Open Journal of Biophysics
Vol.3 No.4(2013), Article ID:38468,6 pages DOI:10.4236/ojbiphy.2013.34034
Binding of Quinoline-Based Inhibitors to Plasmodium falciparum Lactate Dehydrogenase: A Molecular Docking Study
1School of Natural Sciences, Indiana University Southeast, New Albany, USA
2Department of Chemistry, Purdue University, West Lafayette, USA
3Department of Chemistry & Biochemistry, University of Notre Dame, Notre Dame, USA
Email: *vwaingeh@ius.edu
Copyright © 2013 Victor F. Waingeh et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received August 2, 2013; revised September 9, 2013; accepted October 5, 2013
Keywords: Quinoline-Based Inhibitors; Molecular Docking; Plasmodium falciparum
ABSTRACT
Development of new antimalarial drugs continues to be of great importance due to the resistance of the malaria parasite to currently used drugs. Glycolytic enzymes have emerged as potential targets for the development of new drugs due to the reliance of the parasite on glycolysis for energy. In this study, molecular docking was used to study the binding of some quinoline-based drugs to the glycolytic enzyme lactate dehydrogenase. The docking studies identified two potential binding sites for each ligand, one of them being the cofactor-binding site. For all ligands studied, there was the comparable binding to the cofactor-binding site as well as the secondary binding site when the cofactor was absent. All ligands showed significantly lower binding affinity than NADH for the cofactor binding site. The alternative site was the site of preference when docking was done in the presence of the cofactor. While binding to the cofactor site may support other studies suggesting potential for competitive inhibition, the fact that the binding affinities of all the ligands are significantly lower than that for NADH in this site suggests that these ligands will be ineffective competitive inhibitors. The identification of an alternative binding site with comparable affinity that is not affected by the presence of the cofactor may suggest the possibility of non-competitive inhibition that requires further exploration.
1. Introduction
Malaria is a major infectious disease that kills millions of people worldwide yearly, with the majority of fatalities occurring in poor countries. Plasmodium falciparum (P. falciparum) is the most virulent of the malaria parasites and its resistance to currently available drugs continues to grow, and presents an impediment to attempts to successfully deal with the disease. There is, therefore, great need and challenge to continuously develop new inhibitors, with the goal to overcome parasite resistance. Chloroquine and other quinoline-based compounds such as quinine, mefloquine and amodiaquine (Figure 1) have been used for the treatment of malaria incidents for a long time. However, the mechanism by which these compounds exert their anti-malarial properties is still not fully evident. One suggested mechanism is the formation of a complex with heme within the food vacuole that inhibits hematin polymerization [1,2]. Growing and spreading resistance to these current drugs is of concern and emphasizes the need for continuous work aiming at developing new and better anti-malarials.
