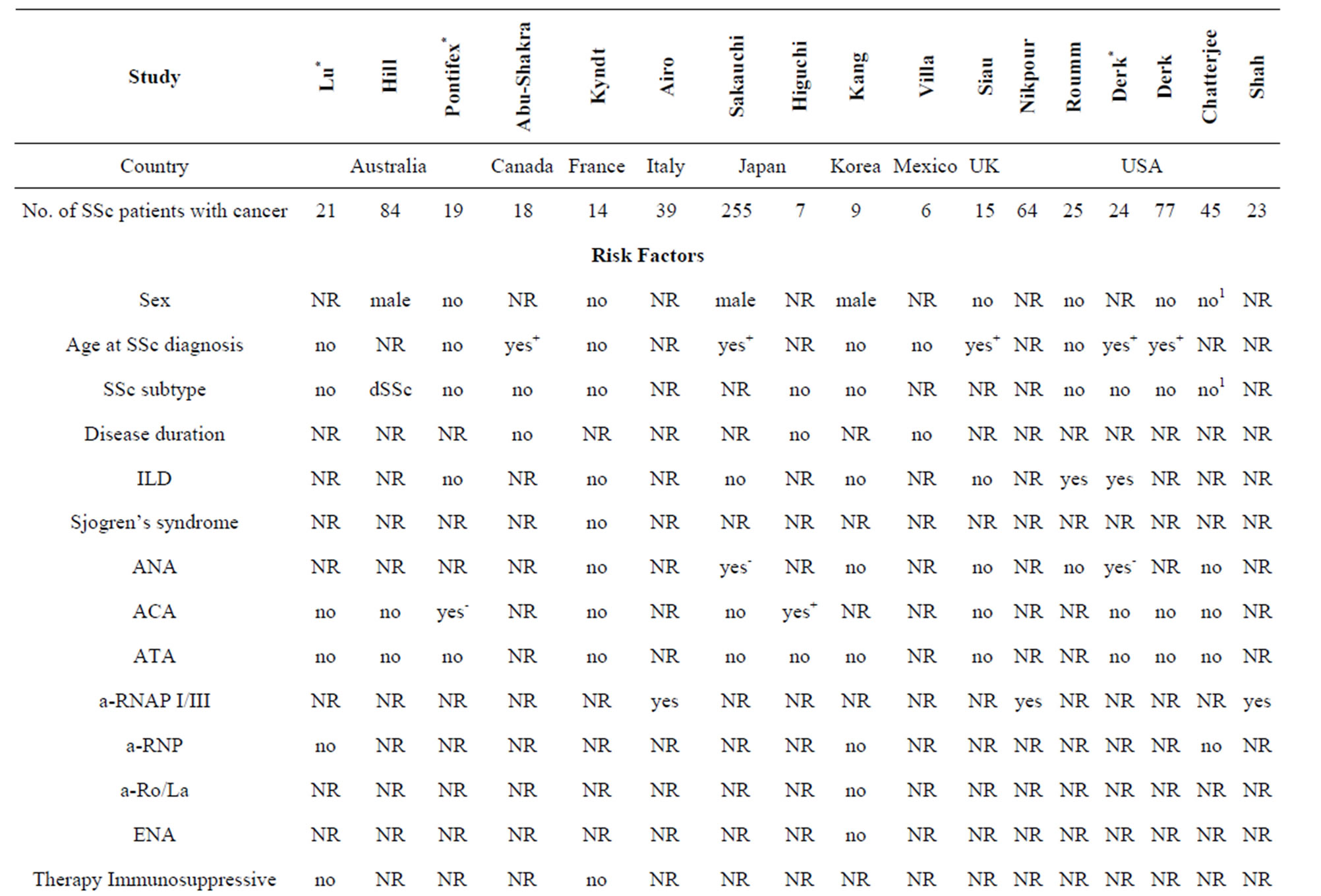

Table 3. Potential associations that may place systemic sclerosis patients at a higher risk for the development of cancer.
carcinoma and adenocarcinoma) appeared earlier, in 5 years at average [20].