Resistance is thought to be a result of mutations in the active sites of the drug target [2]. Therefore, by identifying the main targets of the drugs and fully understanding the mechanism of action with respect to these targets, new and better antimalarials may be developed with a goal to overcome drug resistance. Amongst the emerging targets for antimalarial drug developments, are the enzymes of the glycolytic pathway due to the exclusive dependence of the parasite on glycolysis for energy. One of the important glycolytic enzymes is lactate dehydrogenase (LDH), which is involved in the final step of glycolysis and catalyzes the interconversion of pyruvate to lactate. This step of glycolysis is also important because it regenerates NAD+ which is needed by glyceraldehyde- 3-phosphate dehydrogenase, another glycolytic enzyme.
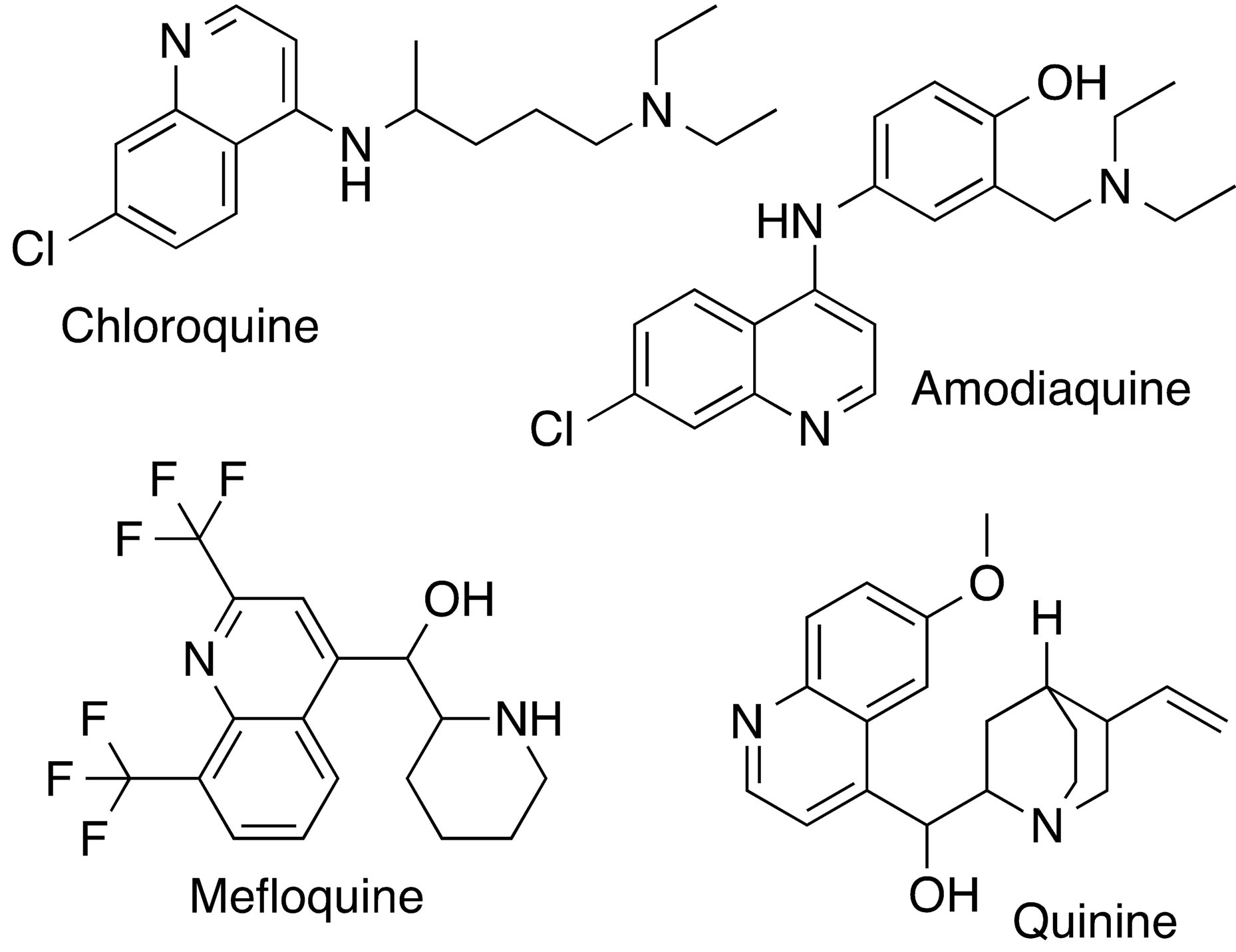
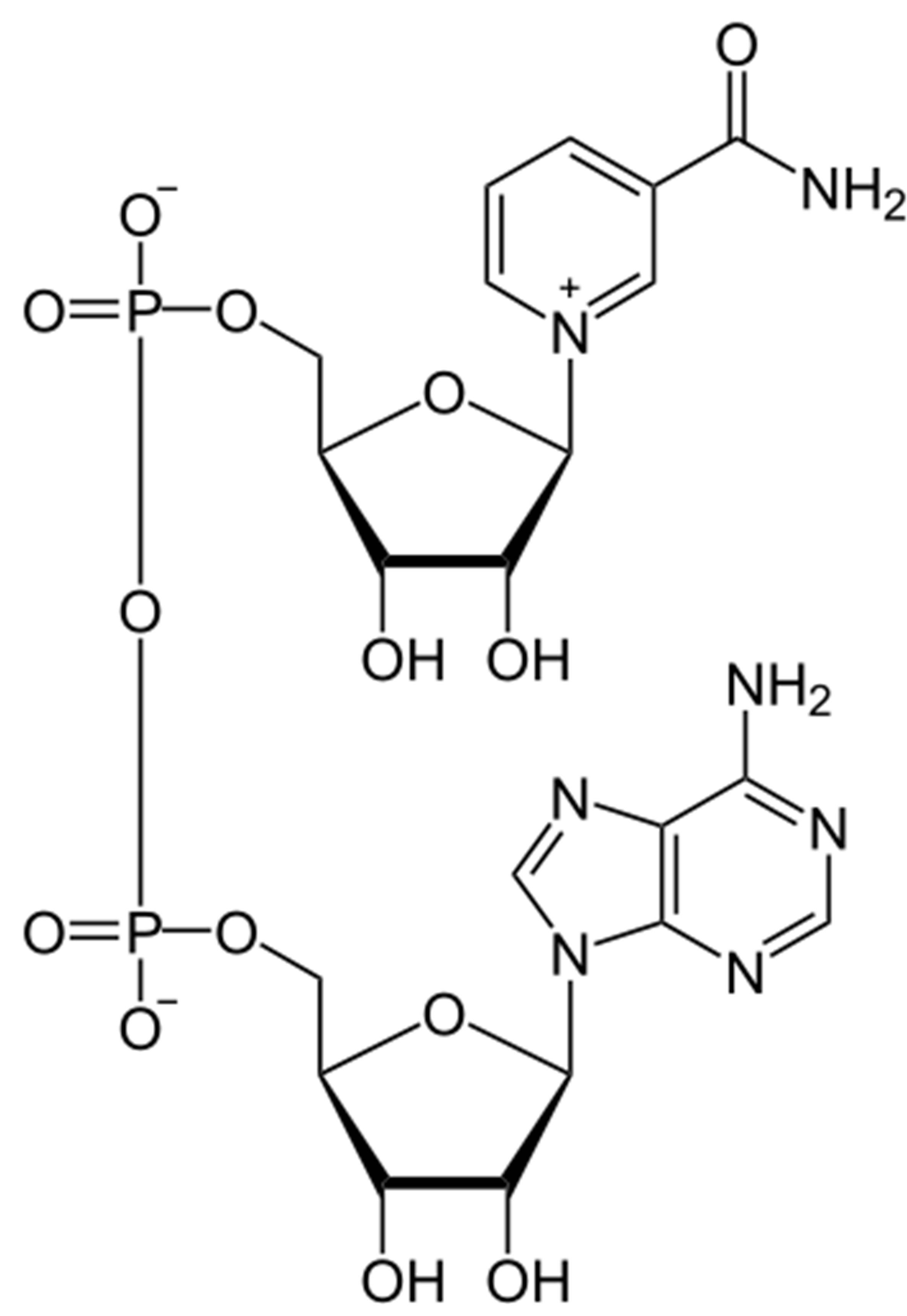
Figure 1. Structures of quinoline-based antimalarial drugs used in this docking study, together with the structure of the enzyme cofactor, NADH.
Inhibitors with anti-malarial activity have been shown to bind to P. falciparum lactate dehydrogenase (pfLDH) [3-7]. Chloroquine binding in or near the cofactor binding site of pfLDH suggests that chloroquine acts as a competitive inhibitor for this enzyme [5]. Similarly, gossypol, which has been shown to exhibit significant antimalarial activity, seems to selectively bind to pfLDH compared to human LDH [8]. However, the exact effect of these interactions or their role in the antimalarial activity of these compounds is still not fully understood.
In this study, the binding of quinoline-based ligands to pfLDH is investigated by molecular docking, with the goal to map potential binding sites and determine the most favorable binding conformations.
2. Methods
2.1. Protein Structure and Setup
For this study, the crystal structure of pfLDH monomer in complex with its cofactor NADH and oxamate was obtained from the Protein Data Bank (PDB entry code 1LDG) [4]. Although the biological functional unit of the enzyme is a tetramer of four identical units, the study was performed using only a single subunit. Accelrys Discovery Studio (DS) Visualizer 2.5 was used to edit the protein structure to remove water molecules together with bound ligands. For docking studies in the absence of cofactor, the bound NADH was also removed.
2.2. Ligand
Ligands used in this study are quinolone-based inhibitors that have been used in the treatment of malaria and include amodiaquine (adq) mefloquine (mfq), quinine (qnn) and chloroquine (clq). Figure 1 shows the structures of the ligands used in this study, including the cofactor NADH for comparison. The ligand structure coordinates were obtained from the Drug Bank as structure data files [9]. DS visualizer was used to rewrite the data files into pdb format. AutodockTools [10] was used to add full hydrogens to the ligands, compute Gasteiger partial atomic charges and save the resulting structure in the required format for use with AutoDock. All possible flexible torsions of the ligand molecules were defined using AUTOTUTORS in AutoDockTools [10,11].
2.3. Docking
Docking simulations were performed with AutoDock 4.2 [10], using the Lamarkian genetic algorithm and default procedures for docking a flexible ligand to a rigid protein were followed. First, blind docking was performed to identify all potential binding sites on the target protein. For this purpose, docking was done on 90 × 90 × 90 cubic grid centered on the protein and enclosing the entire protein. A grid spacing of 1.00 Å and a distance-dependent dielectric constant were used for the calculation of the grid maps using the autogrid module of AutodockTools. For each ligand, two sets of 100 independent runs with 2.5 × 108 energy evaluations steps were performed, one with the enzyme cofactor present and another in the absence of the cofactor.
Once potential binding sites were identified, docking of ligands to these sites was carried out to determine the most probable and most energetically favorable binding conformations. For this more rigorous docking involving a smaller search space limited to the identified binding site, AutodockVina [12] was used. AutodockVina has been shown to significantly improve the accuracy of predicted binding modes compared to Autodock 4 [12]. 10 independent runs were carried out per ligand per binding site, with an exhaustiveness of 100 and an energy range of 3. Docking solutions were analyzed and ranked on the basis of the Vina scoring function. To compliment the Vina results and selected the best representative complexes, additional analyses of docked solutions were done using NNScore 2.0 [13,14].
All calculations were carried out on PC-based machines running red hat Linux 5.0, ×86 operating systems. Resulting structures were visualized and analyzed using a combination of visualization programs including DS visualizer and VMD.
3. Results and Discussion
3.1. Identification of Binding Sites
All four ligands were successfully docked to pfLDH. Docking solutions revealed two major binding sites on the enzymes (Figure 2), one of which was the cofactorbinding site (Site 1).
Site 1, the NADH-binding site, is a binding pocket on the N-terminal end of the enzyme constituted predominantly by the amino acid residues: GLY 27, SER 28, GLY 29, PHE 52, ASP 53, ILE 54, THR 97, ALA 98, GLY 99, PHE 100, THR 139, and ASN 140. These aminoacid residues form a binding pocket that begins on the close the surface (the adenine end of cofactor site) and extends deep into the enzyme (the nicotinamide end of cofactor site). Site 2, is constituted by amino acid residues on the C-terminal end of the enzyme including LYS 198, MET 199, VAL 200, LEU 201, GLU 226, PHE 229, ASP 230, VAL 233, LYS 314, AND GLU 317. These residues form a binding groove near the surface of the enzyme and are part of the substrate binding domain. This binding groove lies on the backside of the substrate active site which lies about 10 Å deep within the enzyme and adjacent to the nicotinamide end of the cofactor site [15].
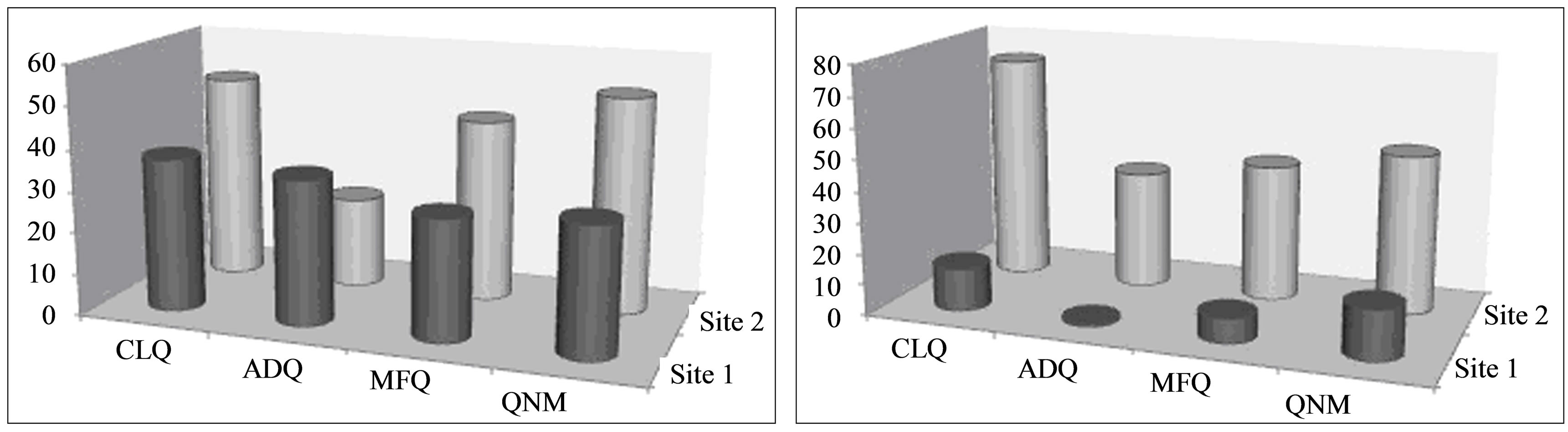
The distribution of docked conformations to the two binding sites was affected by the presence of the cofactor (Figure 3). In the absence of the cofactor, there was comparable distribution of stable docked conformations to both binding sites. However, in the presence of the cofactor, the secondary site (Site 2) was the preferred binding site for all the ligands studied.
3.2. Favorable Docked Conformations
For all ligands studied, there was comparable binding within the cofactor-binding site and the secondary site when the cofactor was absent (Figure 4). When the cofactor-binding site was occupied during docking, the best-docked solutions for each ligand occurred in Site 2 (Figure 5). The best-docked conformation in each binding site was selected based on the binding energies calculated using the AutoDock scoring function as well as
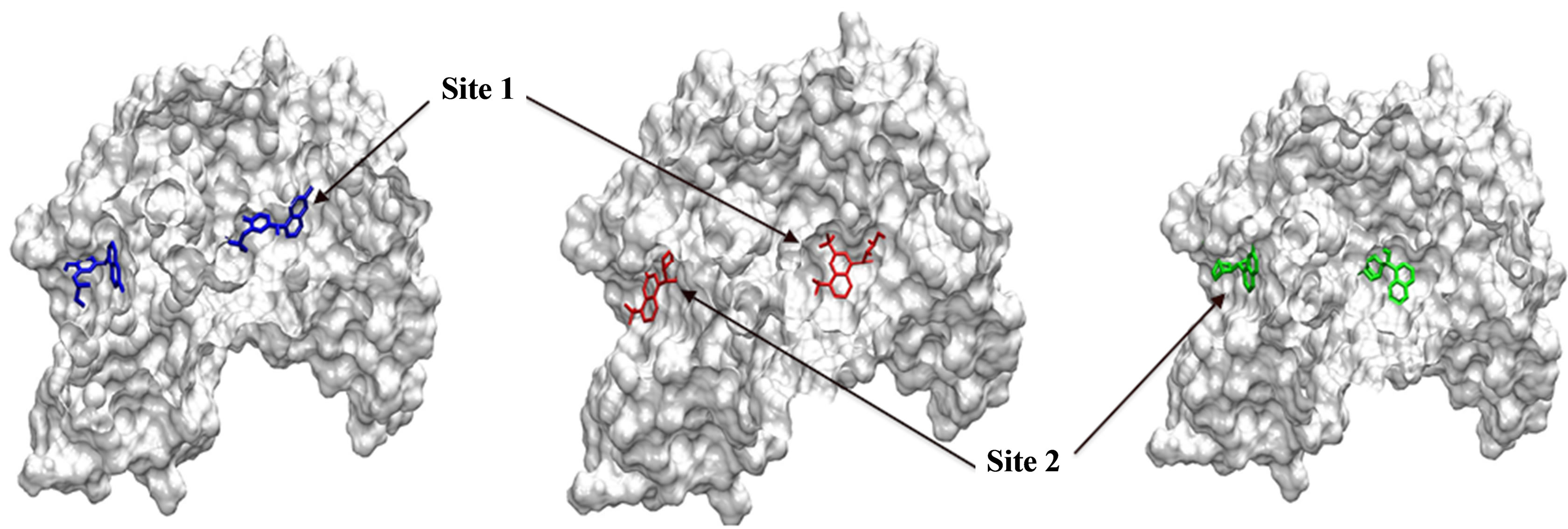
Figure 2. Most probable binding sites identified during docking. Site 1 refers to the cofactor-binding site and Site 2 refers to the secondary binding site.