The most frequent histology is non-small cell lung carcinoma with the great majority of SSc patients (40% - 50%) having adenocarcinoma. The risk appears to be also heightened for haematological malignancies, especially for non-Hodgkin’s lymphoma. Some other sitespecific malignancies, such as melanoma, liver, oropharyngeal and cervical cancers, showed similarly heightened SIR; but paucity of studies preclude any reliable assumption. Based on observed versus expected cases, there was considerable variation in the calculated SIR among the individual studies for breast and esophageal cancers, and the confidence intervals were relatively imprecise. Nevertheless, the random effects metaanalysis did not reveal an increased risk of these types of cancer in SSc patients. A possible explanation of the fact that coexisting breast cancer and SSc often arise in a brief
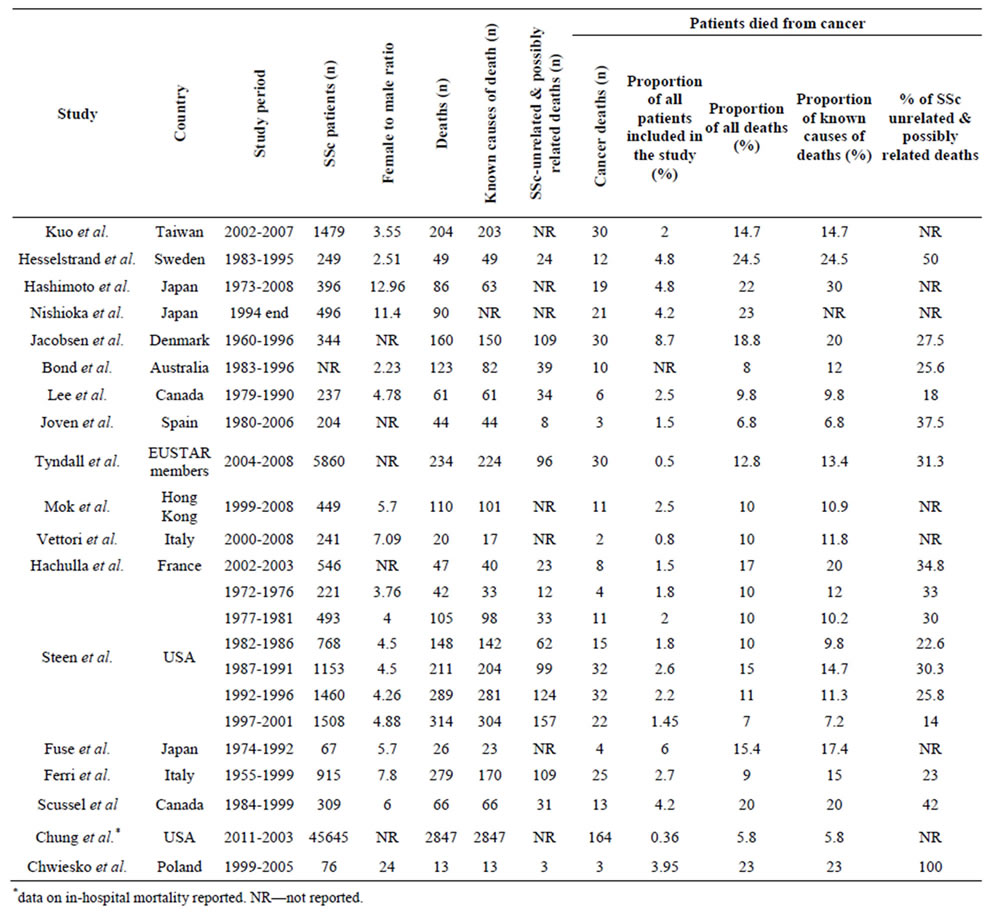
Table 4. Distribution of deaths due to cancer among all deaths, deaths unrelated to systemic sclerosis and known causes of death.
time period is that in these cases the onset of malignancy might trigger the development of clinically apparent autoimmunity [41].
Cancer was the leading cause of deaths not directly related to disease specific organ-based complications in SSc patients. Mortality attributable to lung cancer comprised one-third to a half of all fatal malignancies, followed by breast and haematological malignancies. The proportion of deaths due to cancer in SSc patients varied between 6.8% - 24.5% that may be explained by the differences in overall cancer mortality rates between countries and/or in a study period as cancer care/outcomes have significantly shifted over time, and/or by the different average age of patients enrolled into the studies.
The identification of SSc patients at high risk for neoplasms could have a large impact on survival by improving patient outcomes and decreasing mortality. The most promising candidate for malignancy screening is RNAP I/III seropositivity in SSc patients with rapid progression of skin involvement [19,41,42].
Other risk factors include male sex and late disease onset. Site-specific malignancy risk factors are smoking and long-lasting interstitial lung disease for lung cancer; longstanding gastroesophageal reflux disease with Barrett’s esophagus for esophageal adenocarcinoma, and a positive family history for breast cancer.
Male patients with SSc are at a higher risk for all-site malignancy, and lung cancer, specifically, that was confirmed by meta-analysis. Some inconsistency seen in the published studies was related to a proportion of specificsite cancers in the cohorts which possess a different distribution among women and men. The further studies should be aimed to investigate each site-specific malignnancy separately according to the gender of SSc patients. In this regard, Chatterjee et al. revealed that liver cancer occurred in the Afro-American female SSc patients at a rate nearly 46 times greater than expected in the general Afro-American female population [4].
ILD showed inconsistent results as a risk factor for malignancy across many studies where it was evaluated.
It might be explained by different methods used for lung fibrosis detection which varied significantly between the studies and over the years, even within the same study considering a retrospective design with a long follow-up period [9,20,37]. Alternative reason for controversial findings could be that a long-lasting ILD regardless of its extent in SSc patients may be a risk factor for cancer. Even minor interstitial pulmonary changes that have lasted for a long time entails a chronic local injury with repeated cycles of epithelial cell damage and repair that might lead to genetic mutations. Indeed, lung cancer more often complicates the course of long-standing SSc as the average disease duration from SSc onset to the occurrence of lung cancer was typically 5 - 13 years [9-11].