 (a) (b)
(a) (b)
Figure 3. Distribution of docked conformations to identified binding sites in the absence (a) and presence (b) of NADH cofactor.
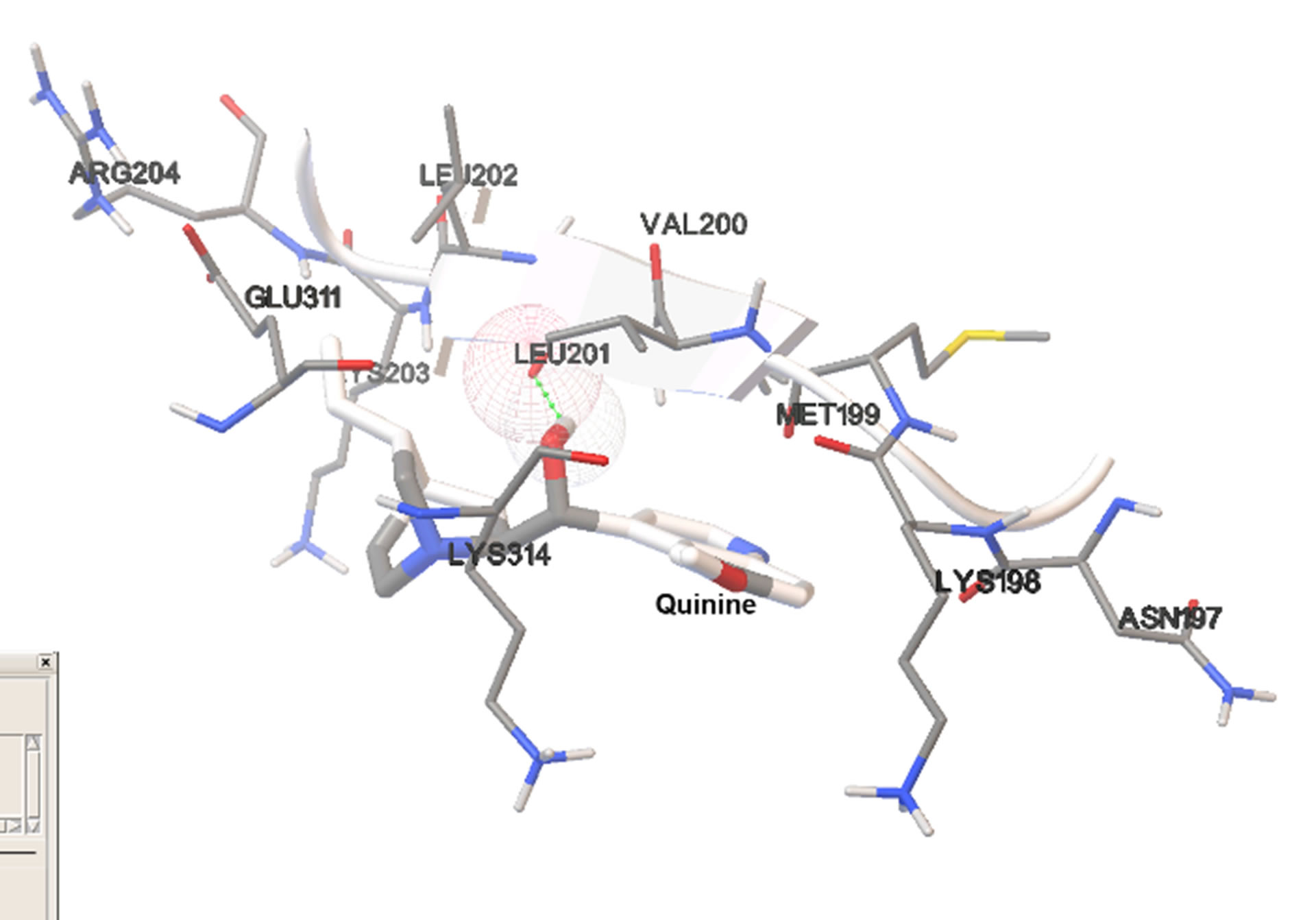
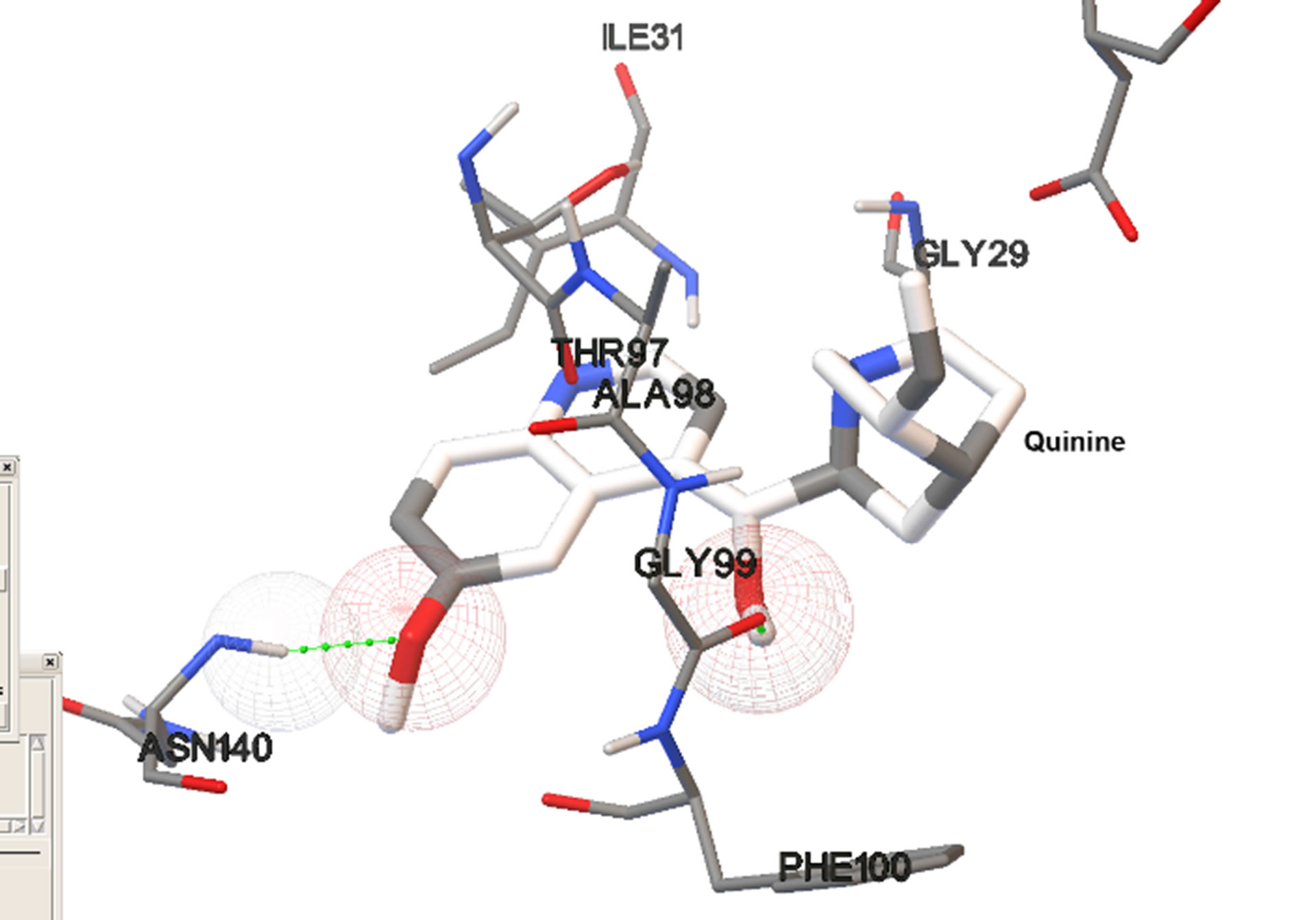
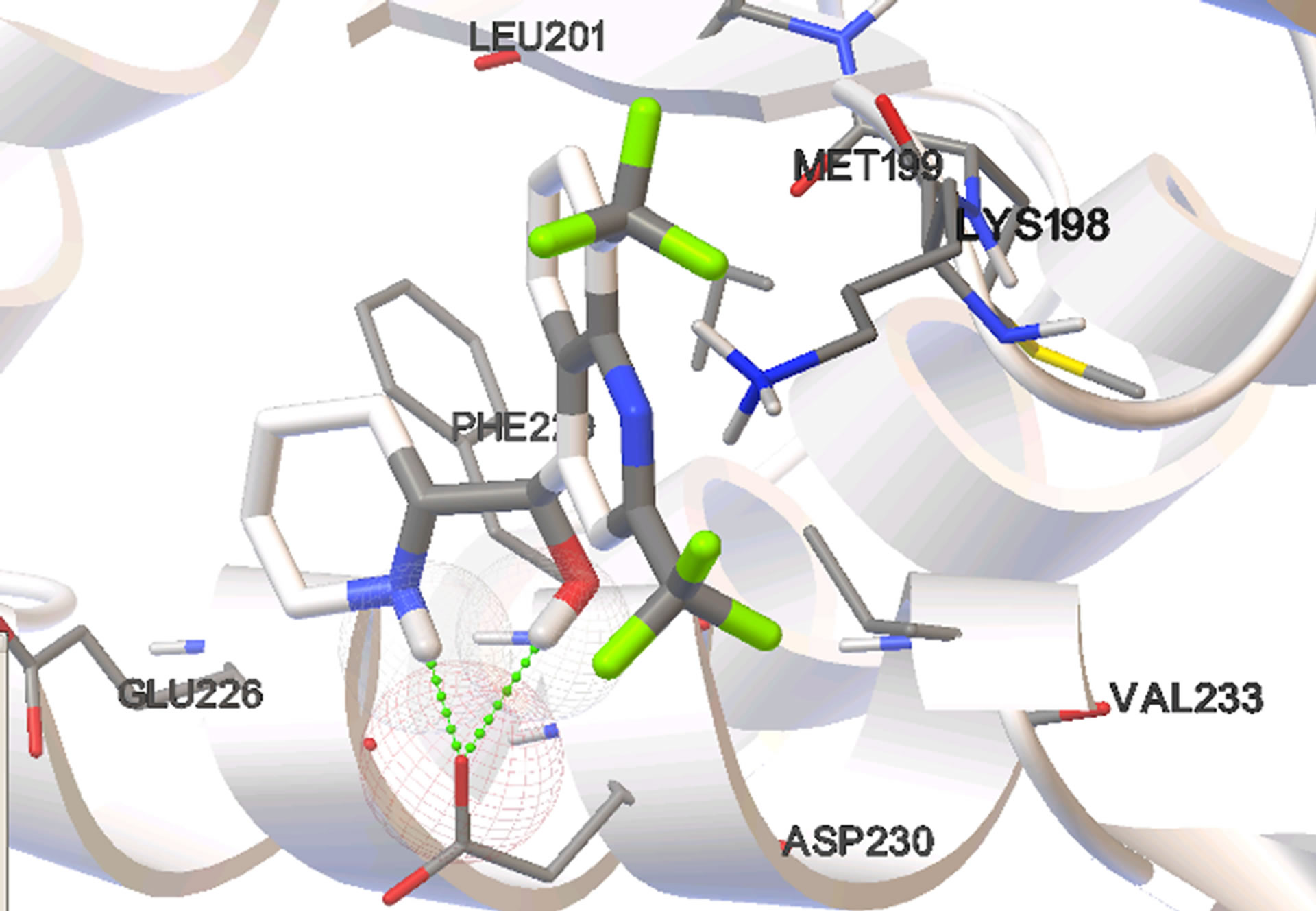
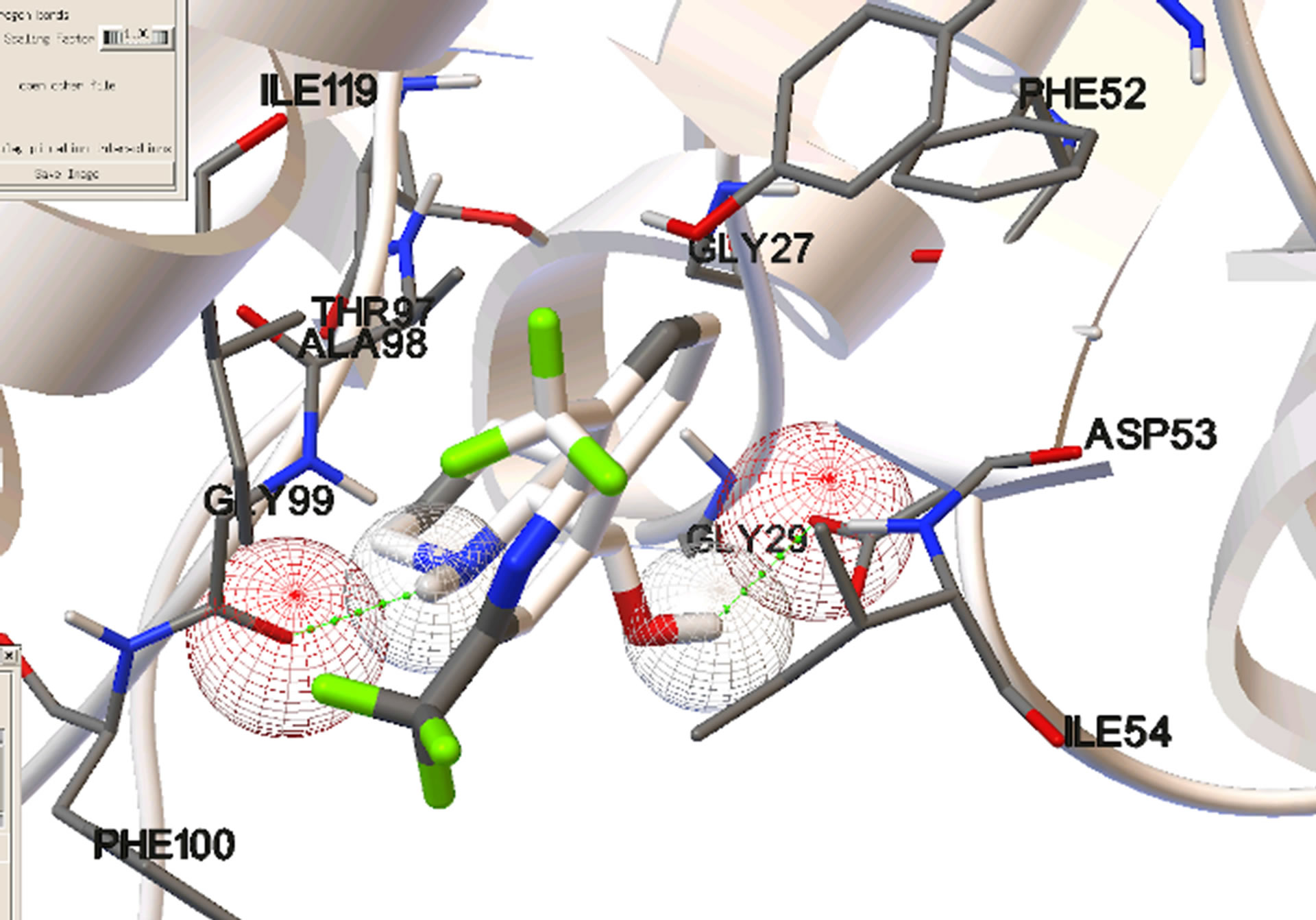
Figure 4. Most stable bound conformations in the binding sites when docking it carried out in the absence of cofactor.
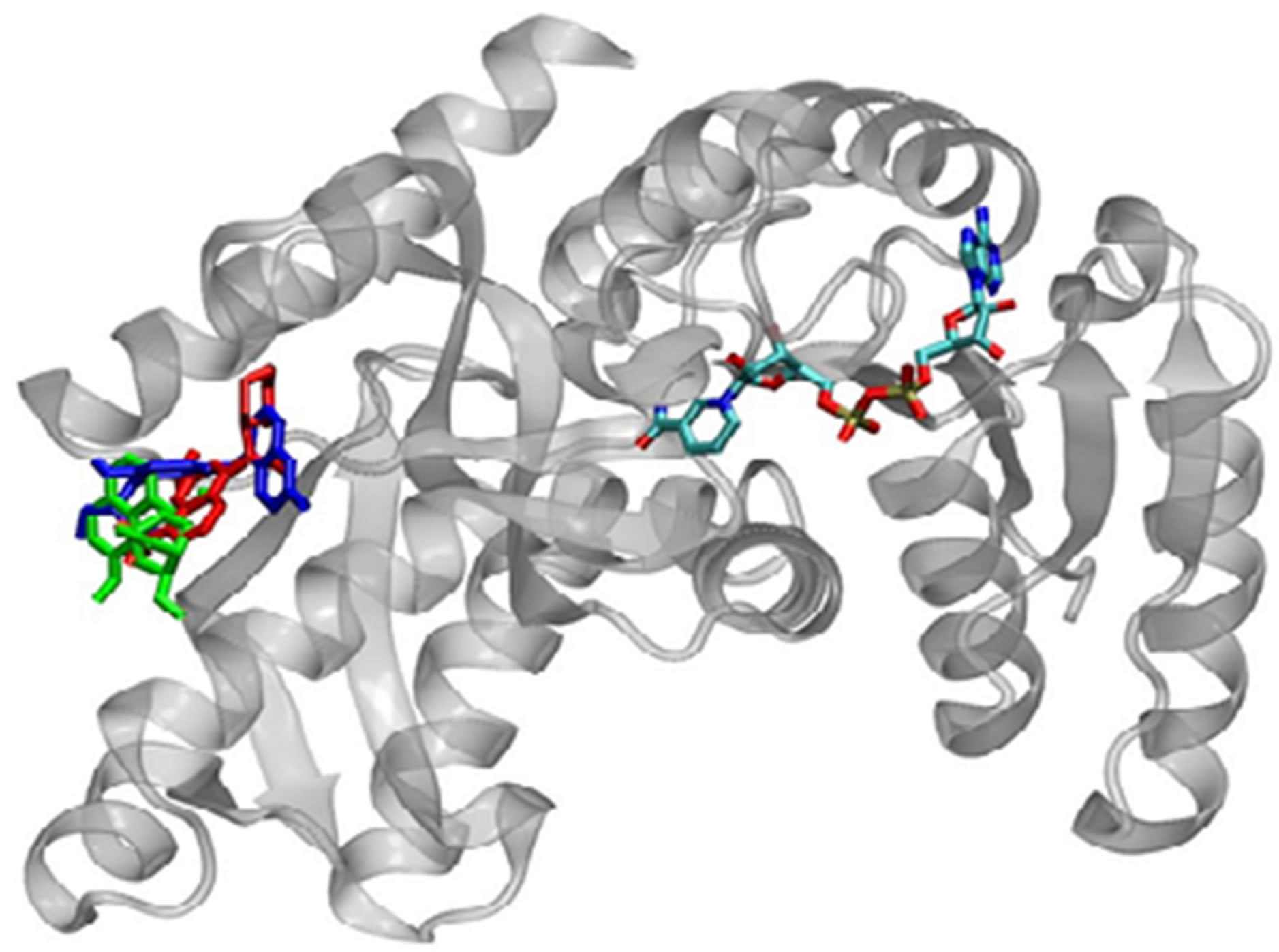
Figure 5. Most stable bound conformations in the binding sites when docking is carried out in the presence of the cofactor.
NNScore ranking. The binding energies for the representative structures as calculated by AutoDock are given in Table 1.