On the molecular level, lung cancer is thought to develop from a stepwise accumulation of multiple acquired genetic alterations [70]. An increase in acquired somatic genetic damage has been recently shown among patients with SSc that might explain the enhanced cancer risk. The number of studies revealed genetic damage not only in SSc patients, but also in their first-degree relatives (increased variable number of random repeat mutations and increased chromosomal breakage rate) [71,72]. This is in accordance with the higher prevalence of cancer among first-degree relatives [73] and with the fact that a positive family history of cancer was one of the risk factors for its development in SSc patients [21,22].
The cause of the increased genetic instability in SSc patients and their relatives is still unknown and should be a priority in further investigation. It likely relates to the altered function of proteins which are responsible for DNA repair and/or which mediate cell apoptosis in response to DNA damage (e.g. tumor suppressor p53 and p21 proteins) [70]. The loss of their functions can be caused by gene polymorphism or chronic DNA damage/repair under the condition of persistent inflammation in SSc. Thus, the studies of tumor suppressor proteins in SSc are merited to provide more information on the mechanism of tumorogenesis.
RNP I/III positivity was reported as a risk factor with a high inter-study consistency. Enhanced nuclear RNA polymerase III antigenic expression by tumor tissues was demonstrated exclusively in SSc patients with RNA polymerase antibodies [41]. Possibly, expression of the same autoantigens in damaged vessels leads to immune response directed against specific host tissues, with consequent tissue damage that generates the SSc clinical phenotype [41]. These data provide strong evidence that anti-cancer immunity and autoimmunity are related.
Further investigations of autoantibodies in SSc patients with cancer are very important. Antinuclear antibodies, usually found in systemic rheumatic diseases (anti-DNA, anti-Sm, anti-RNP, p53, c-myc, anti-Ku, aPL and others), have been reported for decades in patients with various cancers [74]. These autoantibodies are currently considered as extremely promising diagnostic and prognostic biomarkers of cancer (similar to their value in systemic autoimmune diseases) and have the potential to detect cancer early when the treatment has the best chance to affect tumor behavior [75]. As believed, the antibodies to RPA32, annexin XI-A, and EF-2 in the sera of a small proportion of patients with systemic autoimmune diseases may also represent early markers of malignancy [75].
The observed differences in the studies could be real or they could be accounted for by various combinations of the following factors. First, it is difficult to precisely examine the possible increased risk of malignancy in scleroderma patients because of the rarity of this disease and therefore the small number of patients available for a study. We used a broad search strategy to capture evidence from many different settings, and the patient cohorts in some of the studies were relatively small. The heterogeneity among studies is also related to the populations examined, the methodology of case ascertainment and the data sources, as well as to the background incidence of cancer in different geographic areas and the wide variation in follow-up that could potentially bias the results. Besides, it is difficult to separate the underlying risk associated exclusively with the disease from some of the known and unknown confounders: risk factors for cancer (potential treatment effects, dietary factors, cigarette smoking), the genetic, racial, ethnic, behavioral, occupational, and environmental risk factors. Most of these studies excluded cancer cases that may have been diagnosed shortly before scleroderma onset, and therefore the risk may be underestimated. It is possible that the variability observed in the SIRs may result from these differences.
In summary, we found that the average incidence of all-site malignancy in SSc patients was 14 cases per 1000 persons-years. Compared to the general population, SSc was associated with a twofold increase in malignancy risk. Male patients were at a higher risk than female patients. The highest four-fold increased risk was observed for lung cancer, with adenocarcinoma being the most frequent histology. Non-Hodgkin’s lymphoma was associated with a two-fold rise in an overall risk. The risks of breast and esophageal cancers among SSc patients were comparable to the general population. SSc patients may be at a higher risk of leukemia, malignant melanoma, liver cancer, cervical and oropharingeal cancers. Purported risk factors for cancer in SSc include RNAP I/III positivity, male sex, increasing age, a positive family history of breast cancer, smoking and a long-lasting ILD. Vigilance is recommended in this group of patients. Therapy, disease duration and subtype are not among risk factors of cancer in SSc. Further studies of risk factor identification, risk stratification and impact on early diagnosis of cancer are needed to enhance our understanding of malignancy in SSc and to develop preventative strategies to improve survival.
5. Acknowledgements
The authors would like to acknowledge with sincere gratitude the effort put into this paper by Professor Marco Matucci-Cerinic (Department of Medicine, University of Florence, Florence, Italy), Professor Masataka Kuwana, MD PhD (Division of Rheumatology, Department of Internal Medicine, Keio University School of Medicine, Tokyo, Japan) and Professor Kazuhiko Takehara, MD PhD (Kanazawa University, Kanazawa, Japan). We are also grateful to Viola Machel BMSc MLIS, Information Specialist at the University Health Network Library Services for assistance with the literature search.
REFERENCES
- S. Jacobsen, P. Halberg and S. Ullman, “Mortality and Causes of Death of 344 Danish Patients with Systemic Sclerosis (Scleroderma),” British Journal of Rheumatology, Vol. 37, No. 7, 1998, pp. 750-755. http://dx.doi.org/10.1093/rheumatology/37.7.750
- R. Hesselstrand, A. Scheja and A. Akesson, “Mortality and Causes of Death in a Swedish Series of Systemic Sclerosis Patients,” Annals of the Rheumatic Diseases, Vol. 57, 1998, pp. 682-686. http://dx.doi.org/10.1136/ard.57.11.682
- L. Scussel-Lonzetti, F. Joyal, J. P. Raynauld, A. Roussin, E. Rich, J. R. Goulet et al., “Predicting Mortality in Systemic Sclerosis: Analysis of a Cohort of 309 French Canadian Patients with Emphasis on Features at Diagnosis as Predictive Factors for Survival,” Medicine, Vol. 81, No. 2, 2002, pp. 154-167. http://dx.doi.org/10.1097/00005792-200203000-00005
- S. Chatterjee, G. W. Dombi, R. K. Severson and M. D. Mayes, “Risk of Malignancy in Scleroderma: A Population-Based Cohort Study,” Arthritis & Rheumatism, Vol. 52, No. 8, 2005, pp. 2415-2424. http://dx.doi.org/10.1002/art.21225
- K. A. Black, P. J. Zilko, R. L. Dawkins, B. K. Armstrong and G. L. Mastaglia, “Cancer in Connective Tissue Disease,” Arthritis & Rheumatism, Vol. 25, No. 9, 1983, pp. 1130-1133. http://dx.doi.org/10.1002/art.1780250916
- C. T. Derk, M. Rasheed, C. M. Artlett and C. A. Jimenez, “A Cohort Study of Cancer Incidence in Systemic Sclerosis,” Journal of Rheumatology, Vol. 33, No. 6, 2006, pp. 1113-1116.
- A. B. Olesen, C. Svaerke, D. K. Farkas and H. T. Sørensen, “Systemic Sclerosis and the Risk of Cancer: A Nationwide Population-Based Cohort Study,” British Journal of Rheumatology, Vol. 163, No. 4, 2010, pp. 800-806. http://dx.doi.org/10.1111/j.1365-2133.2010.09861.x
- K. Y. Kang, H. W. Yim, I. J. Kim, J. U. Yoon, J. H. Ju, H. Y. Kim et al., “Incidence of Cancer among Patients with Systemic Sclerosis in Korea: Results from a Single Centre,” Scandinavian Journal of Rheumatology, Vol. 38, No. 4, 2009, pp. 299-303. http://dx.doi.org/10.1080/03009740802642062
- A. D. Roumm and T. A. Medsger, “Cancer and Systemic Sclerosis. An Epidemiologic Study,” Arthritis & Rheumatism, Vol. 28, No. 12, 1985, pp. 1336-1340. http://dx.doi.org/10.1002/art.1780281204
- A. K. Rosenthal, J. K. McLaughlin, G. Gridley and O. Nyrén, “Incidence of Cancer among Patients with Systemic Sclerosis,” Cancer, Vol. 76, No. 5, 1995, pp. 910-914. http://dx.doi.org/10.1002/1097-0142(19950901)76:5<910::AID-CNCR2820760528>3.0.CO;2-T
- M. Abu-Shakra, F. Guillemin and P. Lee, “Cancer in Systemic Sclerosis,” Arthritis & Rheumatism, Vol. 36, No. 4, 1993, pp. 460-464. http://dx.doi.org/10.1002/art.1780360405
- C. L. Hill, A. M. Nguyen, D. Roder and P. RobertsThomson, “Risk of Cancer in Patients with Scleroderma: A Population Based Cohort Study,” Annals of the Rheumatic Diseases, Vol. 62, No. 8, 2003, pp. 728-731. http://dx.doi.org/10.1136/ard.62.8.728
- K. Siau, C. J. Laversuch, P. Creamer and K. P. O’Rourke, “Malignancy in Scleroderma Patients from South West England: A Population-Based Cohort Study,” Rheumatology International, Vol. 31, No. 5, 2011, pp. 641-645. http://dx.doi.org/10.1007/s00296-009-1348-y
- Y. Fuse, J. Masuyama, T. Yoshio, A. Mimori, A. Takeda, S. Minota et al., “Progressive Systemic Sclerosis (PSS) and Cancer—Increasing Coincidence Rate of Cancer in 67 PSS Patients,” Ryumachi, Vol. 35, No. 1, 1995, pp. 25-31.
- M. A. Omair, V. Phumethum and S. R. Johnson, “LongTerm Safety and Effectiveness of Tumour Necrosis Factor Inhibitors in Systemic Sclerosis Patients with Inflammatory Arthritis,” Clinical and Experimental Rheumatology, Vol. 30, No. Suppl 71, 2012, pp. S55-S59.