All ligands show lower affinity than NADH for the cofactor binding site. This is attributed the significantly fewer specific interactions between the ligands and the nearby amino acid residues. NADH forms a significantly
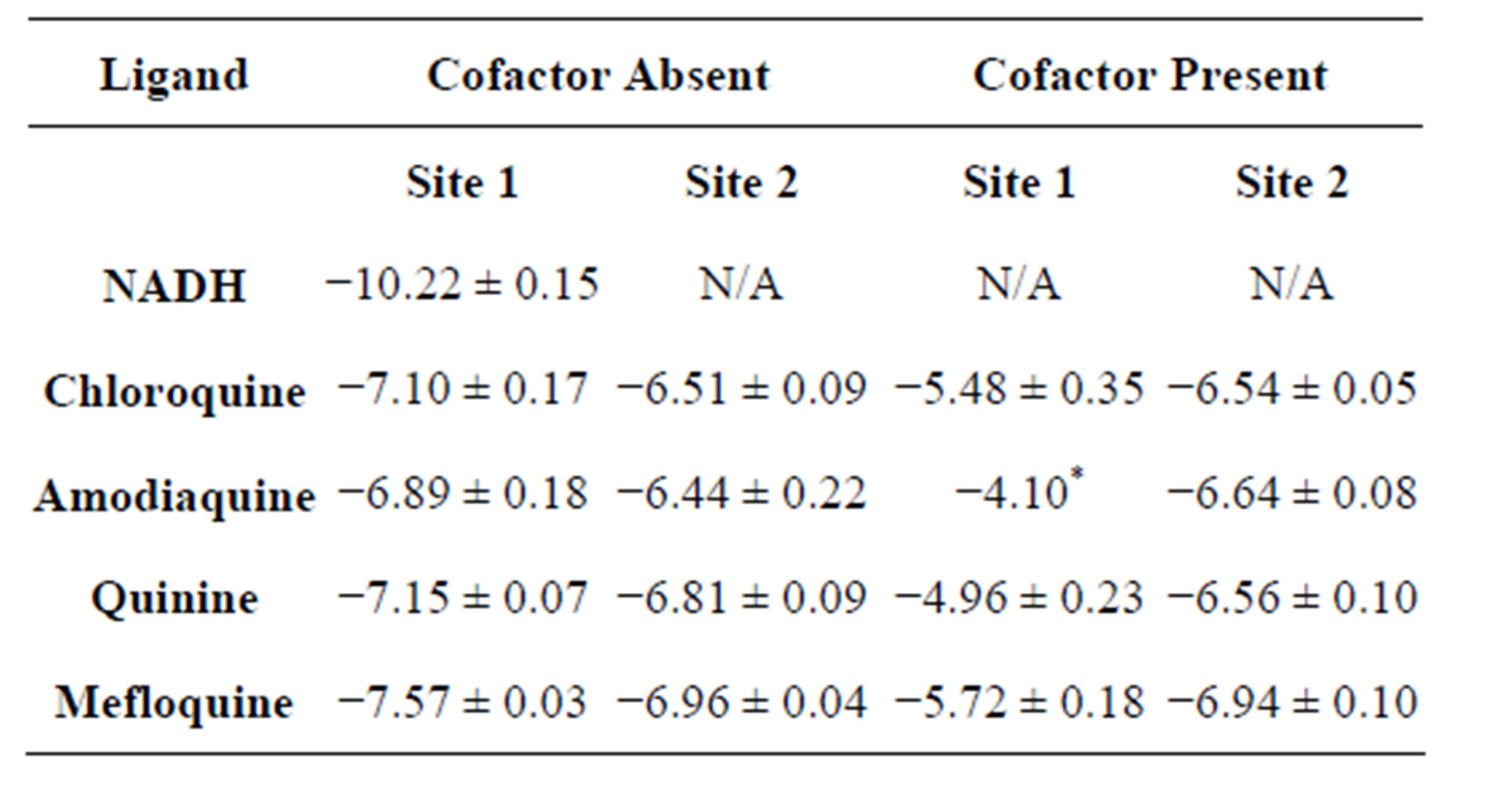
Table 1. Average binding energies (in kcal/mol) of most favorable docked conformations based on AutoDock scoring.
higher number of hydrogen bonds with neighboring residues than any of the compounds studied. The presence of the cofactor significantly diminishes the ability of the ligands to bind to the cofactor binding site both in terms of probability and affinity (Figure 3, Table 1). In the absence of the cofactor, ligands generally bind towards the adenine end of the cofactor binding site, which is closer to the surface. Although the ligands have different structures, they show similar binding affinities at the secondary binding site. This is a, somewhat, surprising observation given the significant structural differences between the ligands. This may be attributed to the fact that this binding site is close to the surface of the enzyme and as such, binding to this is site is not significantly influenced by steric considerations. The flexible ligands (as they are treated in this docking study) can rearrange themselves into favorable conformations without steric overlaps that would be encountered if the binding site were deeper within the enzyme structure. A closer look at the stable conformations reveals that they are generally stabilized by 1 - 2 hydrogen bonds with nearby aminoacid residues.
Although binding to the cofactor-binding site indicate possibility for competitive inhibition, all ligands studied showed significantly lower binding affinity than NADH. A closer look at the complexes formed show that NADH forms a significantly higher number of hydrogen bonds with the neighboring amino acid residues than any of the ligands. The presence of bound NADH during docking significantly reduced the frequency and affinity for binding to the cofactor-binding site. This suggests that ligand binding is not strong enough for any significant competitive inhibition. All ligands showed similar binding affinities for both the cofactor site and the secondary site. This may be significant because this alternative binding site includes residues that form part of the substrate binding domain, and may interfere with substrate binding or catalytic activity of the enzyme. The substrate-binding domain is an alpha/beta structure consisting of residues 163 - 247 and 267 - 331, and located adjacent to the nicotinamide end of the cofactor-binding domain [15]. The amino acid residues involved in binding of the ligands within the secondary binding site are part of this domain. It has been suggested that one of the major substrate binding pathways in LDH catalysis involves the closure of the so-called mobile loop, consisting of 10 surface-residues 98 - 110 [16]. Since the enzyme active site is buried deep, dynamic fluctuations are necessary to facilitate substrate access to the active site [17]. In LDH, the mobile loop is in open conformation prior to ligand binding and closes over the active site after the substrate binds. This involves a transient structural change of 10% - 15% within the substrate-binding domain, which is necessary to bring catalytically necessary aminoacid residues into close proximity with the bound substrate [16]. The quinolone-based ligands in this study do not bind exactly at the active site and, thus, may not be considered competitive inhibitors of the substrate. However, their binding to the secondary binding site, identified in this study, may interfere with the structural changes that should accompany mobile-loop motion and are critical for substrate binding. Formation of significant interactions with residues within the substrate-binding domain and close to the active site such as Lys198, MET199, LEU201, may inhibit the loop movements. The secondary binding site may, therefore, act as an allosteric site, suggesting the possibility of some sort of allosteric inhibition of catalytic activity.
4. Conclusion
In this study, molecular docking was used to study the binding of four quinolone-based ligands to Plasmodium falciparum lactate dehydrogenase. Results show that the molecules were successfully docked to the enzyme and that there are two potential binding sites for these antimalarial drugs on the enzyme, with the cofactor-binding site as one of them. The binding of these ligands to the cofactor-binding pocket of pfLDH suggests that all of these molecules may be potentially competitive inhibitors of the cofactor, NADH as suggested that other studies involving chloroquine [5]. However, any potential inhibition may be ineffective since all ligands show lower binding affinities when compared to NADH. Molecular docking also identifies an alternative binding site with comparable binding affinity to the cofactor site. The fact that these molecules can bind to this site with comparable binding affinities, even in the presence of the cofactor, suggests the possibility of noncompetitive or uncompetitive inhibition. This binding site includes residues that make up the substrate-binding domain and may suggest potential interference with substrate binding or catalytic activity by inhibition of conformational changes that are required for substrate access to the active site, and catalysis. Further investigation, including experimental kinetic studies will be necessary to determine the inhibitory effect, if any, associated with this mode of binding.