- R. A. Nash, P. A. McSweeney, L. J. Crofford, M. Abidi, C. S. Chen, J. D. Godwin et al., “High-Dose Immunosuppressive Therapy and Autologous Hematopoietic Cell Transplantation for Severe Systemic Sclerosis: LongTerm Follow-Up of the US Multicenter Pilot Study,” Blood, Vol. 110, No. 4, 2007, pp. 1388-1396. http://dx.doi.org/10.1182/blood-2007-02-072389
- M. Bonifazi, I. Tramacere, G. Pomponio, B. Gabrielli, E. V. Avvedimento, C. La Vecchia et al., “Systemic Sclerosis (Scleroderma) and Cancer Risk: Systematic Review and Meta-Analysis of Observational Studies,” Rheumatology (Oxford), Vol. 52, No. 1, 2013, pp. 143-154. http://dx.doi.org/10.1093/rheumatology/kes303
- M. Higuchi, T. Horiuchi, N. Ishibashi, S. Yoshizawa, Y. Niho and K. Nagasawa, “Anticentromere Antibody as a Risk Factor for Cancer in Patients with Systemic Sclerosis,” Clinical Rheumatology, Vol. 19, No. 2, 2000, pp. 123-126. http://dx.doi.org/10.1007/s100670050029
- P. Airo, A. Ceribelli, I. Cavazzana, M. Taraborelli, S. Zingarelli and F. Franceschini, “Malignancies in Italian Patients with Systemic Sclerosis Positive for Anti-RNA Polymerase III Antibodies,” Journal of Rheumatology, Vol. 38, No. 7, 2011, pp. 1329-1334. http://dx.doi.org/10.3899/jrheum.101144
- E. K. Pontifex, C. L. Hill and P. Roberts-Thomson, “Risk Factors for Lung Cancer in Patients with Scleroderma: A Nested Case-Control Study,” Annals of the Rheumatic Diseases, Vol. 66, No. 4, 2007, pp. 551-553. http://dx.doi.org/10.1136/ard.2006.056424
- T. Y. Lu, C. L. Hill, E. K. Pontifex and P. J. RobertsThomson, “Breast Cancer and Systemic Sclerosis: A Clinical Description of 21 Patients in a Population-Based Cohort Study,” Rheumatology International, Vol. 28, No. 9, 2008, pp. 895-899. http://dx.doi.org/10.1007/s00296-008-0540-9
- C. T. Derk, “Associations of Breast Cancer Development in Patients with Systemic Sclerosis: An Exploratory Study,” Clinical Rheumatology, Vol. 26, No. 10, 2007, pp. 1615-1619. http://dx.doi.org/10.1007/s10067-007-0546-9
- The PRISMA Statement. http://www.prisma-statement.org
- A. K. Rosenthal, J. K. McLaughlin, M. S. Linet and I. Persson, “Scleroderma and Malignancy: An Epidemiological Study,” Annals of the Rheumatic Diseases, Vol. 52, No. 7, 1993, pp. 531-533. http://dx.doi.org/10.1136/ard.52.7.531
- P. Hissaria, S. Lester, P. Hakendorf, R. Woodman, K. Patterson, C. Hill, et al., “Survival in Scleroderma: Results from the Population-Based South Australian Register,” Internal Medicine Journal, Vol. 41, No. 5, 2011, pp. 381- 390. http://dx.doi.org/10.1111/j.1445-5994.2010.02281.x
- L. Czirják, G. Kumánovics, C. Varjú, Z. Nagy, A. Pákozdi, Z. Szekanecz et al., “Survival and Causes of Death in 366 Hungarian Patients with Systemic Sclerosis,” Annals of the Rheumatic Diseases, Vol. 67, No. 1, 2008, pp. 59- 63. http://dx.doi.org/10.1136/ard.2006.066340
- E. Thomas, D. Symmons, D. Brewster, R. Black, G. Macfarlane, “National Study of Cause-Specific Mortality in Rheumatoid Arthritis, Juvenile Chronic Arthritis, and Other Rheumatic Conditions: A 20-Year-Follow-Up Study,” Journal of Rheumatology, Vol. 30, No. 5, 2003, pp. 958- 965.
- P. G. Vlachoyiannopoulos, U. G. Dafni, I. Pakas, M. Spyropoulou-Vlachou, C. Stavropoulos-Giokas and H. M. Moutsopoulos, “Systemic Scleroderma in Greece: Low Mortality and Strong Linkage with HLADRB1* 1104 Allele,” Annals of the Rheumatic Diseases, Vol. 59, No. 5, 2000, pp. 359-367. http://dx.doi.org/10.1136/ard.59.5.359
- C. T. Derk, C. M. Artlett and S. A. Jimenez, “Morbidity and Mortality of Patients Diagnosed with Systemic Sclerosis after the Age of 75: A Nested Case-Control Study,” Clinical Rheumatology, Vol. 25, No. 6, 2006, pp. 831-834. http://dx.doi.org/10.1007/s10067-005-0177-y
- C. T. Derk, L. I. Sakkas, M. Rasheed, C. Artlett and S. A. Jimenez, “Autoantibodies in Patients with Systemic Sclerosis and Cancer: A Case-Control Study,” Journal of Rheumatology, Vol. 30, No. 9, 2003, pp. 1994-1996.
- C. T. Derk, M. Rasheed, J. R. Spiegel and S. A. Jimenez, “Increased Incidence of Carcinoma of the Tongue in Patients with Systemic Sclerosis,” Journal of Rheumatology, Vol. 32, No. 4, 2005, pp. 637-641.
- E. Szekanecz, S. Szamosi, L. Gergely, P. Keszthelyi, Z. Szekanecz and G. Szucs, “Incidence of Lymphoma in Systemic Sclerosis: A Retrospective Analysis of 218 Hungarian Patients with Systemic Sclerosis,” Clinical Rheumatology, Vol. 27, No. 9, 2008, pp. 1163-1166. http://dx.doi.org/10.1007/s10067-008-0925-x
- D. Launay, R. Le Berre, P. Y. Hatron, J. P. Peyrat, E. Hachulla, B. Devulder and M. Hebbar, “Association between Systemic Sclerosis and Breast Cancer: Eight New Cases and Review of the Literature,” Clinical Rheumatology, Vol. 23, No. 6, 2004, pp. 516-522. http://dx.doi.org/10.1007/s10067-004-0940-5
- M. C. Segel, W. L. Campbell, T. A. Medsger Jr. and A. D. Roumm, “Systemic Sclerosis (Scleroderma) and Esophageal Adenocarcinoma: Is Increased Patient Screening Necessary?” Gastroenterology, Vol. 89, No. 3, 1985, pp. 485-488.
- S. Vettori, S. Staibano, M. Mascolo, G. Ilardi and G. Valentini, “Non-Hodgkin’s Lymphoma in Systemic Sclerosis: Case and Literature Review,” Clinical Rheumatology, Vol. 29, No. 1, 2010, pp. 1-6. http://dx.doi.org/10.1007/s10067-009-1286-9
- E. Retetto, O. Cerrato, N. Spotorno and A. Sorbi, “Clinical Study of the Incidence of Malignant Neoplasms in Some Autoimmune Diseases,” Reumatismo, Vol. 26, No. 2, 1974, pp. 102-108.
- X. Kyndt, M. Hebbar, V. Queyrel, E. Hachulla, P. Y. Hatron and B. Devulder, “Systemic Scleroderma and Cancer. Search for Predictive Factors of Cancer in 123 Patients with Scleroderma,” La Revue de Médecine Intern, Vol. 18, No. 7, 1997, pp. 528-532. http://dx.doi.org/10.1016/S0248-8663(97)80804-1
- J. Kim, S. K. Park, K. W. Moon, E. Y. Lee, J. Y. Lee, Y. W. Song, et al., “The Prognostic Factors of Systemic Sclerosis for Survival among Koreans,” Clinical Rheumatology, Vol. 29, No. 3, 2010, pp. 297-302. http://dx.doi.org/10.1007/s10067-009-1324-7
- F. Sakauchi, M. Mori, O. Ishikawa and H. Endo, “The Association of Systemic Sclerosis with Malignant Neoplasms,” Nihon Rinsho Meneki Gakkai Kaishi (Japanese Journal of Clinical Immunology), Vol. 27, No. 6, 2004, pp. 402-406. http://dx.doi.org/10.2177/jsci.27.402
- M. Peters-Golden, R. A. Wise, M. Hochberg, M. B. Stevens and F. M. Wigley, “Incidence of Lung Cancer in Systemic Sclerosis,” The Journal of Rheumatology, Vol. 12, No. 6, 1985, pp. 1136-1139.
- A. A. Shah, A. Rosen, L. Hummers, F. Wigley and L. Casciola-Rosen, “Close Temporal Relationship between Onset of Cancer and Scleroderma in Patients with RNA Polymerase I/III Antibodies,” Arthritis & Rheumatism, Vol. 62, No. 9, 2010, pp. 2787-2795. http://dx.doi.org/10.1002/art.27549
- M. Nikpour, P. Hissaria, J. Byron, J. Sahhar, M. Micallef, W. Paspaliaris, et al., “Prevalence, Correlates and Clinical Usefulness of Antibodies to RNA Polymerase III in Systemic Sclerosis: A Cross-Sectional Analysis of Data from an Australian Cohort,” Arthritis Research & Therapy, Vol. 13, No. 6, 2011, p. R211. http://dx.doi.org/10.1186/ar3544
- C. F. Kuo, S. F. Luo, K. H. Yu, I. J. Chou, W. Y. Tseng, H. C. Chang, et al., “Cancer Risk among Patients with Systemic Sclerosis: A Nationwide Population Study in Taiwan,” Scandinavian Journal of Rheumatology, Vol. 41, No. 1, 2012, pp. 44-49. http://dx.doi.org/10.3109/03009742.2011.618145
- M. Hudson, A. Sharma, J. Bernstein and M. Baron, Canadian Scleroderma Research Group, “Validity of Self-Reported Comorbidities in Systemic Sclerosis,” Journal of Rheumatology, Vol. 36, No. 7, 2009, pp. 1477-1480. http://dx.doi.org/10.3899/jrheum.081134
- E. Thomas, D. H. Brewster, R. J. Black and G. J. Macfarlane, “Risk of Malignancy among Patients with Rheumatic Conditions,” International Journal of Cancer, Vol. 88, No. 3, 2000 pp. 497-502. http://dx.doi.org/10.1002/1097-0215(20001101)88:3<497::AID-IJC27>3.0.CO;2-J
- J. Wipff, R. Coriat, M. Masciocchi, P. Caramaschi, C. T. Derk, E. Hachulla, et al., “Outcomes of Barrett’s Oesophagus Related to Systemic Sclerosis: A 3-Year EULAR Scleroderma Trials and Research Prospective Follow-Up Study,” Rheumatology (Oxford), Vol. 50, No. 8, 2011, pp. 1440-1444. http://dx.doi.org/10.1093/rheumatology/ker110
- A. R. Villa, A. Kraus, A. Jiménez-Corona, S. Sandino, A. Velázquez-González, J. Granados, et al., “Malignant Neoplasms in Autoimmune Rheumatic Diseases: Examination of the Risk of Developing a Malignancy among Five Different Rheumatic Diseases in One Institution,” Journal of Clinical Rheumatology, Vol. 6, No. 4, 2000, pp. 176-183. http://dx.doi.org/10.1097/00124743-200008000-00002
- C. F. Kuo, L. C. See, K. H. Yu, I. J. Chou, W. Y. Tseng, H. C. Chang, et al., “Epidemiology and Mortality of Systemic Sclerosis: A Nationwide Population Study in Taiwan,” Scandinavian Journal of Rheumatology, Vol. 40, No. 5, 2011, pp. 373-378. http://dx.doi.org/10.3109/03009742.2011.553736
- P. Lee, P. Langevitz, C. A. Alderdice, M. Aubrey, P. A. Baer, M. Baron, et al., “Mortality in Systemic Sclerosis (Scleroderma),” Quarterly Journal of Medicine, Vol. 82, 1992, pp. 139-148.