5. Acknowledgements
This work was supported by the Indiana University Southeast office of the Dean of Research, with a Summer Faculty Research Fellowship and Undergraduate Research Assistant Grants to support undergraduates working on the project.
REFERENCES
- A. Yayon, Z. I. Chabantchik and H. Ginsburg, “Identification of the Acidic Compartment of Plasmodium falciparum-Infected Human Erythrocytes as the Target of the Antimalarial Drug Chloroquine,” EMBO Journal, Vol. 3, 1984, pp. 2695-2700.
- J. Vennerstrom, E. Nuzum, R. Miller, A. Dorn and L. Gerena, “8-Aminoquinolines Active against Blood Stage Plasmodium falciparum in Vitro Inhibit Hematin Polymerization,” Antimicrobial Agents and Chemotherapy, Vol. 43, No. 3, 1999, pp. 598-602.
- L. M. Deck, R. E. Royer, B. B. Chamblee, V. M. Hernandez, R. R. Malone, J. E. Torres, L. A. Hunsaker, R. C. Piper, M. T. Makler and D. L. Vander Jagt, “Selective Inhibitors of Human Lactate Dehydrogenases and Lactate Dehydrogenase from the Malarial Parasite Plasmodium falciparum,” Journal of Medicinal Chemistry, Vol. 41, No. 20, 1998, pp. 3879-3887. http://dx.doi.org/10.1021/jm980334n
- C. R. Dunn, M. J. Banfield, J. J. Barker, C. W. Higham, K. M. Moreton, D. Turgut-Balik, R. L. Brady and J. J. Holbrook, “The Structure of Lactate Dehydrogenase from Plasmodium falciparum Reveals a New Target for AntiMalarial Design,” Nature Structural & Molecular Biology, Vol. 3, No. 11, 1996, pp. 912-915. http://dx.doi.org/10.1038/nsb1196-912
- J. A. Read, K. W. Wilkinson, R. Tranter, R. B. Sessions and R. L. Brady, “Chloroquine Binds in the Cofactor Binding Site of Plasmodium falciparum Lactate Dehydrogenase,” The Journal of Biological Chemistry, Vol. 274, No. 15, 1999, pp. 10213-10218. http://dx.doi.org/10.1074/jbc.274.15.10213
- T. Egan and K. Ncokazi, “Quinoline Antimalarials Decrease the Rate of Beta-Hematin Formation,” Journal of Inorganic Biochemistry, Vol. 99, No. 7, 2005, pp. 1532- 1539. http://dx.doi.org/10.1016/j.jinorgbio.2005.04.013
- J. Menting, L. Tilley, L.W. Deady, K. Ng and R. Simpson, “The Antimalarial Drug, Chloroquine Interacts with Lactate Dehydrogenase from Plasmodium falciparum,” Molecular and Biochemical Parasitology, Vol. 88, No. 1-2, 1997, pp. 215-224. http://dx.doi.org/10.1016/S0166-6851(97)00095-9
- A. Cameron, J. Read, R. Tranter, V. J. Winter, R. B. Sessions, R. L. Brady, L. Vivas, A. Easton, H. Kendrick, S. L. Croft, D. Barros, J. L. Lavandera, J. J. Martin, F. Risco, S. Garcia-Ochoa, F. J. Gamo, L. Sanz, L. Leon, J. R. Ruiz, R. Gabarro, A. Mallo and F. Gomez de las Heras, “Identification and Activity of a Series of Azole-Based Compounds with Lactate Dehydrogenase-Directed Anti-Malarial Activity,” The Journal of Biological Chemistry, Vol. 279, No. 30, 2004, pp. 31429-31439. http://dx.doi.org/10.1074/jbc.M402433200
- C. Knox, V. Law, T. Jewison, P. Liu, S. Ly, A. Frolkis, A. Pon, K. Banco, C. Mak, V. Neveu, Y. Djoumbou, R. Eisner, A. C. Guo and D. S. Wishart, “DrugBank 3.0: A Comprehensive Resource for ‘Omics’ Research on Drugs,” Nucleic Acids Research, Vol. 39, Suppl. 1, 2011, pp. D1035-D1041. http://dx.doi.org/10.1093/nar/gkq1126
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodshell and A. J. Olson, “Autodock4 and AutodockTools4: Automated Docking with Selective Receptor Flexibility,” Journal of Computational Chemistry, Vol. 30, No. 16, 2009, pp. 2785-2791. http://dx.doi.org/10.1002/jcc.21256
- G. M. Morris, D. S. Goodshell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, “Automated Docking Using a Lamarckian Genetic Algorithm and Empirical Binding Free Energy Function,” Journal of Computational Chemistry, Vol. 19, No. 14, 1998, pp. 1639- 1662. http://dx.doi.org/10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B
- O. Trott and A. J. Olson, “AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading,” Journal of Computational Chemistry, Vol. 31, No. 2, 2010, pp. 455-461.
- J. D. Durrant and J. A. McCammon, “NNScore: A Neural-Network-Based Scoring Function for the Characterization of Protein-Ligand Complexes,” Journal of Chemical Information and Modeling, Vol. 50, No. 10, 2010, pp. 1865-1871. http://dx.doi.org/10.1021/ci100244v
- J. D. Durrant and J. A. McCammon, “NNScore 2.0: A Neural-Network Receptor-Ligand Scoring Function,” Journal of Chemical Information and Modeling, Vol. 51, No. 11, 2011, pp. 2897-2903. http://dx.doi.org/10.1021/ci2003889
- J. A. Read, V. J. Winter, C. M. Eszes, R. B. Sessions and R. L. Brady, “Structural Basis for Altered Activity of Mand H-Isozyme Forms of Human Lactate Dehydrogenase,” Protein Structure, Function, Computation, Genetics, Vol. 43, No. 2, 2001, pp. 175-185. http://dx.doi.org/10.1002/1097-0134(20010501)43:2<175::AID-PROT1029>3.0.CO;2-#
- S. McClendon, D. M. Vu, K. Clinch, R. Callender and R. B. Dyer, “Structural Transformations in the Dynamics of Michaelis Complex Formation in Lactate Dehydrogenase,” Biophysical Journal, Vol. 89, No. 1, 2005, pp. L07-L09. http://dx.doi.org/10.1529/biophysj.105.064675
- L. Qiu, M. Gulotta and R. Callender, “Lactate Dehydrogenase Undergoes a Substantial Structural Change to Bind Its Substrate,” Biophysical Journal, Vol. 93, No. 5, 2007, pp. 1677-1686. http://dx.doi.org/10.1529/biophysj.107.109397
NOTES
*Corresponding author.