- A. J. Tyndall, B. Bannert, M. Vonk, P. Airò, F. Cozzi, P. E. Carreira, et al., “Causes and Risk Factors for Death in Systemic Sclerosis: A Study from the EULAR Scleroderma Trials and Research (EUSTAR) Database,” Annals of the Rheumatic Diseases, Vol. 69, No. 10, 2010, pp. 1809-1815. http://dx.doi.org/10.1136/ard.2009.114264
- C. Bond, K. D. Pile, J. D. McNeil, M. J. Ahern, M. D. Smith, L. G. Cleland, et al., “South Australian Scleroderma Register: Analysis of Deceased Patients,” Pathology, Vol. 30, No. 4, 1998, pp. 386-390. http://dx.doi.org/10.1080/00313029800169676
- B. E. Joven, R. Almodovar, L. Carmona and P. E. Carreira, “Survival, Causes of Death, and Risk Factors Associated with Mortality in Spanish Systemic Sclerosis Patients: Results from a Single University Hospital,” Seminars in Arthritis and Rheumatism, Vol. 39, No. 4, 2010, pp. 285-293. http://dx.doi.org/10.1016/j.semarthrit.2009.06.002
- E. Hachulla, P. Carpentier, V. Gressin, E. Diot, Y. Allanore, J. Sibilia, et al., “Risk Factors for Death and the 3-Year Survival of Patients with Systemic Sclerosis: The French ItinérAIR-Sclérodermie Study,” Rheumatology (Oxford), Vol. 48, No. 3, 2009, pp. 304-308. http://dx.doi.org/10.1093/rheumatology/ken488
- L. Chung, E. Krishnan and E. F. Chakravarty, “Hospitalizations and Mortality in Systemic Sclerosis: Results from the Nationwide Inpatient Sample,” Rheumatology, Vol. 46, No. 12, 2007, pp. 1808-1813. http://dx.doi.org/10.1093/rheumatology/kem273
- S. Chwiesko, S. Sierakowski and O. Kowal-Bielecka, “Causes of Death of Patients with Systemic Sclerosis,” Polski Merkuriusz Lekarski, Vol. 21, No. 124, 2006, pp. 341-344.
- S. Vettori, G. Cuomo, G. Abignano, M. Iudici and G. Valentini, “Survival and Death Causes in 251 Systemic Sclerosis Patients from a Single Italian Center,” Reumatismo, Vol. 62, No. 3, 2010, pp. 202-209.
- C. C. Mok, C. L. Kwok, L. Y. Ho, P. T. Chan and S. F. Yip, “Life Expectancy, Standardized Mortality Ratios, and Causes of Death in Six Rheumatic Diseases in Hong Kong, China,” Arthritis & Rheumatism, Vol. 63, No. 5, 2011, pp. 1182-1189. http://dx.doi.org/10.1002/art.30277
- A. Hashimoto, S. Tejima, T. Tono, M. Suzuki, S. Tanaka, T. Matsui, et al., “Predictors of Survival and Causes of Death in Japanese Patients with Systemic Sclerosis,” The Journal of Rheumatology, Vol. 38, No. 9, 2011, pp. 1931-1939. http://dx.doi.org/10.3899/jrheum.100298
- K. Nishioka, I. Katayama, H. Kondo, H. Shinkai, H. Ueki, K. Tamaki, et al., “Epidemiological Analysis of Prognosis of 496 Japanese Patients with Progressive Systemic Sclerosis (SSc). Scleroderma Research Committee Japan,” Journal of Dermatology, Vol. 23, No. 10, 1996, pp. 677-682.
- V. D. Steen and T. A. Medsger, “Changes in Causes of Death in Systemic Sclerosis, 1972-2002,” Annals of the Rheumatic Diseases, Vol. 66, No. 7, 2007, pp. 940-944. http://dx.doi.org/10.1136/ard.2006.066068
- C. Ferri, G. Valentini, F. Cozzi, M. Sebastiani, C. Michelassi, G. La Montagna, et al., “Systemic Sclerosis: Demographic, Clinical, and Serologic Features and Survival in 1012 Italian Patients,” Medicine, Vol. 81, No. 2, 2002, pp. 139-153. http://dx.doi.org/10.1097/00005792-200203000-00004
- S. M. Gadalla, S. Amr, P. Langenberg, M. Baumgarten, W. F. Davidson, C. Schairer, et al., “Breast Cancer Risk in Elderly Women with Systemic Autoimmune Rheumatic Diseases: A Population-Based Casecontrol Study,” British Journal of Cancer, Vol. 100, No. 5, 2009, pp. 817-821. http://dx.doi.org/10.1038/sj.bjc.6604906
- L. M. Brown, G. Gridley, D. Check and O. Landgren, “Risk of Multiple Myeloma and Monoclonal Gammopathy of Undetermined Significance among White and Black Male United States Veterans with Prior Autoimmune, Infectious, Inflammatory, and Allergic Disorders,” Blood, Vol. 111, No. 7, 2008, pp. 3388-3394. http://dx.doi.org/10.1182/blood-2007-10-121285
- A. M. Landgren, O. Landgren, G. Gridley, G. M. Dores, M. S. Linet and L. M. Morton, “Autoimmune Disease and Subsequent Risk of Developing Alimentary Tract Cancers among 4.5 Million US Male Veterans,” Cancer, Vol. 117, No. 6, 2011, pp. 1163-1171. http://dx.doi.org/10.1002/cncr.25524
- L. A. Anderson and E. A. Engels, “Autoimmune Conditions and Hairy Cell Leukemia: An Exploratory Casecontrol Study,” Journal of Hematology and Oncology, Vol. 3, 2010, p. 35. http://dx.doi.org/10.1186/1756-8722-3-35
- L. A. Anderson, S. Gadalla, L. M. Morton, O. Landgren, R. Pfeiffer, J. L. Warren, et al., “Population-Based Study of Autoimmune Conditions and the Risk of Specific Lymphoid Malignancies,” International Journal of Cancer, Vol. 125, No. 2, 2009, pp. 398-405. http://dx.doi.org/10.1002/ijc.24287
- S. Y. Kristinsson, J. Koshiol, M. Björkholm, L. R. Goldin, M. L. McMaster, I. Turesson, et al., “Immunerelated and Inflammatory Conditions and Risk of Lymphoplasmacytic Lymphoma or Waldenstrom Macroglobulinemia,” Journal of the National Cancer Institute, Vol. 102, No. 8, 2010, pp. 557-567. http://dx.doi.org/10.1093/jnci/djq043
- L. Mellemkjaer, R. M. Pfeiffer, E. A. Engels, G. Gridley, W. Wheeler, K. Hemminki, et al., “Autoimmune Disease in Individuals and Close Family Members and Susceptibility to Non-Hodgkin’s Lymphoma,” Arthritis & Rheumatism, Vol. 58, No. 3, 2008, pp. 657-666. http://dx.doi.org/10.1002/art.23267
- National Cancer Institute. Detroit Registry. Metropolitan Detroit Cancer Surveillance System. http://seer.cancer.gov./statistics
- V. Artinian and P. A. Kvale, “Cancer and Interstitial Lung Disease,” Current Opinion in Pulmonary Medicine, Vol. 10, No. 5, 2004, pp. 425-434. http://dx.doi.org/10.1097/00063198-200409000-00017
- G. Rittner, G. Schwanitz, M. P. Baur, C. M. Black, K. I. Welsh, P. Kühnl, et al., “Family Studies in Scleroderma (Systemic Sclerosis) Demonstrating an HLA-Linked Increased Chromosomal Breakage Rate in Cultured Lymphocytes,” Human Genetics, Vol. 81, No. 1, 1988, pp. 64- 70. http://dx.doi.org/10.1007/BF00283732
- C. M. Artlett, C. M. Black, D. C. Briggs, C. Stephens and K. I. Welsh, “DNA Allelic Alterations within VNTR Loci of Scleroderma Families,” British Journal of Rheumatology, Vol. 35, No. 12, 1996, pp. 1216-1222. http://dx.doi.org/10.1093/rheumatology/35.12.1216
- L. I. Sakkas, D. F. Moore and N. C. Akritidis, “Cancer in Families with Systemic Sclerosis,” American Journal of the Medical Sciences, Vol. 310, No. 6, 1995, pp. 223-225.
- M. Zuber, “Positive Antinuclear Antibodies in Malignancies,” Annals of the Rheumatic Diseases, Vol. 51, No. 4, 1992, pp. 573-574. http://dx.doi.org/10.1136/ard.51.4.573-b
- F. F. Madrid and M. Maroun, “Serologic Laboratory Findings in Malignancy,” Rheumatic Disease Clinics of North America, Vol. 37, No. 4, 2011, pp. 507-525. http://dx.doi.org/10.1016/j.rdc.2011.09.006
NOTES
*Sources of support: Dr. Johnson has been awarded a Canadian Institutes of Health Research Clinician Scientist Award. This study was supported by the Norton Evans Fund for Scleroderma Research and the Freda Fejer Fund.
Competing interests: None
#Corresponding author.